The complete genome sequences of Bacillus velezensis B26: a promising biocontrol agent and biofertilizer
Venkatesh Kamath B, Sandeep Mallya, Subrahmanyam Volety Mallikarjuna, Kiran Kumar Kolathur, Diego Sauka, Surajit Basak, Saranya Nallusamy

TL;DR
This paper reports the full genome sequence of Bacillus velezensis B26, a beneficial bacterium that helps control plant diseases and promote growth.
Contribution
The study provides the complete genome sequence of a newly isolated strain of B. velezensis with biocontrol and biofertilizer potential.
Findings
Strain B26 has a 3.95 million base pair genome with 46.3% G+C content.
The genome includes genes related to stress response, nutrient metabolism, and virulence.
The genome data are publicly available for further research and application.
Abstract
Bacillus velezensis is a bacterium widely recognized for its biocontrol properties and ability to promote plant growth. This study presents the whole-genome sequence of B. velezensis B26, a newly identified strain isolated from chicken carcass soil in Udupi, India. The bacterium showed strong activity against fungal pathogens and exhibited diverse enzymatic activities. The whole-genome sequencing was executed using Illumina technologies. Assembly revealed that strain B26 possesses a genome of 3,946,698-bp with a G+C content of 46.3%. Genome annotation identified 3776 protein-coding genes, 1 rRNA gene, 50 tRNA genes, 5 ncRNA genes, and 59 pseudogenes. Functional analysis of the B. velezensis B26 genome revealed 216 genes involved in carbohydrate metabolism, 3 genes in potassium metabolism, 148 genes linked for cofactors, vitamins, prosthetic groups and pigments, 10 genes involved in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
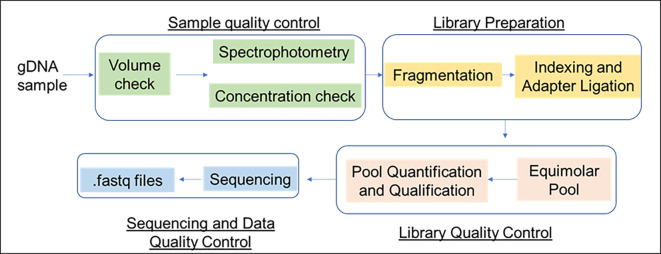 Figure 1
Figure 1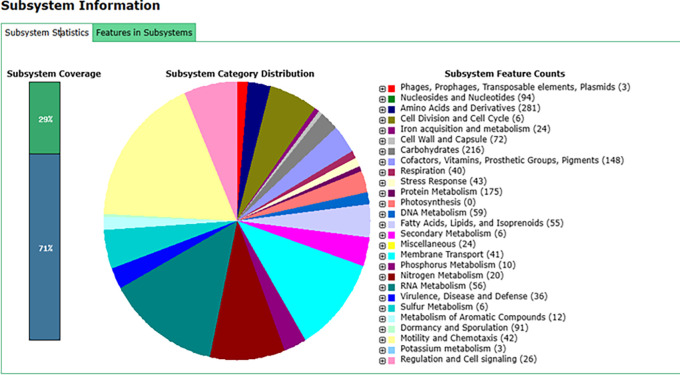 Figure 2
Figure 2| ##Genome-Annotation-Data-START## | |
|---|---|
| Annotation Provider: | NCBI |
| Annotation Date: | 01/04/2024 08:32:37 |
| Annotation Pipeline: | NCBI Prokaryotic Genome Annotation Pipeline (PGAP) |
| Annotation Method: | Best-placed reference protein set; GeneMarkS-2+ |
| Annotation Software revision: | 6.6 |
| Features Annotated: | Gene; CDS; rRNA; tRNA; ncRNA |
| Genes (total): | 3,891 |
| CDSs (total): | 3,835 |
| Genes (coding): | 3,776 |
| CDSs (with protein): | 3,776 |
| Genes (RNA): | 56 |
| rRNAs: | 1 (5S) |
| complete rRNAs: | 1 (5S) |
| tRNAs: | 50 |
| ncRNAs: | 5 |
| Pseudo Genes (total): | 59 |
| CDSs (without protein): | 59 |
| Pseudo Genes (ambiguous residues): | 0 of 59 |
| Pseudo Genes (frameshifted): | 40 of 59 |
| Pseudo Genes (incomplete): | 32 of 59 |
| Pseudo Genes (internal stop): | 8 of 59 |
| Pseudo Genes (multiple problems): | 19 of 59 |
| ##Genome-Annotation-Data-END## | |
| ##Genome-Assembly-Data-START## | |
| Assembly Method: | Megahit v. 2023-07-12 |
| Genome Representation: | Full |
| Expected Final Version: | Yes |
| Genome Coverage: | 100.0x |
| # of Contigs: | 31 |
| # of Proteins: | 3,776 |
| Total length: | 3,947,698 bp |
| BioProject: |
|
| BioSample: |
|
| Keywords: | WGS |
| Annotation: | Contigs |
| Organism: |
|
| Biosource: | /collected_by = Manipal College of Pharmaceutical Sciences (MCOPS)
|
| WGS: |
|
| Reference: | Evaluation of bioactivities of the bacterial strain Bacillus velezensis B26: Unpublished |
| Submission: | Submitted (04-JAN-2024) Pharmaceutical Biotechnology, Manipal College of Pharmaceutical Sciences (MCOPS), MAHE, Madhav Nagar, Manipal, Karnataka 576104, India – Kolathur,K.K. |
| Gene_type | Criteria | Number_of_genes | |
|---|---|---|---|
| 1 | Core genes | (99% <= strains <= 100%) | 3480 |
| 2 | Soft core genes | (95% <= strains < 99%) | 0 |
| 3 | Shell genes | (15% <= strains < 95%) | 1143 |
| 4 | Cloud genes | (0% <= strains < 15%) | 953 |
| 5 | Total genes | (0% <= strains <= 100%) | 5576 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacillus and Francisella bacterial research · Genomics and Phylogenetic Studies · Bacteriophages and microbial interactions
Introduction
Bacillus velezensis is a valuable bacterium that is extensively used for several biotechnological applications to promote plant growth. For instance, the B. velezensis LT1 strain displayed antifungal activity against the soil-borne fungal plant pathogen Sclerotium rolfsii LC1. B. velezensis LT1 secretes a diverse antimicrobial compound that inhibit the growth of S. rolfsii LC1 ( Tang et al. 2024). Likewise, two B. velezensis strains 5YN8 and DSN012 act as biocontrol agents against Botrytis cinerea, a pathogen that causes severe gray mold disease in crops. These strains inhibit spore formation and the growth of B. cinerea through the production of certain secondary metabolites or volatile organic compounds ( Jiang et al. 2018). Several strains of B. velezensis isolated from different sources produce diverse secondary metabolites to control plant pathogens. For example, B. velezensis HNA3 produced several secondary metabolites. Similarly, several antimicrobial metabolites to control plant pathogens were produced by different B. velezensis strains ( Zaid et al. 2022; Rabbee et al. 2023). Besides anti-microbial activity, another B. velezensis SQR9 strain implicated in promoting plant growth by enhancing root colonization through biofilm formation ( Xu et al. 2019). Another strain, B. velezensis Ag75, has been identified as a biocontrol agent, phosphate solubilizer and biofertilizer for maize and soybean crops ( Mosela et al. 2022).
The plant growth promoting and biocontrol capabilities of several B. velezensis strains were deciphered using the genomic sequencing and analysis. These studies were key to understand organism’s capability to produce antimicrobial compounds, enzyme secretion or secondary metabolites that promotes plant growth. For example, whole genome sequencing of B. velezensis HNA3 led to establish several genes clusters associated in promoting plant growth. Among these genes, major percentage of genes are involved in amino acid metabolism, carbohydrate transport, and secondary metabolite biosynthesis (S. et al. 2022). Similarly, the B. velezensis CH1 strain was isolated from high-quality oats, exhibited antimicrobial properties contributing to oat growth and resistance to infections. Comparative analysis of the CH1 strain revealed 13 gene clusters linked with production of 15 secondary metabolites with antimicrobial properties. Furthermore, the strain harboured numerous putative genes for indole acetic acid (IAA) production, spermidine and polyamine synthesis. These results indicate that the possible applications of B. velezensis CH1 as a biofertilizer ( Cheng et al. 2024).
In this study, we report genome sequences of B. velezensis B26, which was isolated from soil samples collected from a chicken carcass, Manipal, Udupi, India (Location: 13.325922, 74.804554). In our screening assay to identify chondroitinase producers, B. velezensis B26 was identified. This strain demonstrated potent antifungal capabilities against the opportunistic fungal pathogen Candida albicans and the emerging fungal pathogen Saccharomyces cerevisiae ( http://doi.org/10.7324/JAPS.2025.200474). This study aims to provide a comprehensive genomic analysis of B. velezensis B26, with a focus on identifying genetic traits that contribute to its biotechnological potential.
Methods
Whole genome analysis of bacteria
The genome of B. velezensis B26 strain was analysed to explore its microbial genomic characteristics. HiMedia Laboratories Pvt Ltd, Maharashtra, INDIA performed the whole-genome analysis.
Sample preparation workflow
Genomic DNA (gDNA) from B. velezensis B26 was isolated using DNA Mini Kit (Qiagen). The integrity of the gDNA was assessed spectrophotometrically to measure the A260/280 ratio and its concentration. For library preparation, 250ng of DNA was processed with the QIASeq FX DNA Kit (Qiagen), following manufacturer’s protocol to generate fragmented, adapter-ligated and indexed libraries. The Illumina NextSeq 550 with 300-cycle paired-end sequencing chemistry was employed for sequencing the finalized library.
Tapestation analysis of NGS libraries
For fragment analysis, 2 μL of high-sensitivity D1000 sample buffer and 2 μL of the final library sample were mixed using vortexer for 1 min. The mixture was then loaded onto the D1000 ScreenTape and analyzed using the Agilent 4200 TapeStation System. This system employs DNA electrophoresis to separate fragments up to 1000 base pairs. The trace analysis revealed an average fragment size of 301 base pairs, which, along with the library concentration, indicates the library’s suitability for next-generation sequencing.
Analysis workflow
In data analysis all reads are checked for quality and then the quality control (QC’)-quality reads are passed through the process flows simultaneously. The detailed steps involved in each process flow are included within each section below.
Reads quality control
Briefly, raw fastq files were verified using FastQc v0.11.9.8 The details of parameters checked are added in a MultiQC report ( Ewels et al. 2016) (Supplementary file 1). The fastp tool (v0.12.4) was utilized to remove adapter contamination from the sequencing data ( Chen et al. 2018). A quality score of Q30 or higher indicates a basecall accuracy of 99.9%, meaning that only 1 in 1000 bases is likely to be incorrect. The resulting files for each read were subjected for genome assembly.
Genome assembly and annotations
A denovo assembly using a De-Brujin graph was performed to organize the short DNA reads into longer contiguous sequences, referred to as contigs. These contigs served as a starting material used for performing a genome annotation, facilitating the assignment of functional roles to various genomic regions of the organism under investigation.
The quality of the genome assemblies produced by the Spades ( Bankevich et al. 2012) and Megahit ( Li et al. 2015) assemblers was evaluated using the Quast tool ( Gurevich et al. 2013). Compared with the megahit assembler, the Spades assembler had longer assembled contigs with better N50 values. Hence, the assembly generated by Spades was selected for further downstream analyses.
RAST Server: Rapid annotations using subsystems technology
The RAST server is an automated platform developed for the bacterial genome annotation. It detects protein-coding regions, along with genes coding for ribosomal and transfer RNAs, allots functional roles to these elements ( Aziz et al. 2008). Additionally, it allows comparative analysis through the SEED environment ( Overbeek et al. 2014). The RAST server remains a viable tool for efficient and reliable genome annotation.
Results
Whole-genome analysis of
B. velezensis B26
We performed a genome analysis of B. velezensis B26 to understand the biochemical potential of the organism. Figure. 1 depicts the sample preparation and sequencing workflow. Importantly, the assembled genomes may not be 100% complete. However, the assembly generated here meets the standards acceptable to general bioinformatics repositories as part of the data gathered for a publication. These whole-genome shotgun data have been provided with the accession number JAYKOV000000000 (NCBI database). The master record data is available from our various Entrez servers. The individual sequences are available from a hyperlink at the bottom of the WGS master record JAYKOV000000000.
Sample preparation and sequencing workflow.
The genome assembly method employed was Megahit v. 2023-07-12 tool, the genome representation was full, the genome coverage was 100.0x and the sequencing technology employed was Illumina. Genome annotation was carried out using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) ( Haft et al. 2018; Li et al. 2021). The annotation results are summarized in Table 1, which provides an overview of the genome features and information. Table 2 summarizes key assembly statistics, including the number of contigs, total length of the sequenced genome, and the number of proteins annotated.
Table 1.: Genome annotation data for Bacillus velezensis B26.
Table 2.: Assembly statistics for B. velezensis B26.
Identification of subsystem features by The RAST Server: Rapid Annotations using Subsystems Technology (RAST)
In addition to utilizing the NCBI PGAP, we also submitted the sequenced genome data of B. velezensis B26 to the RAST server. This service provides fully automated annotation for bacterial genomes. Using the RAST annotation platform, we recognized genes coding for proteins, ribosomal and transfer RNAs, assigned functional roles to these genes, and predicted the subsystems present in the B. velezensis B26 genome (Supplementary Table 1). Furthermore, the annotated genome is presented on a platform that allows for comparative analysis in the SEED environment ( Figure 2).
Subsystem statistics of the B. velezensis B26 genome annotations.The genome of B. velezensis B26 was annotated using the RAST server, and the features in subsystem are compared within the SEED environment. The figure highlights key subsystems identified in the genome, including genes associated with carbohydrate metabolism, stress response, virulence, disease and defense, as well as genes involved in metabolism.
Our analysis revealed the presence of 216 genes involved in carbohydrate metabolism, 3 genes related to phages, prophages, transposable elements, and plasmids, 43 genes for stress response, and 35 genes linked to virulence, disease and defense. The genes related to the stress response and virulence might be responsible for the biocontrol properties of B. velezensis B26 (Supplementary Table 2).
Additionally, several genes involved in metabolic processes were identified, including 59 genes related to DNA metabolism, 10 related to phosphorus metabolism, 20 related to nitrogen metabolism, 6 related to sulfur metabolism, 12 related to metabolism of aromatic compounds, 3 related to potassium metabolism, 10 related to phosphorus metabolism, and 24 related to acquisition and metabolism. These metabolic pathways suggest potential biofertilizer activity (Supplementary Table 2).
B. velezensis B26 pangenome analysis
Pangenome analysis provides valuable understandings into the genome of prokaryotes. Using Roary software, a large-scale pangenomes are constructed by identifying both core and accessory genes. This method aids in understanding the conserved genes within an organism, as well as accessory genome, to understand the fundamentals linked with natural selection and evolutionary dynamics.
Pangenome analysis of B. velezensis B26 identified a total of 5,576 genes. Among these genes, 3,480 genes were considered core genes and were found in 99% to 100% of strains. A total of 1, 143 genes were classified as shell genes, which were present in 15% to 95% of strains, whereas 953 genes were identified as cloud genes, which were found in fewer than 15% of strains ( Table 3). These analyses indicate that the major portion of the genome is conserved across species.
Table 3.: Pangenome analysis of B. velezensis B26.
Conclusion
The whole-genome analysis, genome annotation, and pangenome analysis of B. velezensis B26 deciphered the key genes responsible for its biotechnological applications. A huge number of protein coding genes (3776) responsible for several cellular functions were identified. Importantly, presence of gene cluster coding for stress response, virulence, and defense indicate that B. velezensis B26 can effectively inhibit plant pathogens. Further, presences of genes associated with nitrogen, phosphorus, and sulfur metabolism might suggest the strain’s ability to act as a biofertilizer, supporting plant growth. Altogether, these findings can pave the way for further exploration of B. velezensis B26 in agricultural applications.
Ethics and consent
Ethical approval and consent were not required.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aziz RK Bartels D Best AA : The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. 10.1186/1471-2164-9-75 18261238 PMC 2265698 · doi ↗ · pubmed ↗
- 2Bankevich A Nurk S Antipov D : SP Ades: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012;19:455–477. 10.1089/cmb.2012.0021 22506599 PMC 3342519 · doi ↗ · pubmed ↗
- 3Chen S Zhou Y Chen Y : fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i 884–i 890. 10.1093/bioinformatics/bty 560 30423086 PMC 6129281 · doi ↗ · pubmed ↗
- 4Cheng C Su S Bo S : A Bacillus velezensis strain isolated from oats with disease-preventing and growth-promoting properties. Sci. Rep. 2024;14:12950. 10.1038/s 41598-024-63756-8 38839805 PMC 11153497 · doi ↗ · pubmed ↗
- 5Ewels P Magnusson M Lundin S : Multi QC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. 10.1093/bioinformatics/btw 354 27312411 PMC 5039924 · doi ↗ · pubmed ↗
- 6Gurevich A Saveliev V Vyahhi N : QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. 10.1093/bioinformatics/btt 086 23422339 PMC 3624806 · doi ↗ · pubmed ↗
- 7Haft DH Di Cuccio M Badretdin A : Ref Seq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018;46:D 851–D 860. 10.1093/nar/gkx 1068 29112715 PMC 5753331 · doi ↗ · pubmed ↗
- 8Jiang C-H Liao M-J Wang H-K : Bacillus velezensis, a potential and efficient biocontrol agent in control of pepper gray mold caused by Botrytis cinerea. Biol. Control. 2018;126:147–157. 10.1016/j.biocontrol.2018.07.017 · doi ↗