β‑Selective Addition of Pyrroles to Electron-Deficient Alkenes in Both Catalytic and Stoichiometric Modes on B(C6F5)3
Seina Sekine, Miho Kashiwa, Maho Kawakami, Takumi Sonoda, Arisa Ono, Teruhisa Tsuchimoto

TL;DR
This paper reports a new method for adding pyrroles to electron-deficient alkenes at the beta position using B(C6F5)3.
Contribution
The study introduces a novel electrophilic aromatic substitution system for beta-selective addition of pyrroles.
Findings
A beta-adduct was successfully formed using B(C6F5)3 in both catalytic and stoichiometric modes.
The reaction system enables selective addition at the beta-position of pyrroles.
This contrasts with previous reports that only alpha-adducts were formed.
Abstract
To the best of our knowledge, the addition of pyrroles to electron-deficient alkenes (APEda) via electrophilic aromatic substitution (SEAr) has been reported to occur exclusively at the α-position of the pyrrole without any formation, even in trace amounts, of a β-adduct, β-3. In sharp contrast to the prolonged immutable observation, we established an original SEAr-based APEda (SEAr-APEda) system applicable to both the catalytic and stoichiometric synthesis of β-3.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1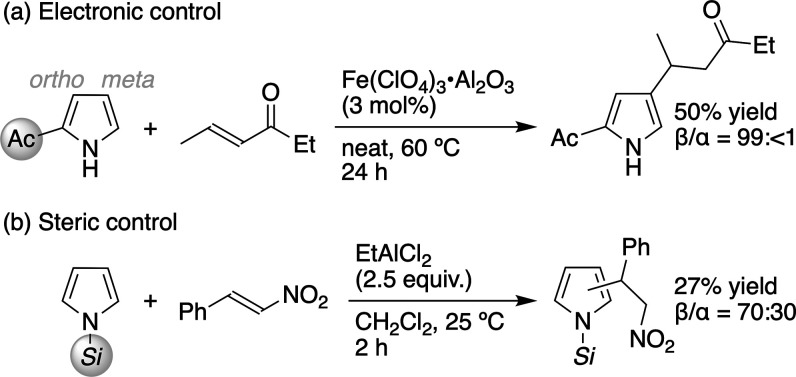 1
1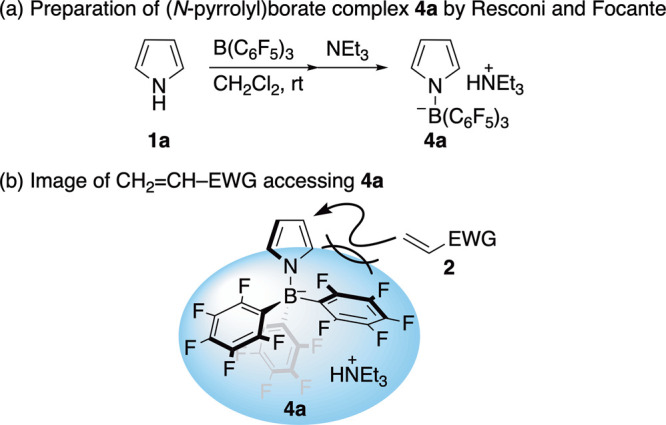 2
2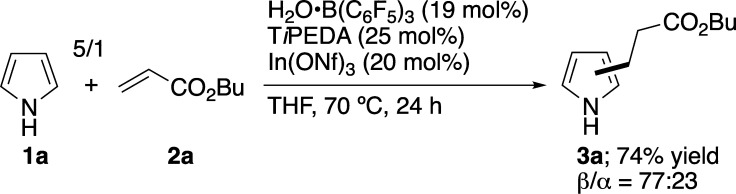 3
3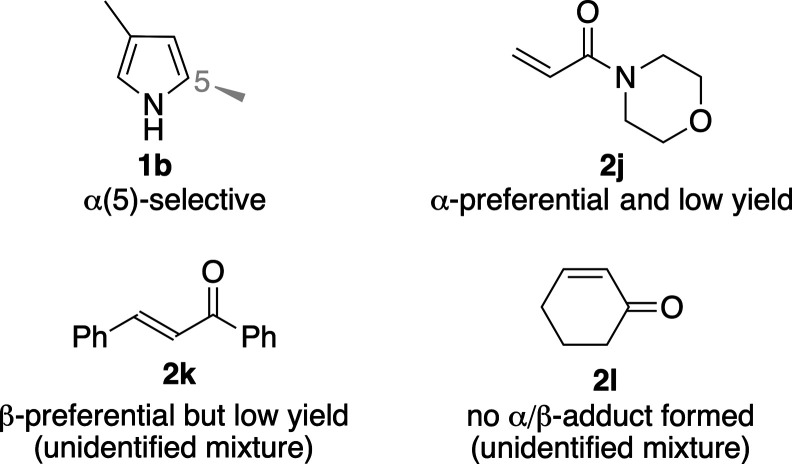 2
2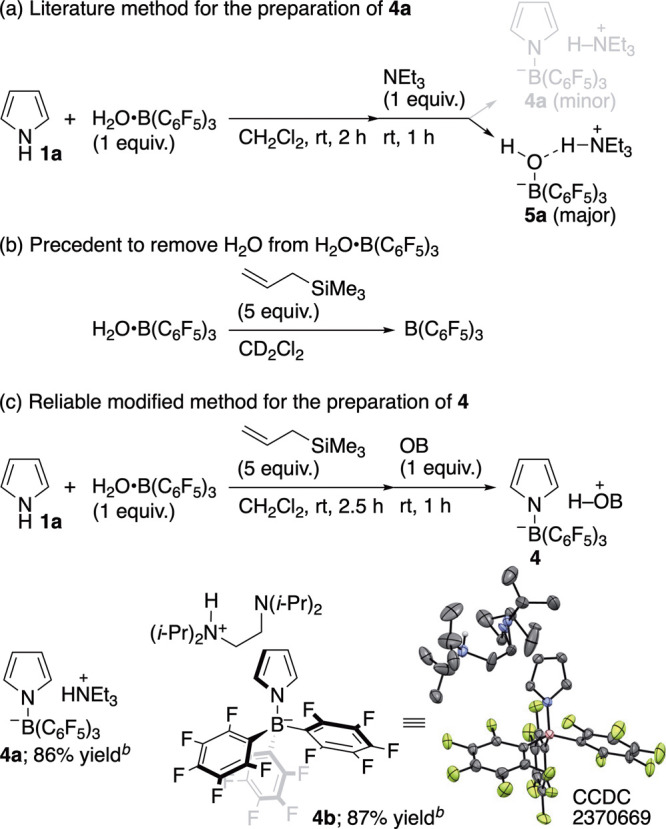 4
4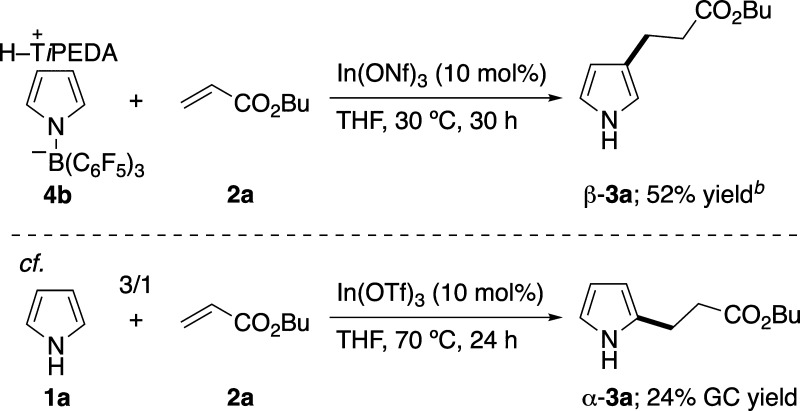 5
5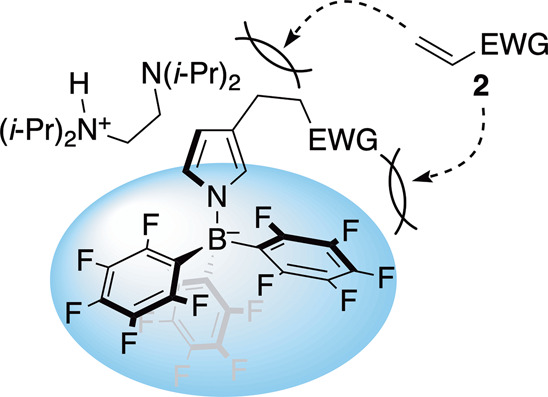 3
3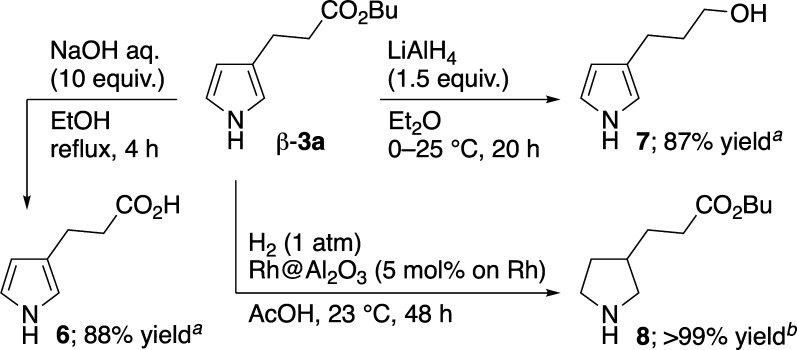 6
6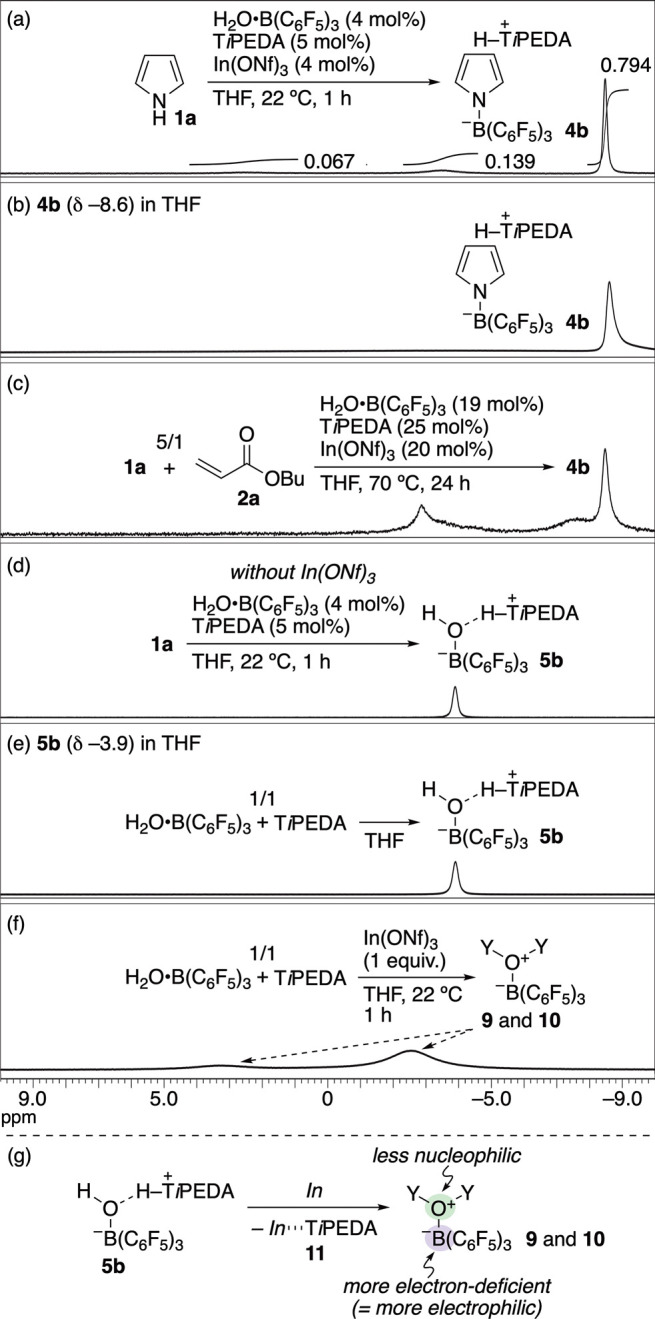 7
7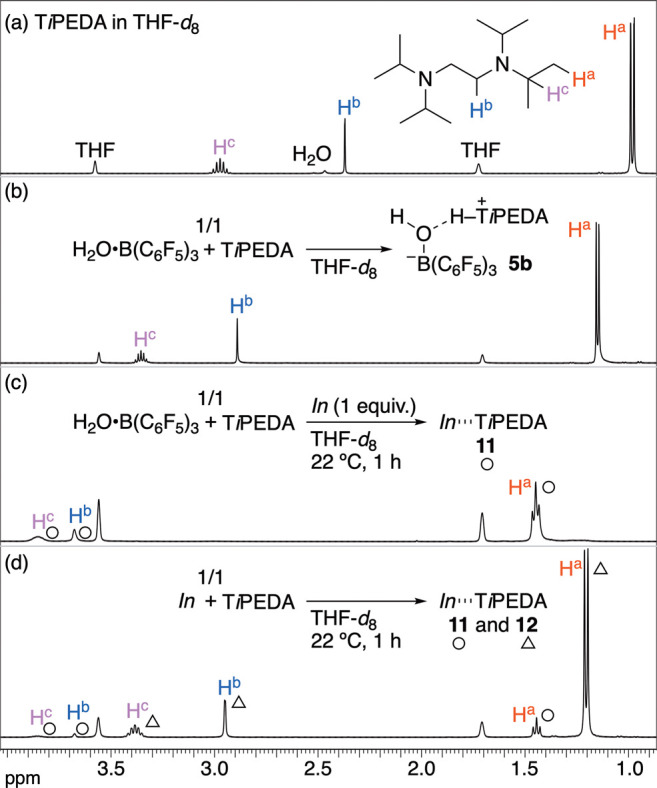 8
8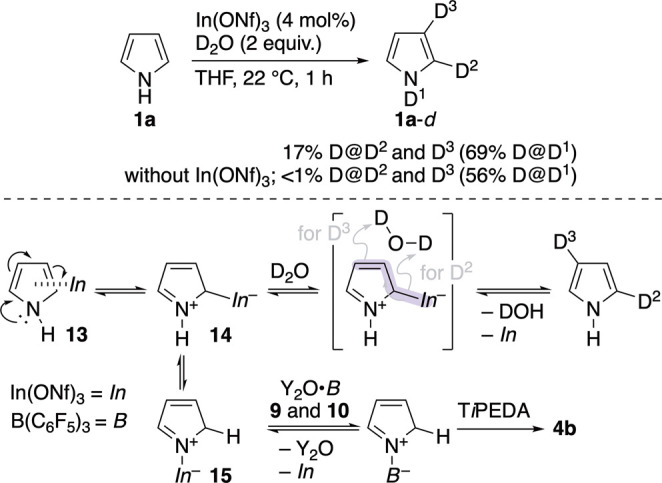 9
9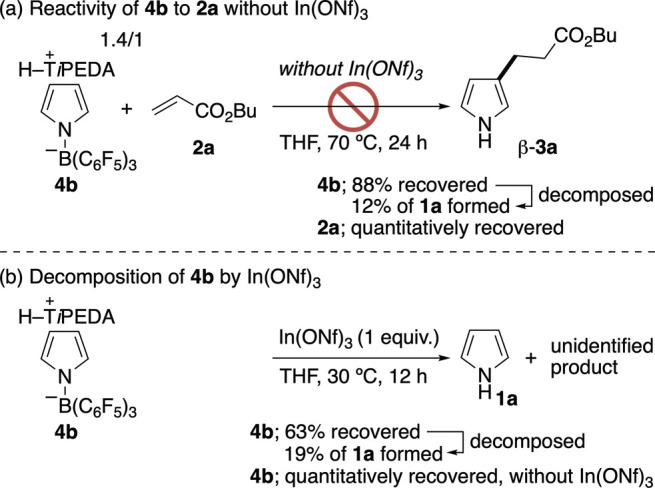 10
10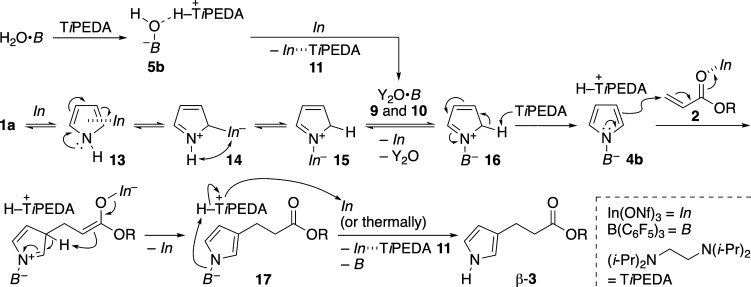 11
11- —Meiji University10.13039/100014424
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFluorine in Organic Chemistry · Chemical Synthesis and Analysis · Asymmetric Synthesis and Catalysis
Introduction
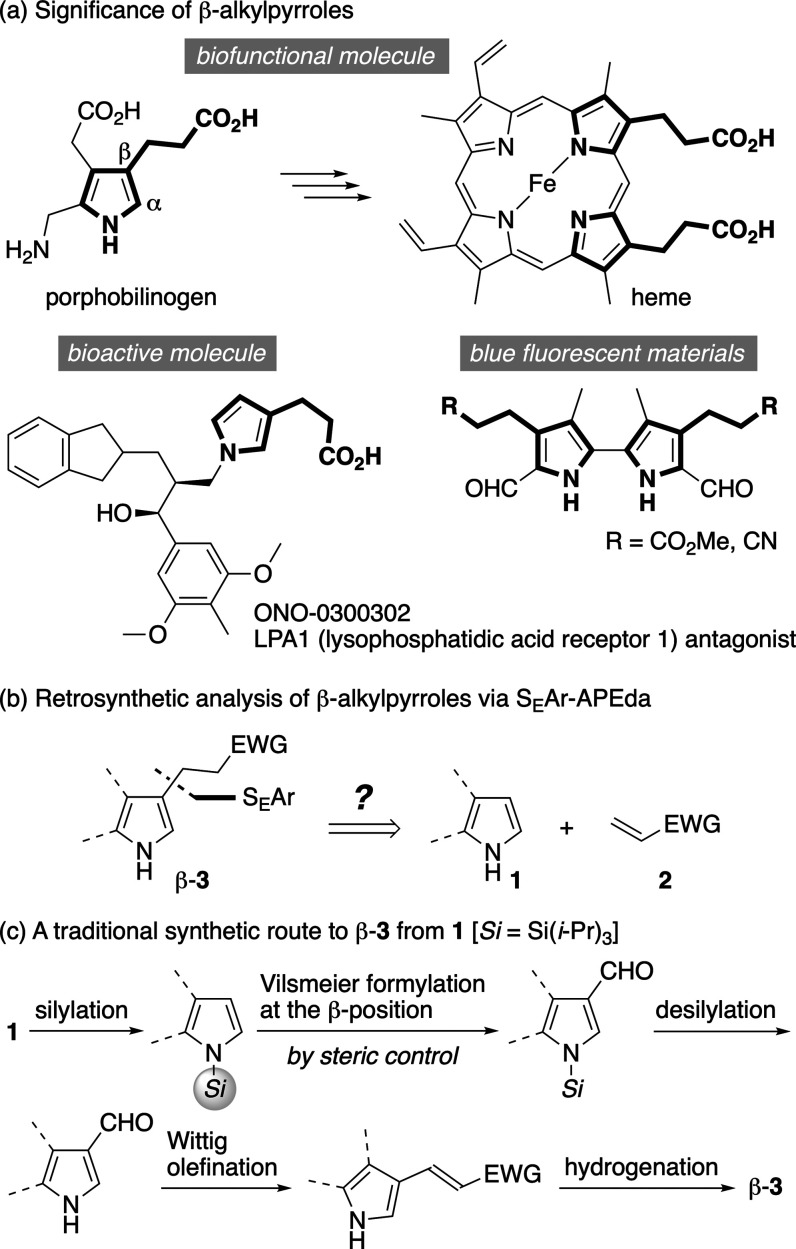
β-Alkylpyrroles, wherein a β-pyrrolyl group is tethered by a two-carbon chain to an electron-withdrawing group (EWG), are crucial units of biofunctional? and bioactive? molecules, as well as optoelectronic materials (Figurea).? This structural motif is hence an attractive synthetic target. Retrosynthetic analysis suggests that the S_E_Ar-APEda should be a straightforward route for obtaining β-alkylpyrroles β-3 in a single step (Figureb). However, due to the inherent α-orientation of pyrroles 1 in the S_E_Ar process, access of electrophiles to the β-position of 1 cannot be a major route.? In fact, to the best of our knowledge, an exclusive α-addition has been observed in the preceding S_E_Ar-APEda, and even contamination by β-3 has thus yet to be observed.? Importantly, to access β-3 from 1 by a traditional strategy, multiple steps have been necessary (Figurec). ?,? Despite the strict α-orientation of 1 in the S_E_Ar-APEda, two strategies for the β-addition controlled electronically and sterically by a preinstalled substituent onto the pyrrole ring have been reported, albeit as only two specific reactions: One uses the pyrrole with the acetyl (Ac) group at the α-position (Schemea),? which can be regarded as a so-called meta-directing EWG in a S_E_Ar reaction.? The other uses the pyrrole with the bulky N-Si(i-Pr)3 (Si) group (Schemeb).? The substrate scope of these strategies is unclear. The precedents in references ? ? ? ? ?–? show that directing-group-free β-selective S_E_Ar-APEda of 1 is undoubtedly challenging.
β-Alkylpyrroles: (a) significance and (b) retrosynthetic analysis.
Precedents on β-Selective SEAr-APEda Using Substituted Pyrroles for Electronic and Steric Control
On the other hand, Resconi, Focante and colleagues have reported that treating pyrrole (1a) with B(C_6_F_5_)3 and NEt_3_ at room temperature (rt) yielded (N-pyrrolyl)borate complex 4a (Schemea).? 4a was attractive to us in that its C_6_F_5_ units could efficiently cover its α-position from the attack of external electrophiles such as CH_2_CH–EWG 2 (Schemeb). We envisaged that 4, formed catalytically in situ, could lead to the β-selective S_E_Ar-APEda and that this will be the first example of the direct synthesis of β-3 from a simple pyrrole like 1a via the S_E_Ar-APEda. Besides the catalytic reaction, we examined how 4 behaves as a stoichiometric reagent. Herein, we report the β-selective S_E_Ar-APEda in both catalytic and stoichiometric modes on B(C_6_F_5_)3. NOTE: During this research, we found that B(C_6_F_5_)3 stored in a sample bottle forms a complex with water. Therefore, the chemical formula is represented as H_2_O**·**B(C_6_F_5_)3 hereafter.?
(N-Pyrrolyl)borate Complex 4a: (a) Preparation and (b) Image of 4a + CH2CH–EWG
Results and Discussion
This study commenced with investigating the reaction of 1a (3 equiv) with butyl acrylate (2a). After careful examination based on our insights gained through the development of a series of Lewis-acid-catalyzed reactions of heteroarenes,? we found the following important aspects and reaction conditions: Boron, organic base (OB), and indium compounds were all required as catalysts to obtain desired product β-3a; if even one was absent, the reaction failed to proceed or yielded only α-3a. As a result, we developed the reaction conditions summarized in Scheme ? [see the Supporting Information (SI) for detailed experimental results with different reaction conditions]. Thus, the reaction of 1a (5 equiv) with 2a in THF at 70 °C for 24 h in the presence of H_2_O**·**B(C_6_F_5_)3 (19 mol %), ?,? TiPEDA (25 mol %), and In(ONf)3 (20 mol %) delivered 3a in a good yield of 74% with 77% β-selectivity. Notably, this is the first achievement in which a simple pyrrole without a preinstalled β-directing group undergoes the S_E_Ar-APEda reaction to yield a β-adduct and, furthermore, to yield the β-adduct preferentially over the α-adduct.
Reaction Conditions for Catalytic Reaction
We investigated the scope of the catalytic reaction on H_2_O**·**B(C_6_F_5_)3 using the reaction conditions in Scheme. The results are summarized in Table, where the results of both before and after the purification of 3 are presented, and regioisomers of α-3 and β-3 were successfully separated and characterized in all reactions. Similarly to that of 2a, the reactions of alkyl acrylates 2b–2e proceeded with β-selectivities of over 70%, indicating that the length and size of R^3^ does not significantly affect the β-selectivity (3a–3e). The yield and selectivity before and after the purification process varied depending on the difficulty in isolating β-3 from a crude product including α-3 and a regioisomeric mixture of 1/2 adducts of 1 and 2. Cyclic ester 2f also led to β-preferential addition (3f). Besides esters 2a–2f, acrylonitrile (2g) and N-phenylmaleimide (2h) afforded the corresponding β-adducts 3g and 3h in moderate yields. Other than the reaction of 2h, 1/2 adducts of 1 and 2 were formed as major byproducts, directly affecting the yields of β-3. Unlike the findings of Table, usage of 3-methylpyrrole (1b), N-acryloylmorpholine (2j), (E)-chalcone (2k), and 2-cyclohexen-1-one (2l) as the starting substrates yielded poor results (Figure): The reason is stated below each substrate.
1: Substrate Scope of Catalytic Reaction Favoring β-Alkylpyrroles
Unsuccessful substrates in catalytic reaction.
Scaling up the catalytic mode was feasible, albeit with somewhat reduced yield and β-selectivity of 3. For example, a 5.00 mmol scale reaction of 2g with 1a (25.0 mmol) delivered 3g in 58% yield (348 mg) with 57% β-selectivity.
Historically, the typical S_E_Ar-APEda reaction has exclusively yielded α-adducts, as already stated. Hence, it is necessary to uncover why β-3 is formed in the current ternary system consisting of H_2_O**·B(C_6_F_5_)3, an organic base (e.g., TiPEDA), and In(ONf)3. To confirm whether, as initially expected, (N-pyrrolyl)borate complex 4 is an active species for delivering β-3, we aimed to construct 4 in preparation for a control experiment with 2. We first attempted to prepare the known borate complex 4a from 1a, H_2_O·**B(C_6_F_5_)3, and NEt_3_, following a reported procedure.? However, the reaction failed, mainly yielding [(F_5_C_6_)3_BOH]^−^HN^+^Et_3 (5a) (Schemea).? We considered that 4a would not be obtained unless H_2_O coordinating to B(C_6_F_5_)3 is removed. Allyltrimethylsilane is reported to be a reagent for removing the H_2_O, as shown in Schemeb.? We therefore attempted to prepare 4a by conducting the first step in the presence of allyltrimethylsilane. As expected, 4a was isolated in 86% yield after recrystallization from hexane/CH_2_Cl_2_ (Schemec). The modified recipe was reproducible and reliably applied to the synthesis of TiPEDA-based (N-pyrrolyl)borate complex 4b. The molecular structure of 4b was confirmed by single-crystal X-ray diffraction analysis.
Preparation of (N-Pyrrolyl)borate Complexes
Having successfully obtained 4b, a stoichiometric control experiment was performed with 2a, along with catalyst In(ONf)3 (Scheme). Compared to the catalytic reaction on H_2_O**·**B(C_6_F_5_)3 (Scheme), this reaction proceeded smoothly with a lower loading of In(ONf)3 (10 mol %) and at a much lower reaction temperature of 30 °C. Moreover, only β-3a was produced as a single regioisomer.? By contrast, the indium-catalyzed S_E_Ar-APEda of 1a instead of 4b gave only α-3a as a single regioisomer (Scheme). These results are entirely opposite, clearly suggesting that (N-pyrrolyl)borate complex 4 is a crucial active species in the formation of β-3.
Stoichiometric Control Experiment of TiPEDA-Based (N-Pyrrolyl)borate Complex with CH2CHCO2Bu under Indium Catalysis
As already presented in Schemeb, sterically controlled S_E_Ar-APEda has been reported by utilizing the N-Si(i-Pr)3 group as a bulky β-directing group.? However, its β-selectivity is insufficient, promoter EtAlCl_2_ is necessary in more than a stoichiometric amount, and its scope remains unclear. In contrast to this precedent, the reaction with stoichiometric boron but catalytic indium in Scheme occurred with complete β-selectivity. Inspired by this result as the first example of the sterically controlled β-exclusive S_E_Ar-APEda, we explored the scope of the stoichiometric reaction (Table). First, N-phenylmaleimide (2h) added exclusively at the β-position of 4b, giving β-3h in a much higher yield than that of the corresponding catalytic reaction. N-Methylmaleimide (2i) similarly furnished only β-3i. However, using noncyclic amide 2j resulted in a low yield of β-3j, which could not be isolated in pure form (see the SI for details). In contrast to their incompatibility with the catalytic mode (Figure), (E)-chalcone (2k) and 2-cyclohexen-1-one (2l) worked well, regioselectively giving β-3k and β-3l, respectively. Since keto groups easily react with pyrroles under indium catalysis,? the compatibility of the ketone substrates is noteworthy. trans-β-Nitrostyrene (2m) also gave β-3m exclusively in 87% yield. Unlike the other Michael acceptors listed in Table, the use of acrylonitrile (2g) led to 3g with a similar β/α ratio as that in the catalytic reaction, albeit in a much higher yield. The stoichiometric reaction is also applicable to other (N-pyrrolyl)borate complexes 4c and 4d, derived from 3-methyl- and 2-phenylpyrroles, respectively. Remarkably, even 4c,? in which the β-site of the pyrrole ring is premethylated, reacted with 2i at the sterically congested β-position to give β-3n. Exclusive β-addition was also observed when using 4d, giving a mixture of 2,3- and 2,4-disubstituted pyrroles 3o in high yields.
2: Substrate Scope of Stoichiometric Reaction
In a S_E_Ar alkylation, a large excess of (hetero)arenes to alkyl electrophiles is generally used to ensure a high yield of the monoalkylation product, as the reaction otherwise results in multialkylation. ?,? Despite this inherent issue, a large excess of 4 to 2 is unnecessary in the stoichiometric reaction, being likely attributed to the good stability of a β-3-based (N-pyrrolyl)borate complex under the reaction conditions (see the SI for details). This is because multialkylation of the complex seems unfavorable, likely due to the steric congestion around the complex, as shown in Figure. In fact, no multialkylation products were observed in the stoichiometric reactions of Table, except when using 2a (see the SI for further details).
Image of restricted multialkylation in stoichiometric reaction.
The present catalytic S_E_Ar-APEda reaction is the first example of a one-step synthesis of β-3 from pyrrole. Among the products obtained in Table, only the synthetic method for β-3a has been disclosed in patents, albeit in three steps. ?,? β-3f and β-3g are known compounds, but there are no reports on their preparation. Meanwhile, β-3b–β-3e and β-3h are thus far unknown. Accordingly, the accessibility of β-3 in our strategy encouraged us to synthesize other compounds that can be obtained only from β-3. For instance, carboxylic acid 6, alcohol 7, and pyrrolidine 8 ? were obtained from β-3a via simple transformations (Scheme).
Transformation of β-3a
To gain insight into the mechanism of the catalytic reaction on H_2_O**·B(C_6_F_5_)3, several experiments were conducted. First, ^11^B NMR experiments were performed to verify whether 4b could be formed in situ during the catalytic reaction (Scheme). Upon treating 1a with catalytic amounts of H_2_O·**B(C_6_F_5_)3, TiPEDA, and In(ONf)3 in THF, 4b was the main boron-containing product (79%) even at a low reaction temperature of 22 °C (Schemea); The ^11^B NMR spectrum of 4b in THF is given in Schemeb. 4b was also present in the crude reaction mixture after the catalytic reaction (Schemec). Importantly, In(ONf)3 was indispensable for yielding 4b (Schemea vs 7d). The ^11^B NMR peak observed at – 3.9 ppm in Schemed is likely ascribed to [(F_5_C_6_)_3_BOH]^−^(HTiPEDA)^+^ (5b) in which no 1a is contained (Schemed vs 7e). These results confirm that 4b is formed in situ during the catalytic reaction.
11B NMR Analysis for Validation of Formation of (N-Pyrrolyl)borate Complex 4b in Catalytic Reaction
Based on the results in Schemed, the role of In(ONf)3 in the formation of 4b was examined. Adding In(ONf)3 to the THF solution of 5b caused the appearance of the two downfield-shifted broad signals at 3.6 and – 2.6 ppm (Schemef). While the structural assignments of these signals are unclear,? this result reveals that, compared to 5b coordinated by a nucleophilicity-enhanced oxygen atom due to deprotonation by TiPEDA, more electron-deficient B(sp ^3^)-hybridized species 9 and 10 coordinated by less nucleophilic oxygen atoms were formed.? It was assumed that this spectral change might be due to the liberation of TiPEDA from 5b owing to the coordination of TiPEDA to In(ONf)3 (In) to give 11, as proposed in Schemeg.
To verify the above hypothesis, we continuously monitored the behavior of TiPEDA in THF-d 8 by ^1^H NMR spectroscopy (Scheme). The ^1^H NMR spectrum of 5b is shown in Schemeb, where all proton signals of H^a^, H^b^ and H^c^ were shifted downfield, compared to the original H^a–c^ signals of TiPEDA (Schemea vs 8b). Adding In to the solution of 5b gave one TiPEDA-based species and led to a further downfield shift in the H^a–c^ signals (Schemec). This result reveals the generation of distinct species (○) that should be formed by the migration of TiPEDA from mild acid 5b to stronger acid In and is well consistent with the result of Schemef as well as the assumption of Schemeg. A control experiment by mixing In and TiPEDA also yielded the same species (○), proposing that the structure is the complex of In and TiPEDA: In···TiPEDA (11), while giving another In···TiPEDA species 12 (△) (Schemed). Therefore, all the NMR studies support that In works to convert 5b to more electrophilic boron species 9 and 10, thereby probably inducing the generation of 4b in the presence of 1a.
1H NMR Analysis for Exploration of Role of In(ONf)3
Based on our insights on the indium–heteroarene π-complex, ?,?–? ?,?,?,?,? we predicted that the coordination of 1a to In(ONf)3 may be another trigger for the formation of 4b. Hence, 1a was exposed to catalyst In(ONf)3 and D_2_O in THF at 22 °C for 1 h, giving 1a-d with 17% D at both the C2 and C3 positions (Scheme). By contrast, no C2/3-deuterations occurred without In(ONf)3 (In). Note that the H–D exchange on the nitrogen atom of 1a occurred even in the absence of In. A possible route for the observed C2/3-deuterations is proposed in Scheme, which refers to some precedents regarding: (i) the In–heteroarene π-complex, (ii) the α-nucleophilicity of pyrroles,? (iii) the reaction of pyrroles with Lewis acids, ?,?,? and (iv) the deuteration of allylindium species (highlighted in pale purple). ?,? Moreover, the proposed route may be extended to the route leading to 4b, as shown in Scheme. Thus, In activates 1a via the formation of π-complex 13 and then reacts with 1a at the more nucleophilic α-site to yield 14, followed by isomerization to 15. The nitrogen atom of 15 should be more nucleophilic than that of 1a and be able to attack the boron center of Y_2_O**·** B species 9 and 10 (Schemesf and ?g), followed by the deprotonation of the C(2)–H by TiPEDA to provide 4b. This route fits well with the proposed route for the C2/3-deuterations of 1a, similarly starting with the activation of 1a by In.
Deuteration of Pyrrole by In(ONf)3 and Its Possible Route
Lastly, we examined the reactivity of 4b and found that, without In(ONf)3 at 70 °C for 24 h, 4b itself does not have nucleophilicity to add to 2a (Schemea). Meanwhile, part of 4b was thermally decomposed into pyrrole (1a) (12%), and 2a was quantitatively recovered. Since 4b adds to 2a even at 30 °C in the presence of In(ONf)3 (Scheme), it is considered that In(ONf)3 would act to increase the electrophilicity of 2a, as previously reported. ?,? As presented in Schemeb, the decomposition of 4b was promoted by In(ONf)3 (1 equiv) at 30 °C. This result proposes that In(ONf)3 also affects the decomposition of a β-3-based (N-pyrrolyl)borate complex generated during the reaction, thereby regenerating B(C_6_F_5_)3. This becomes one of the roles of the indium catalyst in the catalytic reaction.
Reactivity and Decomposition of 4b
Summarizing the results in Schemes–?, the indium salt is currently thought to uniquely have four roles in the catalytic reaction: activation of 5b, activation of 1a, activation of 2a, and decomposition of (N-pyrrolyl)borate complexes.
Considering all the above experimental results and key references, we propose a catalytic reaction mechanism, exemplified by the reaction of 1a with acrylic acid ester 2 (Scheme). To begin with, 1a π-coordinates to In to form 13 ? and then reacts with In at the α-position to afford 14, due to the inherent α-nucleophilicity of pyrroles.? Intermediate 14 bearing the C–metal bond isomerizes to more stable species 15 with the C–H bond.^10,16,28^ In another route, 5b is generated from the reaction of H_2_O**·** B with TiPEDA and then reacts with In to furnish more electrophilic boron species Y_2_O**·** B 9 and 10. In in 15 can thus be replaced with B of 9 and 10 to yield 16. The C(2)–H of 16 is then deprotonated by TiPEDA for the aromatization, giving (N-pyrrolyl)borate complex 4b. ?,? Because of the effective steric shield around its α-site, key intermediate 4b adds at its β-position to 2 activated by In via S_E_Ar. The resulting (N-pyrrolyl)borate complex 17 decomposes thermally and/or by the action of In, producing β-3 and regenerating TiPEDA and B. At this final stage, a proton (H^+^) arising from the complexation between In and TiPEDA may effectively protonate the N–B ^–^ bond in 17 to form β-3.
Proposed Catalytic Reaction Mechanism
Conclusions
In summary, we disclosed the first example of catalytically proceeding β-preferential S_E_Ar-APEda using the ternary system of B(C_6_F_5_)3/TiPEDA/In(ONf)3 whereas the pyrrole substrate was limited to pyrrole (1a). Unlike the catalytic reaction, when using stoichiometric (N-pyrrolyl)borate complex 4, pyrrole substrates other than 1a were available. A range of electron-deficient alkenes 2 could be also used. The complete β-selectivity is particularly noteworthy. This stoichiometric mode can be achieved using a catalytic amount of an indium salt under mild, room-temperature reaction conditions. The mechanistic studies of the catalytic reaction revealed that 4 is a key intermediate, and, interestingly, the indium catalyst plays multiple roles.
Due to the moderate β-selectivity and limited substrate scope in the catalytic reaction using the β-directing-group-free pyrrole, their improvements, possibly by applying a distinct method or strategy, continue to be a significant challenge for future research.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Layer G.Reichelt J.Jahn D.Heinz D. W.Structure and Function of Enzymes in Heme Biosynthesis Protein Sci.2010191137116110.1002/pro.40520506125 PMC 2895239 · doi ↗ · pubmed ↗
- 2Terakado M.Suzuki H.Hashimura K.Tanaka M.Ueda H.Hirai K.Asada M.Ikura M.Matsunaga N.Saga H.Shinozaki K.Karakawa N.Takada Y.Minami M.Egashira H.Sugiura Y.Yamada M.Nakade S.Takaoka Y.Discovery of a Slow Tight Binding LPA 1 Antagonist (ONO-0300302) for the Treatment of Benign Prostatic Hyperplasia ACS Med. Chem. Lett.201781281128610.1021/acsmedchemlett.7b 0038329259748 PMC 5733272 · doi ↗ · pubmed ↗
- 3Jiao L.Hao E.Vicente M. G. H.Smith K. M.Improved Synthesis of Functionalized 2,2’-Bipyrroles J. Org. Chem.2007728119812210.1021/jo 701310 k 17887702 · doi ↗ · pubmed ↗
- 4a Joule, J. A. ; Mills, K. Heterocyclic Chemistry, 5th ed.; Blackwell: West Sussex, U.K., 2010; pp 295–323 (Chapter 16).
- 5a Paras N. A.Mac Millan D. W. C.New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel–Crafts Alkylation J. Am. Chem. Soc.20011234370437110.1021/ja 015717 g 11457218 · doi ↗ · pubmed ↗
- 6Outlaw V. K.Townsend C. A.A Practical Route to Substituted 7-Aminoindoles from Pyrrole-3-carboxaldehydes Org. Lett.2014166334633710.1021/ol 503078 h 25479249 PMC 4275132 · doi ↗ · pubmed ↗
- 7Kawatsura M.Fujiwara M.Uehara H.Nomura S.Hayase S.Itoh T.Iron Salt-catalyzed Multipoint Alkylation of Pyrrole with Vinyl Ketones Chem. Lett.20083779479510.1246/cl.2008.794 · doi ↗
- 8Smith, M. B. March’s Advanced Organic ChemistryReactions, Mechanisms, and Structure 7th ed.; Wiley: Hoboken, 2013; pp 665–670.