Case Report: Hereditary spastic paraplegia associated with monoallelic variant in the motor domain of KIF1A
Kathryn Sine, David Brodie-Mends, Wafae Chouhani, Lauren Massingham, Saud Alhusaini

TL;DR
A father and son with similar neurological symptoms were found to have a genetic variant in the KIF1A gene, causing hereditary spastic paraplegia.
Contribution
Reports a novel monoallelic variant in KIF1A associated with autosomal dominant hereditary spastic paraplegia.
Findings
A heterozygous variant in KIF1A (p.Arg316Gln) was identified in a father and son with spastic paraplegia.
The variant is likely pathogenic and associated with a slowly progressive HSP phenotype.
The findings expand the genetic spectrum of KIF1A-related neurological disorders.
Abstract
To investigate the genetic etiology of a familial case with spastic paraplegia. Neurological examination, clinical and genetic work-up, including exome sequencing (ES), followed by targeted testing, were performed to determine the underlying etiology of the patients’ phenotype. A 45-year-old man was initially diagnosed with spastic diplegic cerebral palsy in early childhood. He underwent multiple orthopedic interventions for lower extremities spasticity and progressive gait disturbance. His son developed similar neurological symptoms at 2-years of age. Despite unremarkable initial work-up, their relatively similar slowly progressive phenotype was suggestive of hereditary spastic paraplegia (HSP). ES was performed for the son at age 11 years, followed by cascade single testing for the father, which revealed a heterozygous (monoallelic) likely pathogenic variant [NM_001244008.2: c.947G…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
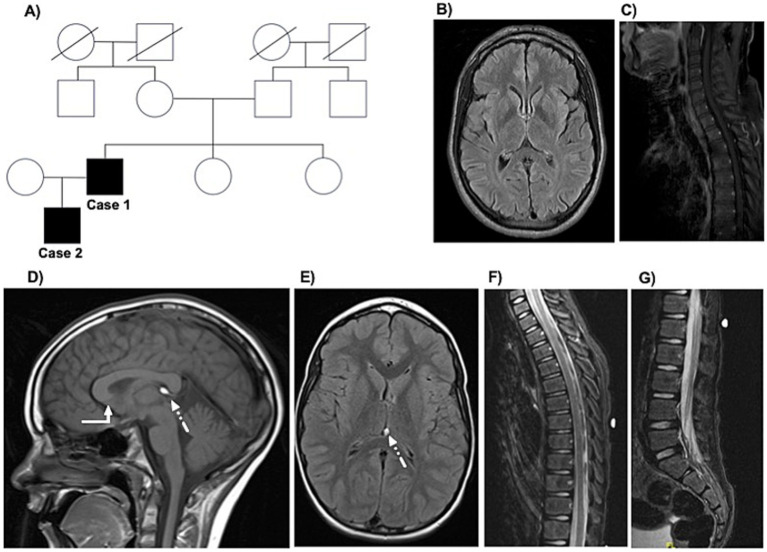 Figure 1
Figure 1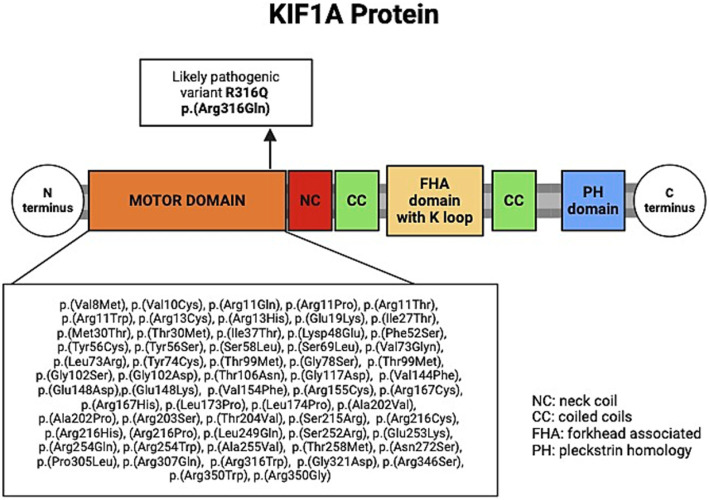 Figure 2
Figure 2| KIF1A variant | Inheritance pattern | Associated phenotype(s) | Reference |
|---|---|---|---|
| p.Met30Thr, p.Tyr56Cys+, p.Ser69Leu+, p.Tyr74Cys, p.Gly78Ser, p.Thr106Asn, p.Arg167His+, p.Leu173Pro, p.Ser252Arg, p.Thr258Met, p.Arg350Trp+ | AD (inherited) | Pure HSP phenotype |
|
| p.Lys48Glu, p.Thr99Met+, p.Gly117Asp., p.Leu174Pro, p.Ala202Val, p.Arg216Cys+, p.Arg216His+, p.Asn272Ser+, p.Arg307Gln+, Arg316Trp+, | AD ( | Complicated HSP phenotype |
|
| p.Arg216Cys+, p.Arg254Trp+, p.Pro305Leu+ | AD ( | Ataxic phenotype: Congenital ataxia |
|
| p.Glu19Lys, p.Arg316Gln | AD (inherited) | Complicated HSP phenotype |
|
| p.Gly321Asp | AD (inherited) | Pure HSP phenotype |
|
| p.Arg13His, p.Val73Gly, p.Thr99Met+, p.Glu148Lys, p.Arg203Ser, p.Arg216Cys+, p.Asn272Ser+, p.Arg316Trp+ | AD ( | Complicated HSP phenotype |
|
| p.Ser58Leu+, p.Thr99Met+, p.Gly102Asp., p.Val144Phe, p.Arg167Cys+, p.Ala202Pro, p.Ser215Arg, p.Arg216Pro, p.Leu249Gln, p.Glu253Lys+, p.Arg316Trp+ | AD ( | Complicated HSP phenotype |
|
| p.Glu148Asp., p.Arg254Gln, p.Arg254Trp+, p.Arg307Gln+, p.Arg316Trp+ | AD ( | Complicated HSP phenotype |
|
| p.Thr99Met+, p.Arg216Cys+, p.Arg216His+, p.Glu253Lys+, p.Arg316Trp+ | AD ( | Complicated HSP |
|
| p.Thr204_Val205delinsLys-Ala, p.Arg155Cys, p.Arg316Trp+ | AD ( | Ataxic phenotype: hypotonia and motor incoordination, progressive ataxia |
|
| p.Ser69Leu+, p.Gly102Ser+, p.Arg167Cys+ | AD (67% | Pure and complicated HSP |
|
| p.Val8Met, p.Ile27Thr | AD (inherited) | Pure HSP |
|
| p.Ile37Thr | AD (inherited) | Pure HSP |
|
| p.Ala255Val | AR | Pure HSP |
|
| p.Arg11Thr | AD ( | Complicated HSP |
|
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHereditary Neurological Disorders · Neurological diseases and metabolism · Endoplasmic Reticulum Stress and Disease
Introduction
Hereditary spastic paraplegia (HSP) refers to a clinically heterogeneous group of inherited conditions characterized by degeneration of the corticospinal tracts and dorsal columns (Shribman et al., 2019; Meyyazhagan et al., 2022). The incidence of HSP is approximately 1–5 per 100,000 individuals (Pennings et al., 2020). HSP presents with progressive and disabling lower extremities spasticity and weakness, and based on the overall phenotype, it can be classified into “pure” or “complicated” forms (Shribman et al., 2019; Meyyazhagan et al., 2022). In contrast to the classic “pure” HSP, which is defined by a slowly progressive lower extremity spastic paraparesis, “complicated” HSP presents with additional neurological or systemic findings, such as seizures, cognitive and neurobehavioral symptoms, peripheral neuropathy, and optic atrophy (Montenegro-Garreaud et al., 2020). Several inheritance patterns have been observed, including autosomal dominant (AD), autosomal recessive (AR), X-linked, or mitochondrial (Shribman et al., 2019). Over 87 genetic loci have been associated with HSP, emphasizing its large genetic heterogeneity (Pennings et al., 2020). Using a targeted multigene panel, a study of 1,550 index cases with HSP identified a causative variant in only 30.7% of cases. Subsequent whole exome sequencing of 42 cases negative for the targeted panel increased the diagnosis rate to about 50% (Méreaux et al., 2022). This indicates that many genetic variants remain undiscovered.
In this report, we describe the clinical phenotype and work-up findings of a familial case of HSP with few complications related to a monoallelic likely pathogenic variant within the kinase motor domain of the kinesin family member 1A (KIF1A) gene (Lee et al., 2015; Ohba et al., 2015).
Case description
Case 1 is a 45-year-old White male who was previously diagnosed with spastic diplegic cerebral palsy. Despite meeting all developmental milestones at an appropriate age, he developed progressive gait disturbance and stiffness of the lower extremities in early childhood. He was able to ambulate independently through late childhood. During adolescence, he underwent multiple orthopedic surgeries, including bilateral Achilles tendon lengthening, to improve lower extremities stiffness and ankle joints’ range of motion. His cognitive development was normal. He completed third-level education and is currently working as a special-needs teacher. More recently, he developed sphincter dysfunction, including urinary frequency and occasional stool incontinence.
He reported no neurological symptoms or gait impairment in three generations of his family (see Figure 1A). He has two healthy younger sisters, aged 38 and 43. However, his son developed similar progressive neurological symptoms at age 2 (see Case 2).
The family tree and MRI findings in the father-son cases. The family tree is displayed in panel A. Parts of the magnetic resonance imaging (MRI) of Case 1 obtained at age 44 at the initial movement disorders clinic appointment are displayed, including axial FLAIR sequences of the brain (panel B) and upper spine (panel C). Selected images of the MRI brain and spine of Case 2 obtained at age 8 at initial child neurology appointment are displayed, including sagittal T1-weighted (panel D; white arrow refers to the lack of visualization of the rostrum and mildly dysmorphic corpus callosum; dotted arrow refers to the 6-mm lipoma in the cavum vellum interpositum) and axial FLAIR sequences (panel E) of the brain and sagittal short tau inversion recovery (STIR) sequences of the upper and lumbar spine (panels F,G).
His neurological examination at age 45 years was notable for spasticity, weakness (ranging from 3–4 out of 5 per the Medical Research Council manual muscle testing scale), and hyperreflexia in the bilateral lower extremities, with positive Babinski sign bilaterally. Impaired vibration and proprioception sensation was detected at the toes bilaterally. His gait was spastic. During work-up, he underwent an MRI of the brain and total spine, which were largely unremarkable (see Figures 1B,C). A formal neuropsychology evaluation revealed intact performance across all cognitive domains. For symptomatic management of spasticity, he is receiving Baclofen 20 mg twice daily. He also started chemo-denervation of the lower extremity muscles with botulinum toxin.
Given the development of similar neurological symptoms in his son, genetic testing was pursued (see detail below).
Case 2 is an 11-year-old White male who was born at full-term (38 weeks of gestation) by elective Cesarian section to a healthy 28-year-old primigravida mother. His birth weight was 6 lbs. 15 oz. The pregnancy was unremarkable, and the only documented perinatal complications were mild jaundice and a weak suck-and-swallow reflex, both of which were managed conservatively and resolved without further intervention. All of his early motor and speech-language developmental milestones were met at the expected age. However, at two years of age, he developed “toe walking.” This progressed to significant gait disturbance and recurrent falls. His initial neurological, metabolic, and genetic work-up was unrevealing. MRI of the brain revealed a mildly dysmorphic corpus callosum without visualization of the rostrum and 6-mm lipoma in the cavum vellum interpositum (see Figures 1D,E). Around age 5, he began to experience significant stiffness in the lower extremities. He later developed nocturnal enuresis, which raised concern for a possible learning disability. A neurodevelopment assessment led to a diagnosis of attention-deficit/hyperactivity disorder (ADHD), impulsivity, and mild executive dysfunction. At 8 years old, based on MRI spine findings (see Figures 1F,G), he was diagnosed with a possible tethered spinal cord syndrome and underwent a release procedure, with some reported gait improvement.
His neurological examination at 11 years of age was notable for spasticity and hyperreflexia in the lower extremities and bilateral weakness of the ankle dorsiflexion. His gait was spastic, with a notable toe walking. For symptomatic management, he received intensive physical therapy and required ankle-foot orthoses and later began chemo-denervation treatment with botulinum toxin for lower extremity spasticity management.
Exome sequencing (ES) was pursued at age 11. This revealed a likely pathogenic heterozygous (monoallelic) variant in exon 10 of KIF1A [NM_001244008.2: c.947G > A; (p.Arg316Gln); chr2-240775862]. His father (Case 1) underwent KIF1A gene sequencing at age 45, revealing the same likely pathogenic variant, confirming that this rare variant (which at the time of submission was not available on varsome.com) is linked to the hereditary spastic paraplegia phenotype in the father and son (Kopanos et al., 2019).
Discussion
Given the genotype–phenotype heterogeneity in HSP, identifying the genetic etiology in many cases with suspect HSP phenotype is becoming increasingly reliant on advanced next-generation sequencing (NGS) technologies, such as ES (Shribman et al., 2019; Meyyazhagan et al., 2022; Méreaux et al., 2022). KIF1A encodes for a kinesin-3 motor protein that is highly expressed in the central nervous system (Gabrych et al., 2019). Kinesins are a diverse group of microtubule-based motor proteins that perform various functions, including intracellular transport of vesicles, organelles, chromosomes, and protein complexes (Lawrence et al., 2004). Originally, they were classified according to their specific functions and other criteria (e.g., the position of the motor core within the protein) (Lawrence et al., 2004). However, at the 2003 meeting of the American Society for Cell Biology, a dedicated subgroup of experts recommended a standardized nomenclature for all kinesins, grouping them into 14 superfamilies according to molecular phylogenetic analysis (Lawrence et al., 2004). Pathogenic variants in the KIF1A gene have been implicated in many neurodevelopmental and neurodegenerative disorders with diverse impacts on the central and peripheral nervous systems. KIF1A pathogenic variants have been associated with four disorders in Online Mendelian Inheritance in Man (OMIM) (Nicita et al., 2021). Two disorders are spastic paraplegia-30 (SPG30) due to pathogenic variants in KIF1A of either AR and AD inheritance patterns with overlapping phenotypes (Krenn et al., 2017; Lo Giudice et al., 2014). The third disorder, neurodegeneration and spasticity with or without cerebellar atrophy or cortical visual impairment (NESCAV syndrome), formerly named Mental Retardation Type 9 (MRD9), is a complex AD condition (Paprocka et al., 2023). NESCAV syndrome is characterized by childhood onset of moderate to severe intellectual disability with a broad range of other clinical features, which may represent an early onset of complicated HSP phenotype (Nicita et al., 2021). The fourth disorder is hereditary sensory and autonomic neuropathy type 2 (HSAN2), which has an AR inheritance pattern (Nicita et al., 2021). KIF1A variants have also been linked with progressive encephalopathy with brain atrophy, progressive neurodegeneration, PEHO (progressive encephalopathy with edema, hypsarrhythmia, optic atrophy)-like syndrome, and Rett-like syndrome (Paprocka et al., 2023).
Most HSP-related KIF1A variants are missense variants in the kinesin-3 motor domain of the KIF1A protein (residues 1 to 365, see Figure 2), and are often associated with “pure” or “complicated” HSP phenotype with de novo or AD inheritance (see Table 1) (Shribman et al., 2019; Montenegro-Garreaud et al., 2020; Lee et al., 2015). In this report, we describe the phenotype of a familial case with HSP due to a likely pathogenic monoallelic variant in exon 10 of the KIF1A gene. The same KIF1A variant, p.Arg316Gln, and another missense variant at the same residue, p.Arg316Trp, were previously reported in individuals with “complicated” HSP (Lee et al., 2015; Ohba et al., 2015). In the report by Hsu et al., carriers of the p.Arg316Gln variant exhibited axonal sensorimotor polyneuropathy and abnormal MRI findings, including thinning of the posterior corpus callosum and atrophy of the thoracic cord (Hsu et al., 2022). In contrast, the p.Arg316Gln variant identified in our cases appears to be associated with a slowly progressive “pure” HSP phenotype with few complications.
Schematic of the KIF1A protein, showing previously reported variants within the motor domain in KIF1A-related HSP. Created by BioRender.
As evident in the reported cases of HSP-related KIF1A variants (see Table 1), the phenotypic heterogeneity associated with these variants likely represents a spectrum of HSP rather than many disorders converging at a single locus (Montenegro-Garreaud et al., 2020). Therefore, the presence of other neurological symptoms and findings in cases with classic HSP phenotype should warrant additional work-up and expert evaluation as clinically indicated. For example, the presence of visual symptoms or optic nerve atrophy should warrant an ophthalmologic evaluation. Similarly, neurocognitive testing, electroencephalography, and nerve conduction studies should be considered in patients with cognitive symptoms, concerns about a seizure disorder, or neuropathy symptoms. The AD inheritance is important for consideration of recurrence risk in preconception discussions.
Genetic identification of HSP variants is crucial and will guide the search for molecular targets for future disease-modifying therapies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Citterio A.Arnoldi A.Panzeri E.Merlini L.D’Angelo M. G.Musumeci O.. (2015). Variants in KIF 1A gene in dominant and sporadic forms of hereditary spastic paraparesis. J. Neurol. 262, 2684–2690. doi: 10.1007/s 00415-015-7899-9, PMID: 26410750 · doi ↗ · pubmed ↗
- 2Dong E.-L.Wang C.Wu S.Lu Y. Q.Lin X. H.Su H. Z.. (2018). Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 13:36. doi: 10.1186/s 13024-018-0269-1, PMID: 29980238 PMC 6035405 · doi ↗ · pubmed ↗
- 3Erlich Y.Edvardson S.Hodges E.Zenvirt S.Thekkat P.Shaag A.. (2011). Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF 1A in hereditary spastic paraparesis. Genome Res. 21, 658–664. doi: 10.1101/gr.117143.110, PMID: 21487076 PMC 3083082 · doi ↗ · pubmed ↗
- 4Esmaeeli Nieh S.Madou M. R.Sirajuddin M.Fregeau B.Mc Knight D.Lexa K.. (2015). De novo mutations in KIF 1A cause progressive encephalopathy and brain atrophy. Ann. Clin. Transl. Neurol. 2, 623–635. doi: 10.1002/acn 3.198, PMID: 26125038 PMC 4479523 · doi ↗ · pubmed ↗
- 5Gabrych D. R.Lau V. Z.Niwa S.Silverman M. A. (2019). Going too far is the same as falling short(†): Kinesin-3 family members in hereditary spastic paraplegia. Front. Cell. Neurosci. 13:419. doi: 10.3389/fncel.2019.00419, PMID: 31616253 PMC 6775250 · doi ↗ · pubmed ↗
- 6Hsu S. L.Liao Y. C.Lin K. P.Lin P. Y.Yu K. W.Tsai Y. S.. (2022). Investigating KIF 1A mutations in a Taiwanese cohort with hereditary spastic paraplegia. Parkinsonism Relat. Disord. 103, 144–149. doi: 10.1016/j.parkreldis.2022.09.001, PMID: 36155026 · doi ↗ · pubmed ↗
- 7Iqbal Z.Rydning S. L.Wedding I. M.Koht J.Pihlstrøm L.Rengmark A. H.. (2017). Targeted high throughput sequencing in hereditary ataxia and spastic paraplegia. P Lo S One 12:e 0174667. doi: 10.1371/journal.pone.0174667, PMID: 28362824 PMC 5375131 · doi ↗ · pubmed ↗
- 8Kopanos C.Tsiolkas V.Kouris A.Chapple C. E.Albarca Aguilera M.Meyer R.. (2019). Var some: the human genomic variant search engine. Bioinformatics 35, 1978–1980. doi: 10.1093/bioinformatics/bty 897, PMID: 30376034 PMC 6546127 · doi ↗ · pubmed ↗