High-Risk Neuroblastoma Stage 4 (NBS4): Developing a Medicinal Chemistry Multi-Target Drug Approach
Amgad Gerges, Una Canning

TL;DR
This paper explores a new approach to treating high-risk stage 4 neuroblastoma in children by identifying multi-target drug candidates that could inhibit key disease-related proteins.
Contribution
The novelty lies in identifying potential multi-target inhibitors for four key proteins in high-risk neuroblastoma using computational drug design methods.
Findings
Eight compounds were identified as potential inhibitors for four neuroblastoma-related targets using computational methods.
The four targets (BRD, HDAC, HH, TRK) share high amino acid similarity in their binding sites, suggesting multi-target drug potential.
Two additional targets (RA and c-Src) showed lower binding site similarity compared to the primary four.
Abstract
Childhood neuroblastoma (NB) is a malignant tumour that is a member of a class of embryonic tumours that have their origins in sympathoadrenal progenitor cells. There are five stages in the clinical NB staging system: 1, 2A, 2B, 3, 4S, and 4. For those diagnosed with stage 4 neuroblastoma (NBS4), the treatment options are limited with a survival rate of between 40 and 50%. Since 1975, more than 15 targets have been identified in the search for a treatment for high-risk NBS4. This article is concerned with the search for a multi-target drug treatment for high-risk NBS4 and focuses on four possible treatment targets that research has identified as having a role in the development of NBS4 and includes the inhibitors Histone Deacetylase (HDAC), Bromodomain (BRD), Hedgehog (HH), and Tropomyosin Kinase (TRK). Computer-aided drug design and molecular modelling have greatly assisted drug…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
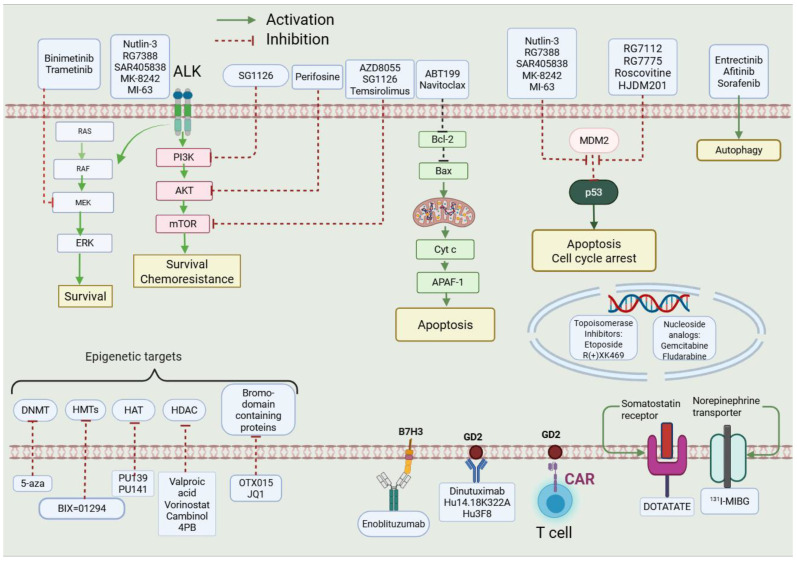 Figure 1
Figure 1 Figure 2
Figure 2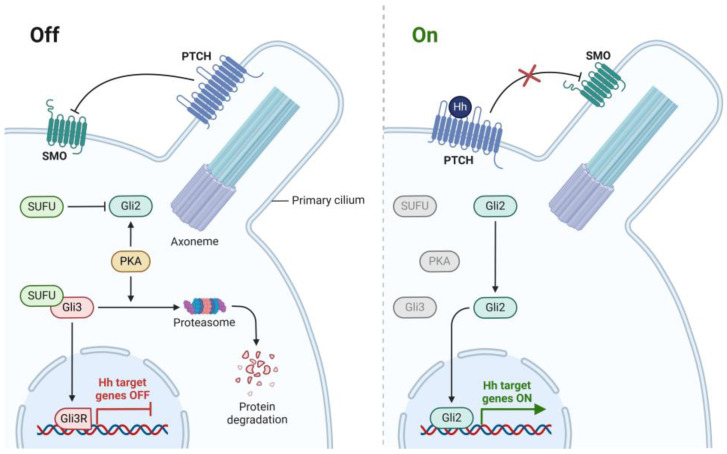 Figure 3
Figure 3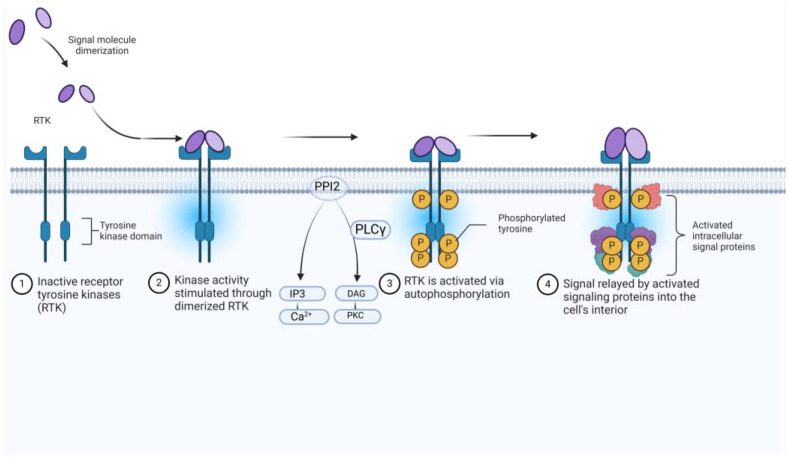 Figure 4
Figure 4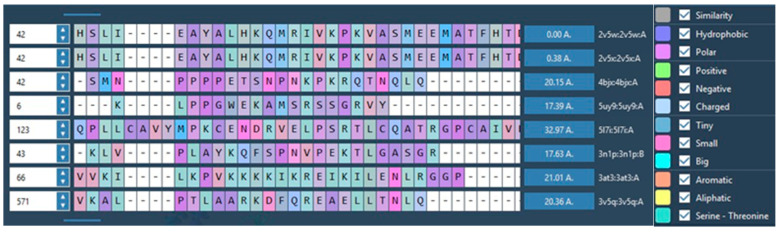 Figure 5
Figure 5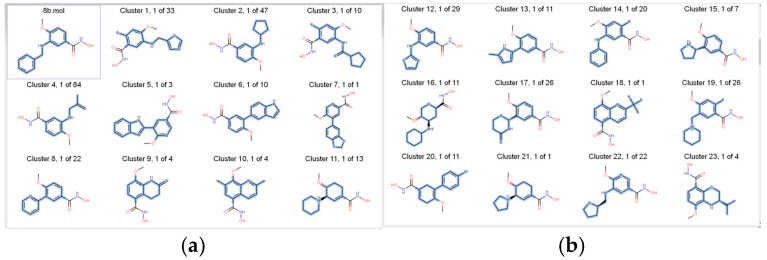 Figure 6
Figure 6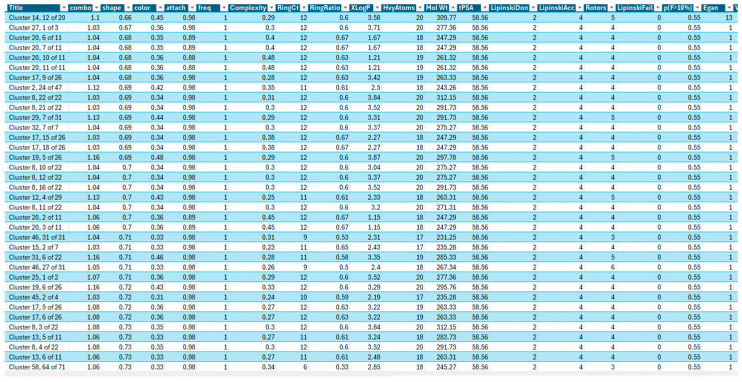 Figure 7
Figure 7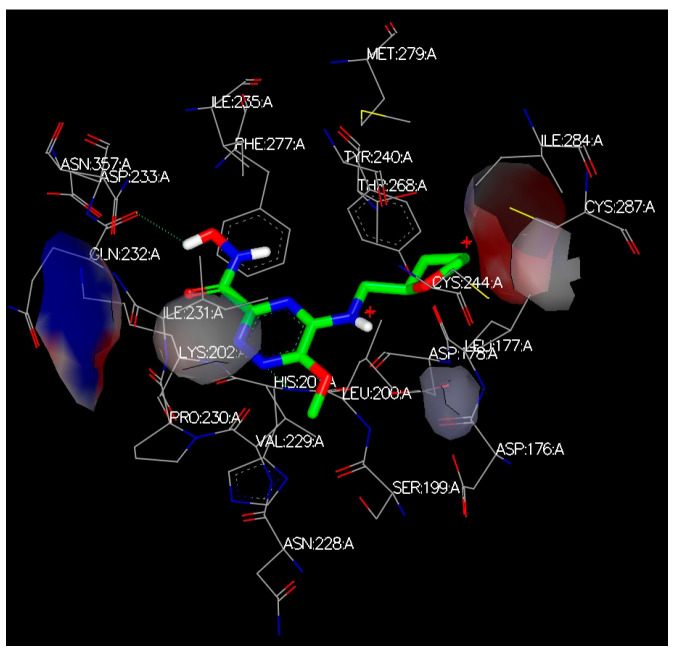 Figure 8
Figure 8 Figure 9
Figure 9 Figure 10
Figure 10 Figure 11
Figure 11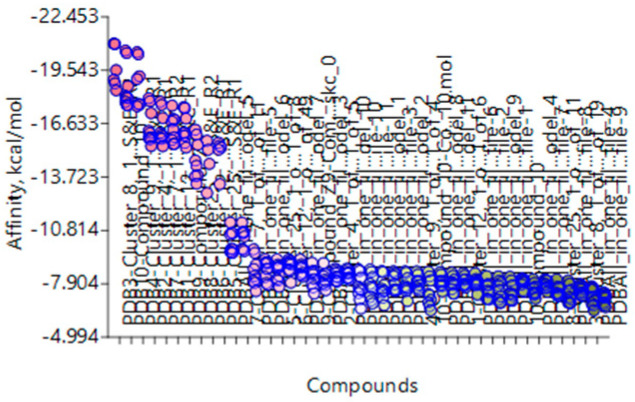 Figure 12
Figure 12 Figure 13
Figure 13 Figure 14
Figure 14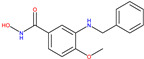 Figure 15
Figure 15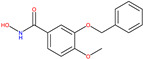 Figure 16
Figure 16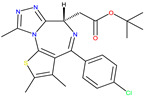 Figure 17
Figure 17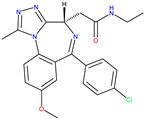 Figure 18
Figure 18 Figure 19
Figure 19 Figure 20
Figure 20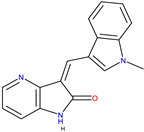 Figure 21
Figure 21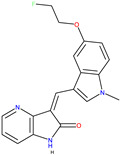 Figure 22
Figure 22 Figure 23
Figure 23 Figure 24
Figure 24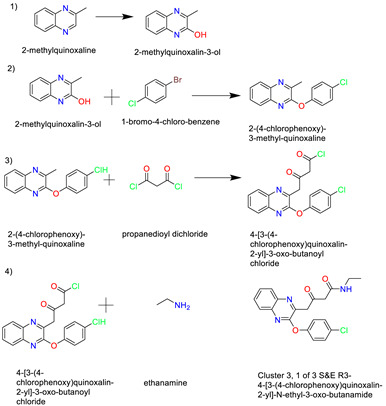 Figure 25
Figure 25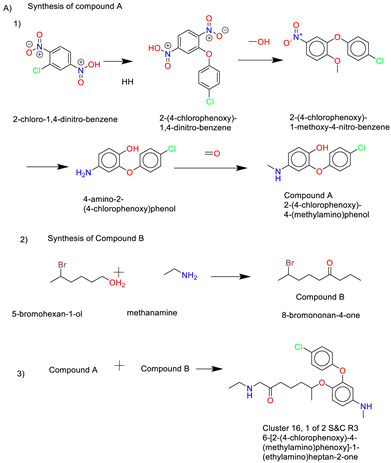 Figure 26
Figure 26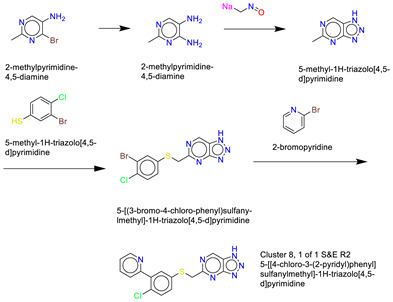 Figure 27
Figure 27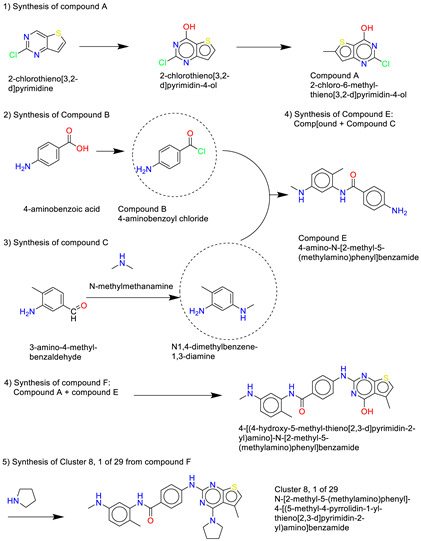 Figure 28
Figure 28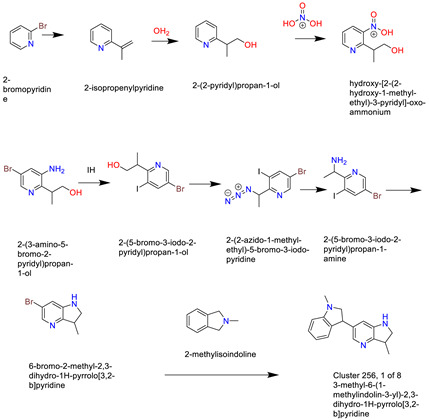 Figure 29
Figure 29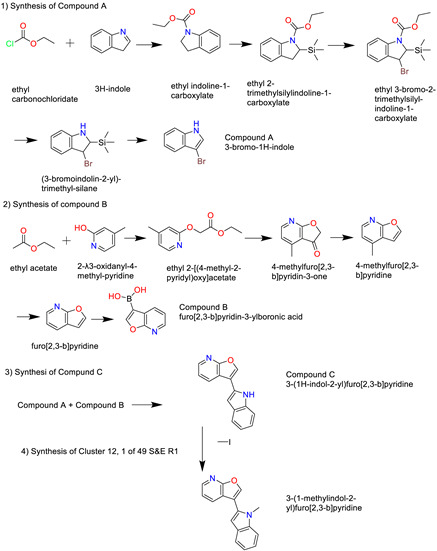 Figure 30
Figure 30Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeuroblastoma Research and Treatments · Protein Degradation and Inhibitors · Ubiquitin and proteasome pathways
1. Introduction
Childhood neuroblastoma (NB) is a malignant tumour that is a member of a class of embryonic tumours that have their origins in sympathoadrenal progenitor cells [1]. There are five stages in the clinical NB staging system: 1, 2A, 2B, 3, 4S, and 4 [1]. For those diagnosed with stage 4 neuroblastoma (NBS4) the treatment options are limited with a survival rate of between 40 and 50% [2]. This article is concerned with the search for a multi-target drug treatment for high-risk NBS4 and focuses on four possible treatment targets. Since 1975, more than 15 targets have been identified in the search for a treatment for high-risk NBS4 [3]. Six targets were originally selected and of these, on the basis of the most promising research (Figure 1) [4] Four demonstrated amino acid sequence similarity of 80–100% in the receptors binding sites and are the focus of this paper. The other two targets showed similarity across their receptors of 80–100% but lower than 80% for the other four targets.
The four targets were selected because of promising results in previous research for NBS4. The four have been investigated separately for the role they play in the development of NB and include: Histone Deacetylase (HDAC), Bromodomain (BRD), Hedgehog (HH), and Tropomyosin Kinase (TRK). Of the four BRD, HH and HDAC are epigenetics, a term used to describe the study of heritable traits or a stable change in cell function, that happen without changes to the DNA sequence. In the case of cancers, it is nearly impossible to reverse genetic alterations, whereas epigenetic changes “can dynamically respond to signals from the physical, biological and social environment” [6]. Other targets investigated for NBS4 include Tyrosine Kinases such as MYCN that is involved in gene amplification in NB.
Current treatment for NBS4 involves immunotherapy combined with anti-cancer drugs [7]. With therapeutic agents meeting with little success in treating NBS4, the search for a new target remains urgent [4]. Currently, treatment agents focus on a one-drug-one-target approach and/or combination therapy, which has had little success in improving survival rates. An alternative approach is to develop a multi-target drug that interacts with multiple targets with high efficacy to change the disease network. Further perceived benefits to developing a multi-target drug offer the possibility of making “cocktail therapies” or drug combinations redundant [8], leading to less pharmacokinetic and safety profile testing, as the risk of drug–drug interactions would be reduced [9]. Since it is uncommon for multiple targets to mutate simultaneously in different pathways or at various locations within a single cascade pathway, multi-target drugs may also avoid drug resistance brought on by single-target mutations or changes in expression [10].
This article presents a developmental, multi-targeted drug design approach using computational methods available to medicinal chemistry. Computer-aided drug design and molecular modelling have greatly assisted drug development within the field of medicinal chemistry [11]. Computational methods such as molecular docking, homology modelling, molecular dynamics, and quantitative structure–activity relationships (QSAR) are frequently used as part of the process of finding new therapeutic drug targets [12]. Using these methods, eight compounds (inhibitors) were identified as possible inhibitors for four targets. Results revealed that all four targets BRD, HDAC, HH, and TRK share similar amino acid sequencing in the binding site that ranges from 80 to 100%, offering the possibility of further testing for their suitability for multi-target drug use. Two additional targets were also tested as part of this work, Retinoic Acid (RA) signalling pathway and c-Src kinase (Csk), which showed similarity (of the binding pocket) across their receptors of 80–100% but were lower that 80% for the other four targets.
1.1. Reasoning Behind Selected Targets
The four targets were selected due to their downregulation of NBS4:
- (1)Histone Deacetylase (HDAC)
The Histone Deacetylase (HDAC) family comprises 18 enzymes divided into four classes (I, II, III, and IV) according to their enzymatic activities, subcellular localisation, and homology to yeast HDCA [13]. In the case of HDAC 8 (class I), it was found to be downregulated in NBS4 (Figure 1) [14]. Two compounds, 8b and 20a [15], and two receptors, 2V5X and 2V5W [16], were selected for this target (see Table 1).
(2)Bromodomain (BRD)
Early in the 1990s, the Brahma gene of Drosophila Melanogaster was found to contain a family of evolutionarily conserved motifs known as Bromodomains (BRD) [17]. Numerous studies have been conducted on the Bromodomain and extra terminal (BET) family. It consists of BRDT, BRD2, BRD3, and BRD4, all of which are widely expressed, with the exception of BRDT, which is only expressed in the testis [18]. BRDs bind histone tail acetylated lysines, recognizing the acetyl group is essential for recruiting additional chromatin factors and transcriptional machinery, which controls gene transcription [18]. The BET family also functions as a cell cycle regulator with BRD4 regulating the expression of genes necessary for the transition from the M to early G1 phase [18]. Research found that the compound JQ1, a BRD inhibitor, upregulated p27 and the proapoptotic gene BIM while suppressing MYC expression, resulting in G1 cell cycle arrest [19,20]. Studies on the Bromodomain inhibitor BET762 in vivo have also shown that it has anti-cancer properties [21]. Two compounds, JQ1 and BET762, and two receptors, 4BJX and 5UY9 [22], were selected for this target (see Table 1).
(3)Hedgehog (HH)
Hedgehog Inhibitors (HHIs) have become a promising new target for cancer therapy [23]. The signalling pathway identified in 1980 [24] was a crucial regulator of growth, patterning formation, and cell migration during embryonic development [25]. The components of the HH signalling pathway are involved in signalling to the transcription factors [26]. One study found that signalling deregulation was observed with Gorlin syndrome and cancers (Figure 2) [27]. Two compounds, BMS-833923 [28] and Vismodegib [29], and two receptors, 5L7I [30] and 3N1P [31], were selected for this target (see Table 1).
(4)Tropomyosin Receptor Kinase (TRK)
The neurotrophin family of peptide hormones activates three related tyrosine kinases known as tropomyosin receptor kinases (TRK) [32] that along with various forms of cancer, are also essential in neurodegenerative diseases. TRK inhibitor development to target cancers driven by NTRK fusion has gained attention in the past ten years (Figure 3) [33].
TRK activation results in the autophosphorylation of an intracellular tyrosine residue [34]. This phosphorylation is a crucial step in activating the TRK receptor and initiating downstream signalling cascades [35]. Two compounds, GW441759 and Compound 10 [36], and two receptors 4AT3 [36] and 3V5Q [36], were selected for this target. molecules-30-02211-t001_Table 1Table 1Selected lead compounds and receptors for each target type by literature review.TargetLead Compound 1Lead Compound 2Protein/Receptor
-
1-Histone Deacetylase 8 Inhibitors 2V5X and 2V5W [16] 8b [15]20a [15]
-
2-Bromodomain Inhibitors JQ1 [18,20]BET762 [18]4BJX [18] and 5UY9 [22]
-
3-HH inhibitors BMS-833923 [28]Vismodegib [29]5L7I [30] and 3N1P [31]
-
4-Tropomyosin Receptor Kinase Inhibitors GW441759 [36]Compound 10 [36]4AT3 [36] and 3V5Q [36]
1.2. Medicinal Chemistry Approaches to Drug Design
Rational multi-target drug design strategies in recent years have varied from pharmacophore combination, screening [37], and similar scaffold structure [38]. Each strategy has advantages and disadvantages, along with varying challenges. The purpose of this work is to suggest a possible modified approach. In the search for a multi-target drug for NBS4 using advanced medicinal chemistry software, this approach assesses the possibility of different targets by assessing receptor similarities (in the binding pocket) and the selection of two lead compounds for each of the four target (eight compounds in total) that have demonstrated an inhibitory effect on NBS4 cells, to form the basis of a search for a multi-target drug.
Having identified four targets that share similar amino-acid sequencing, two receptors from each target were selected (see Table 1) and a Protein Aligner tool from Samson was used to check for their suitability, with results reporting 80–100% similarity for the four targets (Figure 4). Protein Aligner checks for amino acid sequence similarity between receptors with high similarity between receptors indicating their suitability for use in cross-docking.
The selection of ligands was based on literature research in relation to NBS4. Two compounds were selected for each target (see Table 1), representing a total of eight compounds.
For the purposes of this work docking involves docking a compound to a receptor known for that target (i.e., BRD) whereas cross-docking is docking the same compound to a receptor that belongs to a different target (i.e., TRK) and vice versa. The aim of cross-docking is to explore the suitability of the selected compound for use across different targets to determine its suitability as a multi-target drug. This is a process that involves selecting known lead compounds to produce hitlists of compounds and was performed using BROOD [39] as part of the OpenEye suite.
Several BROOD rounds were completed on the 2 lead compounds for each target to produce hitlists (Figure 5) using “Shape and Colour” and “Shape and Electrostatics” (see Table 2). On completion, BROOD ranked each of the hitlists according to BROOD hitlist parameters (Figure 6), the top 25 of each hitlist was selected, and the best 8–10 compounds of those 25 were selected (see Table 3) to run on OEDocking [40] using FRED (Figure 7), ROCS [41] (Figure 8), AFITT [42] (Figure 9) (see Table 4), and VIDA [43]. In addition, the Samson docking suite, including AutoDock Vina Extended (Figure 10, Figure 11 and Figure 12)) and Fitted (Molecular Forecaster) [44], with another docking programme, Molegro [45], acted as confirmation for both the docking and cross-docking procedures (see Table 5). BIOVIA Discovery Studio was also used for visualization (Figure 13) [46]. From this process, 8 to 10 compounds were selected for each target (see Table 6), all of which showed improved parameters compared to the original lead compounds. The compounds from each individual target were then cross-docked with the receptors from the other three using AFITT. From the 35 compounds run only 8 showed potential suitability for multi-target use (two from each list) (see Table 6).
Selected clusters from all targets were also docked with the Samson Suite, including AutoDock Vina Extended (V 5.1.3) and Fitted by Molecular Forecaster (V 1.7.2).
To check and compare the compounds (clusters) for toxicity and mutagenicity, Toxicity Estimation Software Tools (TEST) (Version 5.1.2) were used to provide the prediction mechanisms of the toxic action of the clusters [48]. The results from TEST showed some similarity with the original lead compounds (Table 7) which also provided their suitability for use. Further work was performed to explore possible synthesis routes for each of the eight compounds using the retrosynthesis programme Spaya [49].
2. Results
2.1. Medicinal Chemistry Results
Table 1 below contains the selected targets, protein/receptors (Protein Data Bank), and the identified lead compounds.
The next stage was to compare the similarity of the proteins (receptors) binding sites for all four targets. This was achieved using the Samson Protein Aligner tool, showing that the similarities between the selected proteins ranged from 80% to 100% (Figure 4).
By selecting an active group in the lead compound, the programme can produce a hitlist using shape and colour and shape and electrostatics. Table 2 shows how many rounds were performed for each lead compound according to target. The top 25 compounds from the hitlists were selected for the docking studies from each round. Figure 5 shows some of the compounds.
BROOD [50] hitlist parameters (see Figure 6) include the following:
- (1)AroRingCt: Number of aromatic rings in the molecule;
- (2)ClusterID/IdeaGroup: ClusterID of the molecule;
- (3)Colour: The replacement fragment’s colour Tanimoto score in comparison to the query fragment;
- (4)Combo: Tanimoto combo score for the replacement fragment’s shape and colour in comparison to the query fragment;
- (5)Egan: The Boolean indicates if the molecule satisfies the Egan bioavailability model;
- (6)Fragment: SMILES string of the replacement fragment;
- (7)Freq: The replacement fragment’s frequency;
- (8)fsp3C: The molecule’s fraction of sp3 hybridized carbon atoms;
- (9)HvyAtoms: Number of heavy atoms in the molecule;
- (10)LipinskiDon: Number of Lipinski donors in the molecule;
- (11)LipinkskiAcc: Number of Lipinski acceptors in the molecule;
- (12)LipinskiFail: Boolean specifying whether the molecule fails Lipinski’s rule of five;
- (13)Local strain: Calculated local strain of the molecule;
- (14)Molecular TanimotoCombo: Shape + colour Tanimoto combo score of the molecule against the query molecule;
- (15)MolWt: Molecular weight of the molecule;
- (16)p (active): Belief score of the molecule;
- (17)RingCt: Number of ring atoms;
- (18)RingRatio: Ratio of the number of ring atoms to the total number of heavy atoms;
- (19)Rotors: Number of rotatable bonds in the molecule;
- (20)Shape: Compare the replacement fragment’s Shape Tanimoto score to that of the query fragment;
- (21)Source Mols: SMILES strings of the molecules the replacement fragment is part of;
- (22)Source Mol Labels: Labels of the molecules the replacement fragment is part of;
- (23)tPSA: Calculated topological polar surface area of the molecule;
- (24)Veber: Boolean specifying whether the molecule passes the Veber bioavailability model;
- (25)XlogP: Calculated LogP of the molecule [50].
The next stage was to dock each hitlist with their relevant receptors; Figure 7 shows one of the docking outcomes using FRED (OpenEye suite) at the top of the list cluster 22, 1 of 1.
Having completed all the docking using FRED, Molegro, AutoDock Vina, and Fitted only the top compounds for each target were selected (range 8 to 10 see Table 3) to be run for validation on ROCS a programme that scores and aligns a database of molecules with a query. The score assigns a number to molecules according to their likelihood of having biological characteristics in common with the query molecule (Figure 8).
Another crystallographic tool used from the OpenEye suite is AFITT (Figure 9). AFITT creates a new combined forcefield that fits small molecules into crystallographic density while preserving superior chemistry by combining the shape and MMFF technologies of OpenEye. In order to verify the refinement, it also offers an interface to external refinement programmes, such as real space correlation coefficient calculation (RSCC) and interactive Ramachandran plots.
All possible clusters (Table 3) were docked with all docking tools: FRED, AFITT, AutoDock Vina Extended, Molegro, and Fitted. Table 4 provides an example.
Table 5 shows the docking of selected clusters from each target with more than one receptor of the same target.
The next step was cross-docking, which involved docking receptors with various cluster types and comparing the results. Figure 14 shows some of the results from the cross-docking of each target.
Having completed all cross-docking for the receptors and the selected clusters, eight compounds were identified as possible multi-target compounds (see Table 6 below).
2.2. Retrosynthesis Results Using Spaya
(1)Synthesis of Cluster 22, 1 of 22 R1 S&C [49]
(2) Cluster 10, 1 of 86
(3) Cluster 3, 1 of 3
(4) Cluster 16, 1 of 2
(5) Cluster 8, 1 of 1
(6) Cluster 8, 1 of 29
(7) Cluster 25, 1 of 8
(8) Cluster 12, 1 of 49
All data are available in the Supplementary Materials, see link below.
3. Discussion
The development of multi-target drugs is described as the process of “taking a well-validated primary target for a given disease and adding secondary activities to enhance efficacy” [37]. Designing such a drug involves fusing the inhibitory actions of two or more drugs into one molecule [38]. Typical approaches used for designing multi-target drugs include “Pharmacophore” and “Screening”. The pharmacophore approach can include processes such as merged-pharmacophore mode, fused-pharmacophore mode, non-cleavable linked pharmacophore, and cleavable pharmacophore. Pharmacophore modelling aims to “strip” functional groups of their true chemical nature in order to categorize them into a small number of pharmacophore types based on their predominant physicochemical characteristics [51]. Difficulties with this method occur due to inadequate or inaccurate conformational sampling, ambiguities in pharmacophore typing (primarily because of uncertainty regarding the tautomeric/protonation status of compounds), computer time limitations in complex molecular overlay calculations, and the selection of inappropriate anchoring points in active sites when ligand cocrystal structures are unavailable [51]. Along with pharmacophore the technique of screening is also used for drug discovery and has four identifiable categories: Fragment-based Drug Discovery (FBDD), High-throughput Screening (HTS), High-content Screening (HCS), and Virtual Screening (VS). For the work described here, a modified version of the Virtual Screening (VS) programme is used to enable the discovery of multi-target compounds (inhibitors). Using computational methods available to medicinal chemistry, computer-aided drug design and molecular modelling have greatly assisted drug design in the field [11]. Computational methods such as molecular docking, homology modelling, molecular dynamics, and quantitative structure–activity relationships (QSAR) are frequently used as part of the process for finding new therapeutic drug targets using computational methods [12]. For this work, eight compounds (inhibitors) were identified as possible inhibitors for four targets: HDAC, BRD, HH, and TRK.
Having selected four targets used in the study of NBS4, two receptors from each of the four targets were selected and the similarities of the receptors were compared as representations of the targets (Figure 4). Results indicated 80–100% similarities (using the Protein Aligner programme from Samson) confirming the possibility for multi-target use. High similarity between receptors indicates their suitability for use in cross-docking and is a process that involves selecting known lead compounds to produce hitlists of compounds (clusters). Using BROOD [39] as part of the OpenEye suite, several BROOD rounds were completed on the lead compounds to produce hitlists with the top 25 of each hitlist selected to run on OEDocking [40], AFITT [42], ROCS [41], and VIDA. The hitlists of compounds (clusters) were docked and the selected compounds, cross-docked. The clusters were docked with more than one docking programme as a means to validating the result.
The final ranking of the selected clusters was performed on AFITT as the receptor preparation with the tool MakeReceptor gives the user more control over the receptor-creating process. AFITT also has the advantage of real-space fitting of ligands in density, integrated with REFMAC, PHENIX, BUSTER, CNX, and COOT, and also fragment and cocktail fitting. In AFFIT, it is possible to select more than one ligand to fit generation of high-quality refinement dictionaries for use. This can be performed during reciprocal space refinement that includes the following: the use of forcefield (MMFF); semi-empirical (AM1, PM3) methods during reciprocal space refinement for BUSTER and Phenix; real space fitting of protein residues; proper handling of covalently bonded ligands, and proper handling of multiple occupancy ligands.
The ranking is based not only on the best results but also on the ability of the identified compounds to cross-dock on the receptors of other targets, and it was this process that led to the selection of the eight compounds. Using the tool ROCS provided cluster validation and is based on a large database search. Toxicity and mutagenicity of the eight compounds were tested using Toxicity Estimation Software Tools (TEST) [48], and the results showed some similarity with the original lead compounds (Table 7). With the aid of the retro-synthesis programme Spaya (IKTOS), the possibility of synthesizing all eight compounds was demonstrated (see the Results section) [49]. These results point the way to future work that will focus on preparing and testing the eight compounds in vitro and in vivo [52].
4. Materials and Methods
4.1. Materials
Computer programmes:
- OpenEye Scientific programmes, which include various applications, were used. The suite comprises BROOD, MakeReceptor, FRED, and AFITT.
- Molegro Virtual Docker.
- The Samson suite includes Autodock Vina Extended, the Fitted suite by Molecular Forecaster, and Protein Aligner.
- Toxicity Estimation Software Tools (TEST).
- BIOVIA Discovery Studio Visualizer.
- Spaya retrosynthesis software.
4.2. Method
Identifying drug targets.Selection of two proteins (receptors) for each target and downloading the PDB files and their electron density map from the Protein Data Bank database.Comparing the binding/active sites similarities of the receptors. Run protein similarity on Samson (Protein Aligner) to determine suitability.Selection of two lead compounds from each type.Run the lead compounds on BROOD (from the OpenEye suite) and produce hit lists using Shape and Colour and Shape and Electrostatics.Receptor preparations using MakeReceptor from the OpenEye suite.Docking the hit compounds with OpenEye suite (FRED), Molegro, and Samson suite (AutoDockVina and Fitted).Run cross-docking; each hitlist clusters from one target to the other 3 targets (using their protein/receptor).Run hits with AFITT to rank the compounds according to their fitting probabilities.Run selected clusters on ROCS.Run selected clusters on Toxicity Estimation Software Tools (TEST)Run clusters on Spaya to find the best synthesis route.
5. Conclusions
Using Virtual Screening with some modifications, eight compounds were identified as potential inhibitors across four targets (HDAC, BRD, HH, TRK) for the development of multi-target drug treatment of NBS4. The next stage is for the eight compounds to undergo single molecule testing in vivo and in vitro [52].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cullinane C.J. Burchill S.A. Jeremy A.S. O’Leary J.J. Lewis I.J. Molecular Biology and Pathology of Paediatric Cancer Oxford University Press New York, NY, USA 2003360
- 2Paraboschi I. Privitera L. Kramer-Marek G. Anderson J. Giuliani S. Novel Treatments and Technologies Applied to the Cure of Neuroblastoma Children 2021848210.3390/children 806048234200194 PMC 8226870 · doi ↗ · pubmed ↗
- 3Cohen S. Carpenter G. Human epidermal growth factor: Isolation and chemical and biological properties Proc. Natl. Acad. Sci. USA 1975721317132110.1073/pnas.72.4.13171055407 PMC 432524 · doi ↗ · pubmed ↗
- 4Gerges A. Canning U. Neuroblastoma and its Target Therapies: A Medicinal Chemistry Review Chem Med Chem 202419 e 20230053510.1002/cmdc.20230053538340043 · doi ↗ · pubmed ↗
- 5Zafar A. Wang W. Liu G. Wang X. Xian W. Mc Keon F. Foster J. Zhou J. Zhang R. Molecular targeting therapies for neuroblastoma: Progress and challenges Med. Res. Rev.202141961102110.1002/med.2175033155698 PMC 7906923 · doi ↗ · pubmed ↗
- 6Inomistova M. Khranovska N. Skachkova O. Role of Genetic and Epigenetic Alterations in Pathogenesis of Neuroblastoma Academic Press Cambridge, MA, USA 20192341
- 7Bhoopathi P. Mannangatti P. Emdad L. Das S.K. Fisher P.B. The quest to develop an effective therapy for neuroblastoma J. Cell Physiol.20212367775779110.1002/jcp.3038433834508 · doi ↗ · pubmed ↗
- 8Zhang W.L. Pei J.F. Lai L.H. Computational Multitarget Drug Design J. Chem. Inf. Model.20175740341210.1021/acs.jcim.6b 0049128166637 · doi ↗ · pubmed ↗