Still's disease in sub-Saharan Africa through ten observations from the internal medicine department of Libreville University Hospital (Gabon)
Josaphat IBA BA, Annick MFOUMOU, Ingrid NSENG-NSENG ONDO, Arthur KANGANGA EKOMY, Léonie Esther LEDAGA LENTOMBO, Marielle IGALA, Ulrich Davy KOMBILA, Jean Bruno BOGUIKOUMA

TL;DR
This paper reports 10 cases of Still's disease in Gabon, confirming its existence and highlighting diagnostic challenges in sub-Saharan Africa.
Contribution
The study confirms the presence of Still's disease in Gabon and describes its clinical features in a region where it is rarely reported.
Findings
Still's disease in Gabon presents with fever, joint, and skin involvement, similar to Caucasian populations.
Hepatic, lymphatic, and cardiac involvement were less frequent compared to other regions.
Diagnostic delays and high costs are significant challenges in sub-Saharan Africa.
Abstract
La maladie de Still (MS) est une affection inflammatoire systémique rare, plus fréquente dans la population infantile. Dans la forme de l'adulte, elle peut être primitive ou la résurgence de forme infantile. Nous rapportons 10 observations de maladie de Still dans la population gabonaise dans le but de confirmer son existence dans ce pays, et d'en rechercher les particularités. Il s'agit d'une étude rétrospective, descriptive et analytique réalisée dans le service de médecine interne du CHU de Libreville du 01/12/2003 au 31/12/2021, et prenant pour support les dossiers de patients hospitalisés dans ce service et/ou suivis en ambulatoire. Les patients retenus répondaient aux critères de Yamaguchi et Fautrel. Les données épidémiologiques, socio-économiques, cliniques, biologiques, morphologiques, immunologiques, thérapeutiques, évolutives, et saisonnières ont été détaillées. Dix…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
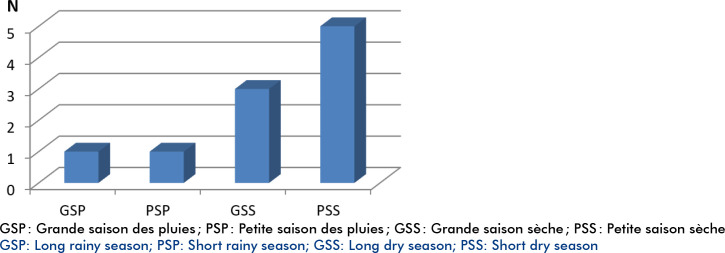 Figure 1
Figure 1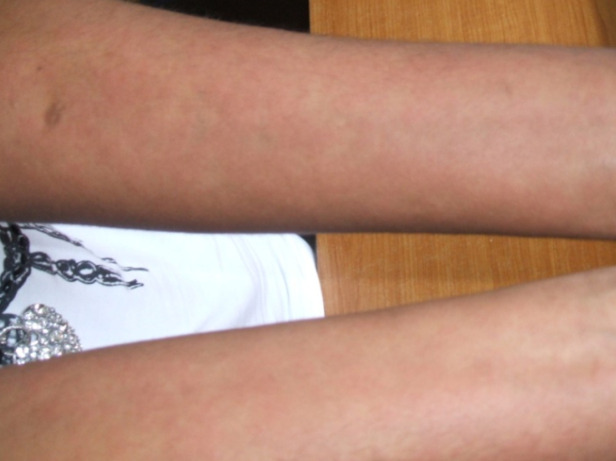 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research
Introduction
La maladie de Still (MS) est un rhumatisme inflammatoire systémique rare, d’étiologie inconnue, à début infantile, sans caractère familial, pouvant survenir ou se réactiver à l’âge adulte. Elle a été décrite pour la première fois en 1987 par le pédiatre anglais Georges Frederick Still [17] devant l'existence de polyarthrites séronégatives, non érosives, ne répondant pas aux critères classiques de la polyarthrite rhumatoïde. En 1971, Bywaters, interniste anglais, émettait l'hypothèse d'une entité clinique similaire chez 14 jeunes adultes présentant un tableau clinique semblable à celui de la forme systémique des arthrites juvéniles [2]. Les nouvelles recommandations européennes en 2023, présentées au congrès de l’EULAR (European League of Associations for Rheumatism) [8], considèrent que la forme pédiatrique et la forme de l'adulte représentent une seule et même entité, qu'il convient de nommer « maladie de Still ». Le diagnostic de MS repose sur les critères de Yamaguchi de 1992 [19], révisés par Fautrel et al. en 2002 [9]. L'association fièvre hectique, arthralgies et/ou arthrites, rash cutané, et hyperferritinémie > 1 000 ng/l, est habituellement suggestive de la maladie, après l'exclusion préalable d'une étiologie infectieuse, tumorale ou inflammatoire. En Afrique subsaharienne, la MS (comme de façon générale les maladies autoimmunes) demeure peu connue des praticiens, ce qui peut expliquer les données parcellaires dans cette région. Nous rapportons 10 observations de MS dans la population gabonaise pour confirmer l'existence de cette maladie dans ce pays, et en rechercher les particularismes.
Matériel et méthodes
Il s'agit d'une étude rétrospective, descriptive, analytique et monocentrique réalisée dans le service de médecine interne du CHU de Libreville sur une période de 18 ans, du 01/12/2003 au 31/12/2021. Elle a pris pour support les dossiers de patients hospitalisés dans le service et/ou suivis en ambulatoire. Tous les patients inclus devaient avoir d'une part, un diagnostic de MS répondant aux critères de Yamagushi et al. [19] et/ou de Fautrel et al. [9] (Tableau I), réalisé en cours d'hospitalisation ou en ambulatoire, et avoir d'autre part fait l'objet d'un suivi. Les dossiers incomplets et les patients ne répondant pas aux précédents critères, étaient exclus. Les variables de l’étude détaillaient les données épidémiologiques, socio-économiques (identité, âge, sexe, profession), cliniques (fièvre, atteinte cutanée, articulaire et ORL), biologiques (NFS, CRP, transaminases), morphologiques (en fonction de l'orientation), immunologiques (autoanticorps antinucléaires, facteurs rhumatoïdes), thérapeutiques (antalgiques, et/ou antiinflammatoires non stéroïdiens, corticothérapie, immunosuppresseurs), évolutives (suivi, perdu de vu, décès), et saisonnières (en précisant la saison du diagnostic : grande saison sèche, de mai à septembre, petite saison sèche, de décembre à janvier, grande saison des pluies, de février à avril, et petite saison des pluies, d'octobre à novembre). Toutes les données étaient répertoriées sur une fiche de recueil de données, saisies à l'aide du logiciel Epi Info. Les données quantitatives étaient décrites à l'aide de moyenne et de médiane, et les données qualitatives à l'aide d'effectifs et de pourcentages.
Tableau I: Critères de Yamagushi et de Fautrel de la maladie de Still
Résultats
Dix patients (4 hommes et 6 femmes), de 26 ans d’âge médian (interquartiles IQ : 12-33; extrêmes : 2 et 45 ans) ont été inclus dans l’étude. L’âge médian chez les adolescents était de 11 ans (IQ : 10-12; extrêmes : 2 et 13 ans). Chez les adultes, l’âge médian était de 28 ans (IQ : 23-39; extrêmes : 19 et 45 ans). La tranche d’âge supérieur ou égal à 23 ans représentait 70 % de la population d’étude. La population estudiantine au sens large (élèves + étudiants) prédominait avec 6 cas, suivie des patients sans emploi (n=3), et de salariés (n=1). Une patiente signalait une tante avec polyarthrite rhumatoïde, et une seconde un asthme allergique dans une forme infantile. Huit cas ont été diagnostiqués en saison sèche (Fig. 1).
Période saisonnière de diagnostic
Sur le plan clinique, les données pertinentes se résumaient à une fièvre présente chez tous les patients, une atteinte articulaire et cutanée chez 8 d'entre eux, et une atteinte ORL chez 7 patients (Tableau II). Le délai diagnostic moyen de la MS était de 940 jours (extrêmes : 7 et 7 300 jours), avec un diagnostic inférieur ou égal à 14 jours chez 5 patients.
Tableau II: Données cliniques des patients
Sur le plan biologique, une leucocytose existait chez 8 patients (taux moyen de 25 500/mm^3^, extrêmes : 7 500 et 60 000, médiane : 32 500/mm^3^) avec une prédominance de neutrophiles (9 cas), couplée à une anémie constante. Il existait un syndrome inflammatoire dans tous les cas où la CRP avait été réalisée (n=5), une perturbation du bilan hépatique chez 2 des 5 patients chez lesquels elle a été recherchée, et une hyperferritinémie dans 9 cas avec une moyenne de 23,3 N (extrêmes : normal et 9,6 N, médiane 3,7 N) (Tableau III). Les données morphologiques confirmaient une atteinte des séreuses dans 4 cas sur 7, chez les patients dont le bilan morphologique a été réalisé, une absence d'atteinte cardiaque et radiologique osseuse, et un tableau de polyadénopathies infra et centimétriques chez un patient (Tableau IV). Le bilan immunologique était négatif chez tous les patients, détaillé dans le Tableau V. Sur le plan thérapeutique, tous les patients avaient bénéficié d'une corticothérapie à base de prednisone (1 mg/ kg/jour), précédée dans 3 cas de bolus de méthyl prednisolone, couplé à du méthotrexate per os d'emblée chez un patient, ou en cas de corticorésistance (n=4) à la dose de 0,3 mg/kg/semaine soit 15 à 20 mg/semaine (Tableau VI), permettant une issue favorable chez 8 patients. Dans aucun cas, nous n'avons pu sevrer les patients en corticothérapie, avec une dose minimale efficace d'entretien journalière de 7,5 à 10 mg. Nous déplorons un décès en réanimation par syndrome d'activation lympho-histiocytaire (altération de l’état général fébrile, pancytopénie arégénérative, hyperferritinémie, augmentation de lactate déshydrogénase (LDH), et hémophagocytose au myélogramme), et un patient perdu de vue après 6 mois de suivi régulier malgré un contrôle satisfaisant sous traitement médical.
Tableau III: Données biologiques des patients
Tableau IV: Données morphologiques des patients
Tableau V: Données immunologiques des patients
Tableau VI: Données thérapeutiques des patients
Discussion
La MS demeure certainement sous-estimée en Afrique subsaharienne devant le peu d'observations retrouvées, contrastant avec une absence de prédisposition génétique clairement établie de la maladie [11]. En effet, les données disponibles sont surtout issues de populations caucasiennes ou asiatiques et il n'existe que très peu de données dans la population noire ou d'ascendance africaine. À cela, il faut rajouter le coût élevé du bilan concourant au diagnostic de la maladie dans un continent où le système d'assurance maladie n'est pas encore établi pour le plus grand nombre. Le Gabon a la chance de bénéficier d'une assurance maladie nationale depuis 2007 prenant 80 % du coût de prise en charge ambulatoire et hospitalière [14], ce qui nous a permis d'aboutir au diagnostic de maladie de Still. Dans l'itinéraire diagnostic des patients, nous n'avons pas retrouvé de consultation chez des tradithérapeutes, dont le traitement est à base de scarifications et de décoctions traditionnelles. La MS constitue un diagnostic d'exclusion, imposant avant d’être évoquée, d’écarter préalablement une étiologie infectieuse, tumorale et auto-immune.
La MS provoque une inflammation systémique, au stade précoce de la maladie ou lors de rechutes et de récurrences, résultant de l'activation et de la sécrétion de cytokines pro-inflammatoires, notamment d'interleukine (IL)-1, d’IL-18 et d’IL-6 [4, 10, 13]. Neuf de nos patients (un étant lors du diagnostic sous acide acétylsalicylique) avaient une hyperferritinémie comme classiquement retrouvée dans cette maladie. Ce taux très élevé de ferritine (mais également d’IL-8, IL-6, IL-18 et TNF-α) rend compte d'une activation marquée des macrophages lesquels, lorsqu'ils sont activés, synthétisent de l’IL-18 qui jouerait un rôle clé dans la pathogénie de la maladie [1].
De façon générale, pour les mêmes critères diagnostics [19], type d’étude (rétrospective), et type de population [5, 16], notre population demeure plus jeune que celle observée par d'autres auteurs au Cameroun, où l’âge moyen des patients est de 28 ans [16], au Sénégal où il est de 43 ans [5] et en Tunisie où il est de 35,4 ans [3]. Deux patients sur 3 sont diagnostiqués avant 40 ans [15], alors que notre série en compte 9 sur 10. Dans 10 à 15 % des cas, la MS peut être la résurgence d'une maladie de Still de l'enfance. Nous n'avons pas eu dans l'histoire de nos patients, d'observation de MS survenue dans l'enfance, qui aurait pu faire évoquer cette résurgence de forme infantile. Nous retrouvons 8 observations sur 10 de MS en saison sèche, mais l'influence saisonnière sur l'apparition de cette maladie n'a jamais été étudiée en Afrique subsaharienne.
Actuellement, les sociétés savantes s'accordent pour retenir la MS de l'adulte et sa forme juvénile comme une seule et même maladie, avec une même présentation clinique [8]. On distingue les MS à tropisme articulaire et les MS avec atteinte systémique. Sur le plan clinique, la MS se caractérise par une triade associant : a) fièvre élevée et hectique, b) éruption cutanée évanescente, et c) atteinte articulaire à type d'arthralgies ou d'arthrites. La fièvre d'allure septique avec frissons demeure une constante dans les études d’Afrique subsaharienne [5, 16], de même que l'atteinte articulaire (8 cas dans notre série), cutanée (8 cas) et ORL, retrouvée chez 7 de nos patients. Dans sa forme typique, l'atteinte cutanée au cours de la maladie de Still associe sur peau caucasienne de petites macules ou maculo-papules, de couleur rose saumonée, non prurigineuses, fugaces, siégeant à la racine des membres et apparaissant au moment des poussées fébriles. Cette atteinte cutanée est plus difficile à mettre en évidence sur peau noire, et peut prendre à défaut mêmes des spécialistes aguerris (Fig. 2). Ceci pourrait expliquer le faible nombre d'atteintes cutanées retrouvées dans la série de Singwe-Ngandeu et al. de 16,6 % [16].
Lésions cutanées de maladie de Still sur peau noire
Nous notons dans notre série d'une part une plus faible prévalence des atteintes hépatiques et ganglionnaires (rattachées à la MS devant leur régression sous corticothérapie), et, d'autre part, une absence d'atteinte cardiaque. Ces atteintes sont pourtant fréquentes au cours de la MS.
Biologiquement, aucune perturbation n'est spécifique de la MS, mais trois sont très évocatrices : 1) un syndrome inflammatoire, 2) une hyperleucocytose à polynucléaires neutrophiles et 3) une hyperferritinémie avec une fraction glycosylée inférieure à 20 %. À l’état normal, la forme glycosylée représente 60 à 80 % de la forme circulante. Au cours de la maladie de Still, le pourcentage de la fraction glycosylée est plus bas comparé aux autres pathologies inflammatoires et auto-inflammatoires [7]. Cette fraction apparaît diminuée en phase active mais aussi durant les phases de rémission. La combinaison hyperferritinémie 5 fois supérieure à la normale, couplée à une fraction glycosylée < 20% correspond à une spécificité de 93 % et une sensibilité de 40 % pour la maladie de Still [18]. Malheureusement, dans notre étude, du fait de son coût élevé par rapport aux possibilités financières des patients, ce marqueur biologique n'a pu être dosé que chez un seul patient. De ce fait, il nous semble que les critères de Yamaguchi et al. [19], demeurent plus accessibles pour le diagnostic de MS en Afrique subsaharienne que ceux de Fautrel et al. [9].
Sur le plan thérapeutique, les anti-inflammatoires non stéroïdiens (AINS), efficaces dans la MS chez l'enfant, ne semblent efficaces que chez 20 % des adultes atteints de MS dans les séries européennes [12]. C'est pourquoi notre choix s'est tourné d'emblée vers une corticothérapie isolée pouvant être associée au méthotrexate. La corticothérapie a été utilisée à la dose de 0,5 à 1 mg/ kg/jour de prednisone chez tous nos patients, avec introduction de méthotrexate à la dose de 0,3 mg/ kg/jour, dans un but d’épargne cortisonique, mais également dans les atteintes articulaires du fait de son efficacité démontrée, en cas de corticorésistance (4/10 patients). Les récurrences sont moins importantes lorsque le méthotrexate est d'emblée associé à la corticothérapie (8 % dans la série de Diallo et al.) [5]. En cas d’échec thérapeutique, la discussion se porte sur l'instauration de biothérapies. Ces thérapeutiques, quoique novatrices et très efficaces, trouvent leurs limites en Afrique subsaharienne du fait de leur coût élevé d'une part, et de la résurgence de tuberculose qu'elles engendrent dans un continent où cette affection sévit à l’état d'endémie, d'autre part [6].
Dans tous les cas, nos patients n'ont pas pu être sevrés des thérapeutiques (corticothérapie et méthotrexate). Quatre de nos patients ont présenté une corticorésistance imposant l'adjonction de méthotrexate, et nous déplorons un décès par syndrome d'activation lympho-histiocytaire.
Conclusion
Les données de la MS en Afrique subsaharienne demeurent parcellaires. Notre préférence pour le diagnostic va vers les critères de Yamagushi et al. [19]. Notre étude confirme un faible nombre d'atteinte hépatique et ganglionnaire, ainsi qu'une absence d'atteinte cardiaque. Elle nécessite d’être confortée par d'autres études sur une cohorte plus importante. De même, la fréquence du diagnostic en saison sèche (saison de faibles températures) devrait être étudiée sur une plus grande cohorte.
Source de financement
Ce travail n'a bénéficié d'aucune source de financement.
Contribution des auteurs et autrices
Iba Ba Josaphat : conception et rédaction Mfoumou Annick Flore : rédaction et relecture Nseng Nseng Ondo Ingrid : relecture et collecte de données
Kanganga Ekomy Arthur : recherche bibliographique, relecture
Ledaga Lentombo Léonie Esther, Igala Marielle, et Kombila Ulrich Davy : relecture Boguikouma Jean Bruno : conception, relecture, approbation de la version finale
Liens d'intérêts
Les auteurs et autrices déclarent ne pas avoir de liens d'intérêts.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arlet JB Le Thi Huong DB Pouchot J Piette JC Physiopathologie de la maladie de Still de l'adulte Rev Med Interne 2005 Jul 2675495610.1016/j.revmed.2004.11.02115996569 · doi ↗ · pubmed ↗
- 2Bywaters EG Still's disease in the adult Ann Rheum Dis 1971 Mar 3021213310.1136/ard.30.2.1215315135 PMC 1005739 · doi ↗ · pubmed ↗
- 3Cheikhrouhou Abdelmoula L Tekaya R Ben Hadj Yahia C Chaabouni L Zouari R La maladie de Still de l'adulte : à propos de 11 cas Tunis Med 2007 Jun 856461417644897 · pubmed ↗
- 4de Jager W Hoppenreijs EP Wulffraat NM Wedderburn LR Kuis W Prakken BJ Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study Ann Rheum Dis 2007 May 6655899810.1136/ard.2006.06185317170049 PMC 1954617 · doi ↗ · pubmed ↗
- 5Diallo S Pouye A Ndongo S Fall S Diop TM Maladie de Still de l'adulte : étude de 10 observations sénégalaises Rev Rhum 20087510-111105
- 6du Toit T Esterhuizen TM Tiffin N Abulfathi AA Reuter H Decloedt EH Incident tuberculosis disease in patients receiving biologic therapies in the Western Cape, South Africa from 2007 to 2018 BMC Infect Dis 2020 Nov 3035190010.1186/s 12879-020-05624-0PMC 770624033256634 · doi ↗ · pubmed ↗
- 7El Jabri Z Maladie de Still de l'adulte à propos de 8 cas Thèse de Médecine, 2019, n°40, Faculté de médecine et de pharmacie, université Sidi Mohamed Ben Abdellah, Fès (Maroc)
- 8Fautrel B Mitrovic S De Matteis A Bindoli S Antón J Belot A Bracaglia C Constantin T Dagna L Di Bartolo A Feist E Foell D Gattorno M Georgin-Lavialle S Giacomelli R Grom AA Jamilloux Y Laskari K Lazar C Minoia F Nigrovic PA Oliveira Ramos F Ozen S Quartier P Ruscitti P Sag E Savic S Truchetet ME Vastert SJ Wilhelmer TC Wouters C Carmona L De Benedetti F EULAR/ P Re S recommendations for the diagnosis and management of Still's disease, comprising systemic juvenile idiopathic arthritis and adult-onset Still's disease Ann Rheum Dis 20 · doi ↗ · pubmed ↗