Amyloid light-chain (AL) amyloidosis and follicular lymphoma: a case report
Amine Benmoussa, Hajar Maatoui-Belabbes, Reda Allali, Meriem Regragui, Meriem Qachouh, Siham Cherkaoui, Mouna Lamchahab, Mohamed Rachid, Abdellah Madani, Nisrine Khoubila

TL;DR
A rare case of a 67-year-old patient with both follicular lymphoma and AL amyloidosis is reported, highlighting the unusual co-occurrence of these two conditions.
Contribution
This case report presents a rare association between follicular lymphoma and AL amyloidosis, expanding understanding of their potential coexistence.
Findings
The patient had follicular lymphoma confirmed by biopsy and later developed AL amyloidosis.
Standard chemotherapy protocols showed no response, but amyloid deposits were later detected.
The patient is currently undergoing treatment with bortezomib, prednisone, and bendamustine.
Abstract
L´association de l´amylose AL et le lymphome folliculaire B est extrêmement rare car le clone secrétant la chaine légère amyloïdogène est généralement plasmocytaire. Nous rapportons le cas d´un patient âgé de 67 ans sans ATCDs pathologiques particuliers ayant présenté une altération de l´état général avec une dysphonie et dysphagie d´aggravation progressive avec une volumineuse masse du cavum dont la biopsie était en faveur d´un lymphome non hodgkinien folliculaire B grade 1 et 2. La tomodensitométrie (TDM) cervico-thoraco-abdomino-pelvienne a objectivé une masse nasopharyngée de 70 mm x 40 mm étendue sur 60 mm. La biopsie ostéomédullaire était normale ainsi que le bilan préthérapeutique. Le malade a reçu 4 cures de protocole Rituximab plus CHOP (cyclophosphamide, adriamycine, prednisone et oncovin) sans réponse puis 3 cures Rituximab plus DHAOX (dexamethasone, aracytine haute dose, et…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
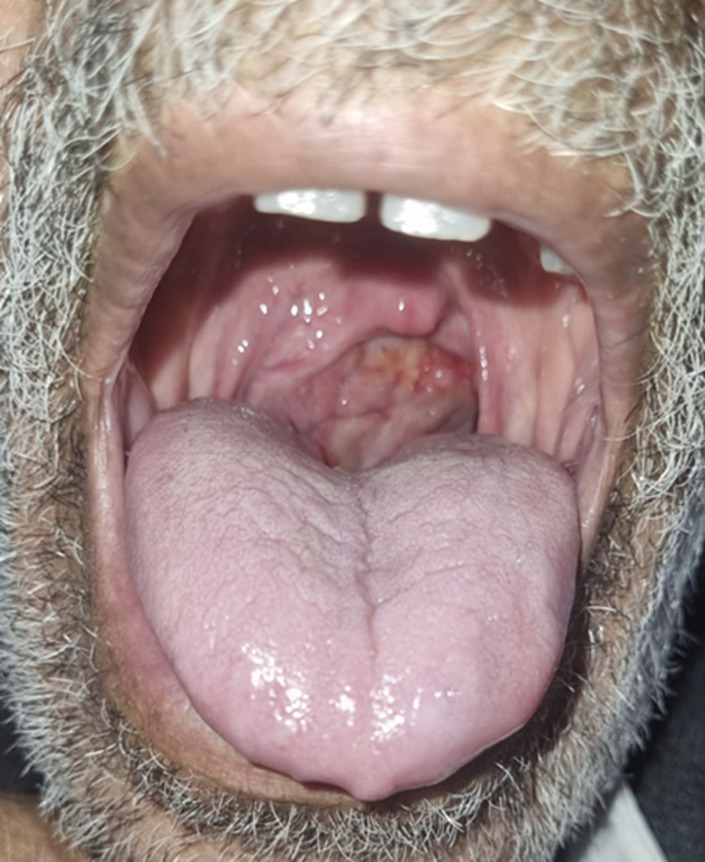 Figure 1
Figure 1 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyloidosis: Diagnosis, Treatment, Outcomes · Dermatological and Skeletal Disorders
Introduction
L´amylose AL est le plus souvent primitive parfois associée à un myélome multiple. Son association à certains types histopathologiques d´un lymphome non hodgkinien (LNH) B (lymphome lymphoplasmocytaire, le lymphome à cellules B de la zone marginale ganglionnaire/le lymphome MALT, la leucémie lymphoïde chronique/lymphome lymphocytique et d´autres) est rapportée dans la littérature [1-3]. Cependant, à notre connaissance, 2 cas d'amylose AL en association avec le lymphome folliculaire B ont été précédemment rapportés [4,5]. Nous présentons les caractéristiques cliniques et l´évolution d´un patient atteint d´une amylose AL associée à un lymphome folliculaire B suivi dans notre service d´hématologie clinique.
Patient et observation
Informations relatives au patient: il s´agissait d´un patient âgé de 67 ans, sans antécédents pathologiques qui a consulté pour une gêne à la déglutition avec une dysphonie et une altération de l´état général.
Résultats cliniques: l'examen clinique initial trouvait un patient conscient avec un score de Glasgow à 15/15, apyrétique, une tension artérielle de 12/07 cmHg, une saturation en oxygène de 100%, une fréquence cardiaque à 80/min, des conjonctives normocolorées avec la présence d´une volumineuse masse au niveau du cavum (Figure 1). Il n´y avait pas d´hépatomégalie ni de splénomégalie, les aires ganglionnaires étaient libres, le reste de l'examen somatique était normal.
volumineuse masse au niveau du cavum
Chronologie: le patient présentait depuis 6 mois une gêne à la déglutition avec une dysphonie, le tableau clinique s´est aggravé par l´installation d´une dysphagie aux solides avec une altération de l´état général (amaigrissement chiffré à 15kg/6 mois).
Démarche diagnostique: la TDM cervico-thoraco-abdomino-pelvienne a objectivé une masse nasopharyngée de 70 mm x 40 mm étendue sur 60 mm. Le bilan biologique du patient était normal (la numération de formule sanguine, le bilan rénal et hépatique, la lacticodéshydrogénase et les sérologies VIH VHC VHB). L´étude histologique et immunohistochimique de la biopsie nasopharyngée était en faveur de LNH folliculaire B grade 1,2 CD20+; CD19+; CD79a+; CD10+ sur 2 lectures dans deux laboratoires différents. La biopsie ostéomédullaire était normale ainsi que le bilan préthérapeutique.
Intervention thérapeutique: le patient a reçu 4 cures RCHOP 21 (rituximab 375mg/m^2^ en intraveineux (iv), cyclophosphamide 750 mg/m^2^ iv, oncovin 2 mg iv, prednisolone 100 mg par voie orale, et doxorubicine 50 mg/m^2^ (iv) sans réponse puis 3 cures RDHAOX (rituximab 375 mg/m^2^ en intraveineux (iv) à J1, aracytine haute dose 2 g/m^2^ x 2 iv à J2, dexamethasone 40 mg de J1 à J4, et oxalipatine 100 mg/m^2^ à J1) sans réponse clinique.
Suivi et résultats des interventions thérapeutiques: la persistance et l´augmentation de la masse nasopharyngée a conduit à la réalisation de la trachéotomie, la biopsie de la masse nasopharyngée a objectivé la disparition de l´infiltration lymphoïde B avec présence des dépôts amyloïdes AL type kappa (Figure 2).
présence des dépôts éosinophiles amorphes exprimant une biréfringence vert pomme à la coloration rouge Congo en lumière polarisée
L´immunoélectrophorèse des protéines plasmatiques a mis en évidence la présence de l´immunoglobuline M kappa, le dosage des chaines légères n´a pas été réalisé par manque de moyens, le myélogramme et une deuxième biopsie ostéomédullaire étaient normaux, la TEP scan a objectivé un processus nasopharyngé hypermétabolique sans autres anomalies, l´évaluation cardiaque (ECG, peptides natriurétiques, troponine, echocoeur) et rénale étaient sans particularités, le malade est actuellement sous protocole bortezomib, prednisone et bendamustine avec bonne évolution clinique après première cure.
Point de vue du patient: il a rapporté une nette amélioration après la première cure du protocole bortezomib, prednisone et bendamustine avec diminution de la taille de la masse nasopharyngée et la disparition de la dysphagie.
Consentement éclairé: le patient a donné son consentement pour publication de son cas.
Discussion
L'amylose à chaînes légères (AL) est une maladie proliférative des plasmocytes monoclonaux rarement des lymphoplasmocytes ou lymphocytes caractérisés par la précipitation dans les tissus intercellulaires de chaînes légères monoclonales d´immunoglobuline sous forme de fibrilles qui résistent à la protéolyse, provoquant des perturbations mécaniques et un stress oxydatif local dans les organes affectés tels que le cœur, les reins, le foie et le tractus gastro-intestinal. Elle est le plus souvent associée à un néoplasme plasmocytaire sous-jacent, mais elle est rarement causée par un lymphome non hodgkinien (LNH) à cellules B. Les cellules lymphoïdes tumorales B en tant que l´origine productrice des chaînes légères d'Immunoglobulin amyloïdogènes n'ont été identifiées que dans environ 2% des cas d'amylose AL [6]. Lorsque l´amylose AL est associée à un LNH, une paraprotéine IgM est souvent présente. L´amylose IgM est le plus souvent associée au lymphome lymphoplasmocytaire/macroglobulinémie de Waldenström (74% des cas). Une association avec d'autres types de LNH est extrêmement rare et n'a été rapportée que dans de petites séries de cas [7].
La présentation des amyloses IgM se distingue de celle des non IgM par la fréquence moindre des atteintes cardiaques (32-45% dans l'amylose IgM contre 70% dans l'amylose non IgM), plus de localisations ganglionnaires ou pulmonaires, et l´implication plus fréquente d´une chaine légère kappa. Le seul fait d´avoir une IgM monoclonale associée à une amylose ne suffit pas pour conclure que le clone sécrétant l´IgM est lymphoplasmocytaire ou lymphocytaire. Il est fondamental de préciser au mieux la nature de la prolifération B monoclonale, par une étude précise de tissu pathologique, la moelle osseuse (et/ou des lymphocytes circulants) comportant un immunophénotypage et, si possible, une étude génétique [6,7]. Ce qui rend notre cas rare, c´est que nous avons découvert que seulement 2 cas de coexistence d´amylose AL et le lymphome folliculaire ont été rapportés dans la littérature, 1 seul cas dans une large série de cas française et un cas japonais avec une amylose AL et lymphome folliculaire avec différenciation plasmocytaire [4,5]. Notre patient est atteint d´amylose AL de type IgM localisée stade I associée au LNH folliculaire de forte masse tumorale.
Les patients atteints d'une amylose IgM à un stade précoce selon le score de la Mayo Clinic (stade I ou II) ont une survie plus faible que les patients atteints d'une forme non IgM à un stade précoce (75% de survie globale à 5 ans pour l´amylose IgM de stade 1 contre >90% pour la forme non IgM) et ont des taux de réponse au traitement plus faibles [8]. Le développement de traitements efficaces pour ce sous-type rare d'une maladie déjà rare est donc un défi, les décisions thérapeutiques reposent principalement sur des données rétrospectives. Notre patient avait une évolution défavorable avec persistance et augmentation en taille de la masse nasopharyngée malgré la disparition de l´infiltration lymphoïde tumorale initiale au niveau du cavum après chimiothérapie.
L'objectif du traitement est d'éliminer la production de la protéine amyloïde en ciblant le LNH à cellules B sous-jacent. Les traitements typiques des amyloses associées aux proliférations plasmocytaires ne sont pas efficaces dans ce sous type, car ils ne ciblent pas le clone tumoral lymphoïde [9].
L´amylose IgM a toujours été traitée par une chimiothérapie conventionnelle seule (par exemple, melphalan, cyclophosphamide), les régimes à base de rituximab associé au bortezomib, aux immunomodulateurs et aux inhibiteurs de Bruton tyrosine kinase (BTKi) ont été introduits dans des études rétrospectives et des séries de cas de petite taille [10]. Le traitement du premier cas de LNH folliculaire avec amylose AL publié dans la série française n'a pas été précisé, tandis que le deuxième cas japonais a subi une résection iléale puis il a reçu 6 cures de chimiothérapie R-THP-COP (rituximab, cyclophosphamide, pirarubicin, vincristine, et prednisone) avec rémission complète.
A l´heure actuelle, il n'existe aucune ligne directrice standard en matière de traitement, d´où l´intérêt de réaliser d´autres études de cas pour déterminer la meilleure stratégie thérapeutique possible.
Conclusion
L´amylose AL est le plus souvent primitive ou associée à un myélome multiple, cependant son association à un lymphome non hodgkinien doit être recherchée. L´association de l´amylose AL et le lymphome folliculaire est rarement décrite dans la littérature, d'où l´intérêt d´autres études de cas pour clarifier la physiopathologie, les caractéristiques cliniques et évolutives et discuter les modalités thérapeutiques de cette association.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sanchorawala V Blanchard E Seldin DCO´Hara C Skinner M Wright DGAL amyloidosis associated with B cell lymphoproliferative disorders: frequency and treatment outcomes Am J Hematol 2006 Sep 81969251679506010.1002/ajh.20635 · doi ↗ · pubmed ↗
- 2Telio D Bailey D Chen C Crump M Reece D Kukreti V Two distinct syndromes of lymphoma-associated AL amyloidosis: a case series and review of the literature Am J Hematol 2010 Oct 851080582087295910.1002/ajh.21814 · doi ↗ · pubmed ↗
- 3Kourelis TV Gertz M Zent C Lacy M Kyle R Kapoor Pet al Systemic amyloidosis associated with chronic lymphocytic leukemia/small lymphocytic lymphoma Am J Hematol 2013 May 88537582350884010.1002/ajh.23413 · doi ↗ · pubmed ↗
- 4Terrier B Jaccard A Harousseau JL Delarue R Tournilhac O Hunault-Berger Met al The clinical spectrum of Ig M-related amyloidosis: a French nationwide retrospective study of 72 patients Medicine (Baltimore)2008 Mar 872991091834480710.1097/MD.0b 13e 31816 c 43b 6 · doi ↗ · pubmed ↗
- 5Matsumoto Y Masuda T Nishimura A Horie H Harada K Yoshida Met al A case of AL amyloidosis associated with follicular lymphoma with plasmacytic differentiation Int J Hematol 2020 Feb 11123173233154285210.1007/s 12185-019-02734-7 · doi ↗ · pubmed ↗
- 6Sachchithanantham S Roussel M Palladini G Klersy C Mahmood S Venner C Pet al European Collaborative Study Defining Clinical Profile Outcomes and Novel Prognostic Criteria in Monoclonal Immunoglobulin M-Related Light Chain Amyloidosis J Clin Oncol 201634172037452711459210.1200/JCO.2015.63.3123 · doi ↗ · pubmed ↗
- 7Basset M Defrancesco I Milani P Nuvolone M Rattotti S Foli Aet al Nonlymphoplasmacytic lymphomas associated with light-chain amyloidosis Blood 2020135429363171495210.1182/blood.2019002762 · doi ↗ · pubmed ↗
- 8Wechalekar AD Chakraborty R Lentzsch S Systemic Amyloidosis due to Low-Grade Lymphoma Hematol Oncol Clin North Am 2020 Dec 346102710393309942110.1016/j.hoc.2020.08.016 · doi ↗ · pubmed ↗