Lymphoproliferation and hyper-IgM as the first manifestation of activated phosphoinositide 3-kinase δ syndrome: A case report
Mónica Fernandes-Pineda,, Andrés F. Zea-Vera

TL;DR
A 15-year-old male presented with lymphoproliferation and hyper-IgM, later diagnosed with a rare immune disorder caused by a mutation in the PIK3R1 gene.
Contribution
This case report highlights a novel splice site mutation in PIK3R1 presenting as hyper-IgM syndrome.
Findings
A heterozygous splice site mutation in PIK3R1 was identified in a patient with lymphoproliferation and hyper-IgM.
The mutation was located in a well-established hotspot of the PIK3R1 gene.
The case underscores the importance of genetic testing for accurate diagnosis of immune disorders.
Abstract
Activated phosphoinositide 3-kinase δ syndrome is an inborn error of immunity due to mutations within the genes responsible for encoding PI3Kδ subunits. This syndrome results in an excessive activation of the phosphoinositide 3-kinase signaling pathway. Gain- of-function mutations in the gene PIK3R1 (encoding p85α, p55α, and p50α) lead to the development of the activated PI3K δ syndrome. Notably, the clinical presentations of this syndrome often closely resemble those of other primary immunodeficiencies. We present a case involving a 15-year-old male who displayed an immunological phenotype that bore a striking resemblance to hyper-IgM syndrome. Whole exome sequencing was undertaken to pinpoint the underlying genetic mutation. Our investigation successfully identified a heterozygous splice site mutation previously reported within the well-established hotspot of the PIK3R1 gene…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
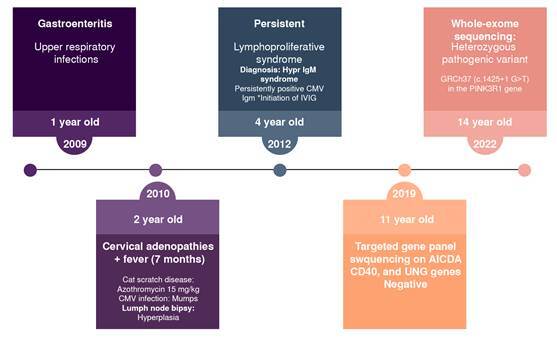 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmunodeficiency and Autoimmune Disorders · Chronic Lymphocytic Leukemia Research · Blood disorders and treatments
Class IA phosphatidylinositol 3-kinases (PI3K) constitute a vital family of heterodimeric enzymes consisting of a 110-kDa catalytic subunit (ρ110α, β, or δ) paired with a regulatory subunit (ρ85α, ρ85β, ρ55α, ρ50α, or ρ55γ). These PI3K enzymes play a pivotal role in immune cell activation, orchestrating essential functions such as cell growth, proliferation, survival, migration, and differentiation 1.
Genetic mutations in genes encoding PI3K subunits can lead to various immune-related disorders. Specifically, mutations in the PIK3R1 gene, responsible for encoding phosphatidylinositol 3-kinase regulatory subunits, underlie the condition known as activated phosphoinositide 3-kinase δ syndrome 2 (APDS2), while mutations in PIK3CD are associated with APDS1 2.
The clinical manifestations of these syndromes encompass symptoms akin to those of other inborn errors of immunity, including recurrent bacterial respiratory infections, heightened susceptibility to herpes virus infections, lymphoproliferation, autoimmunity, enteropathy, and lymphoma 3.
In this case report, we present the clinical profile of a male patient harboring a PIK3R1 mutation, which has led to the manifestation of APDS2, characterized by hyper-IgM and lymphoproliferation, evident since childhood.
Case report
A 15-year-old Colombian boy, born to non-consanguineous healthy parents, was the first child in the family with one unaffected sibling. His medical history revealed an uneventful vaccination with the Calmette- Guérin bacillus (BCG) at birth. At five months of age, he began experiencing recurrent upper respiratory tract infections. A subsequent episode of gastroenteritis at six months necessitated oral antibiotic treatment. At two years old, the patient was admitted to the emergency room due to the bronchoaspiration of a peanut, requiring bronchoscopy for removal.
During a routine physical examination, visible tonsils and bilateral submandibular and cervical lymphadenopathies (greater than 2 cm in diameter) were noted. However, other physical findings were unremarkable, with no abnormalities detected in the lungs, heart, abdomen, or nervous system. Upon reassessment, the patient’s mother reported a persistent high fever (> 38.5 °C), night sweats, and a chronic cough over the previous six months. Subsequently, the patient was admitted to the pediatric infectious disease service, where tuberculosis was ruled out, and additional investigations were conducted.
Serological testing revealed positive IgM and negative IgG for cytomegalovirus (CMV), while tests for Epstein-Barr (EBV) mononucleosis were negative. Viral load assessments for EBV or CMV were unavailable (table 1). A lymph node biopsy demonstrated lymphoid hyperplasia without evidence of malignancy. Infectious complications were considered, and due to suspected acute CMV infection and potential exposure to pets, cat scratch disease was suspected, leading to the prescription of oral antibiotics (azithromycin).
Table 1. Patient’s results of the laboratory testsLaboratory parameterDate 2011 (3 yo)Date 2012 (4 yo)Date 2015 (7 yo)Date 2019 (11 yo)Date 2022 (14 yo)Reference valuesLeukocyte count (cel/pl)7,03015,700
7,3106,000 - 17,500 (3 yo)Neutrophil count (cel/pl)1,3606,140
2,9201,500 - 8,500Lymphocyte count (cel/pl)4,2906,760
3,3903,000 - 9,500Hematocrit (%)27.730.7
32.231 - 43 (3 - 14 yo)Platelet count (cel/pl)692,000440,000
435,000150,000 - 450,000IgG (mg/dl) < 320787415.34480.51700 - 1,600IgA (mg/dl) < 5< 33< 40<4070 - 400IgM (mg/dl) 1,290879.1468.5651.3240 - 230IgE (UI/ml) 15.50< 0.100< 0.100< 200B lymphocytes (CD19+) cel/pl (%)
169 (3.3)200 - 600 (8 - 24)T lymphocytes (CD3+) cel/pl (%)
3,775.5 (80.5)1,088.1 - 2,087.9 (53.3 - 75.3)T lymphocytes (CD3+CD4+) cel/pl (%)
963.0 (20.5)639.5 - 1,278.5 (30.7 - 46)T lymphocytes (CD3+CD8+) cel/pl(%)
2,198 (46.6)377.1 - 876.9 (20.3 - 36.1)NK cells (CD3-CD56+CD16+) cel/pl (%)
690 (13.6)70 - 1,200 (6 - 27)PPD (mm)
0> 10Toxoplasma IgG (UI/ml)0
< 15Toxoplasma IgM (UI/ml)0.288
< 0.4MonotestNegative
Citomegalovirus IgG (UI/ml)00
< 15Citomegalovirus IgM (UI/ml)1.3071.976
< 0.399VIH (index)0.37
< 1AgSHBV (index)0.170.75
< 1.2NK: natural killer; PPD: purified protein derivative test; HIV: human immunodeficiency virus; AgSHBV: hepatitis B surface antigen
At three years of age, the patient was readmitted with suspected lymphoproliferative disease attributed to the enlargement of cervical nodules. Physical examination revealed persistent submandibular and cervical lymphadenopathy but no signs of hepatosplenomegaly. Bone marrow aspirate analysis showed a cellular composition comprising 5% megakaryocytes, 61% myeloid lineage, 31% lymphocytes, and 3% eosinophils, with no evidence of tumorous growth.
Serological assessments indicated persistently negative EBV and CMV IgG, yet CMV IgM remained consistently positive, with escalating titers. Serum immunoglobulin levels revealed elevated IgM (1,290 mg/ dl) with undetectable IgG and IgA, leading to the diagnosis of hyper- IgM immunodeficiency in 2012, according to the European Society for
Immunodeficiencies (ESID) criteria. Consequently, monthly intravenous immunoglobulin treatment (400 - 600 mg/kg) was recommended, along with the prophylactic use of macrolides (azithromycin). Despite these measures, lymphadenopathies persisted.
In 2019, targeted gene panel sequencing for AICDA, CD40, CD40L, and UNG genes yielded negative results, failing to establish a molecular diagnosis during the clinical evaluation and follow-up. A renewed genetic assessment in 2022, when he was 14 years old, employing a comprehensive whole exome sequencing approach, revealed the heterozygous pathogenic variant c.1425+1G>T in the PIK3R1 gene, confirming the diagnosis of APDS2 (figure 1).
Figure 1. Clinical history timelineCMV: cytomegalovirus; IV Ig: intravenous immunoglobulin
Currently, the individual receives monthly intravenous immunoglobulin substitution and is being closely observed for any changes in lung function. Recent scans showed no evidence of bronchiectasis or new masses. It is worth mentioning that the patient was last hospitalized at 11 years old due to pansinusitis. Since then, he has not had an additional decline in their health condition, although he still has persistent splenomegaly and adenomegaly.
Ethical considerations
The authors obtained the written informed consent from the mother and the assent from the patient mentioned in the article. The corresponding author possesses this document.
Discussion
We present the first published case of activated phosphoinositide 3-kinase δ syndrome 2 (APDS2) in Colombia, involving a young boy with a heterozygous splice site mutation in the PIK3R1 gene. The patient’s clinical presentations were chronic lymphadenopathy, recurrent viral and bacterial infections, and hepatosplenomegaly. This case underscores the diagnostic complexities often encountered in patients with APDS1/2, as the initial diagnosis of hyper-IgM syndrome gave way to a genetic diagnosis obtained through subsequent whole exome sequencing.
Phosphoinositide 3-kinases (PI3Ks) are integral enzymes within the PI3K-AKT-mTOR signaling pathway, playing a crucial role in the metabolism, differentiation, proliferation, growth, survival, and migration of immune cells 4. Mutations affecting the ΡΙ3Κδ regulatory subunit lead to the constant hyperactivation of the Akt-mTOR pathway in B and T lymphocytes. This hyperactivation -due to mutations in the PIK3R1 gene coding for regulatory subunits- results in APDS2, marked by progressive lymphopenia and compromised differentiation and function of T and B cells 5.
APDS2 patients frequently suffer from multiple infectious complications, including recurrent upper and lower respiratory tract infections, such as sinusitis, otitis media, and pneumonia 6. These complications arise due to defects in antibody and cytokine production. Our patient experienced multiple hospitalizations for otitis media and sinusitis, which responded well to antibiotic therapy. Despite these recurrent respiratory infections, our patient did not exhibit bronchiectasis in the follow-up thorax computed tomography scan, sometimes developed by APDS2 patients after repeated respiratory infections 7.
Notably, our patient initially presented with CMV infections, ultimately leading to the diagnosis of the hyper-IgM phenotype. The literature has reported that APDS2 patients may experience recurrent infections caused by herpes family viruses, including EBV, CMV, herpes simplex virus, and varicella-zoster virus 6.
Beyond infectious complications, APDS2 patients can encounter non- infectious issues, including lymphadenopathy, hepatosplenomegaly, autoimmune and autoinflammatory disorders, malignancies, and growth problems. In our case, recurrent hospitalizations were attributed to cervical and submandibular lymphadenopathy, often linked to overactive proliferation in lymph node germinal centers, frequently triggered by EBV infections 3. Hepatosplenomegaly, observed in other APDS2 cases, was also evident in our patient 8. Moreover, the lymph nodes exhibited hyperplasia, and bone marrow biopsies ruled out hematological malignancies.
The common occurrence of the hyper-IgM phenotype in APDS2 patients may be a consequence of the dysregulation of PI3K signaling and classswitching recombination in developing B lymphocytes 9. Our patient’s immunoglobulin profile strongly suggested hyper-IgM syndrome despite negative results on molecular diagnostic tests for the AICDA, CD40, and UNG genes. Consequently, the clinical management of the patient was done according to the hyper-IgM syndrome diagnosis.
Notably, the variants localized at c.1425 of the PIK3R1 gene are considered hotspot mutations and affect the donor splice site (c.1425+1G>A, C, T, c.1425+2T>G, A, c.1425+2,3delTG) and the acceptor splice site (c.1300-1G>C) of intron 10 4. Consequently, exon 10 is skipped, leading to the ρ85α regulatory subunit truncation and the activation of PI3K signaling activity in APDS 10. Our patient harbored the previously reported heterozygous pathogenic variant c.1425+1G>T in the PIK3R1 gene 6^,^11.
It is worth noting that APDS was first described in 2014, and since then between 47 and 100 new cases have been reported. Our patient exhibited symptoms in 2010, four years before the disease was described 12 (figure 1). The field of inborn errors of immunity is rapidly evolving, with new genes reported and new diseases defined annually 13. This field requires ongoing assessment of patients being studied for primary immunodeficiency. The importance of characterizing such orphan diseases has led to the development of broad population registries, like the European one, which includes 170 APDS patients and evaluates their heterogeneity 14.
In Latin America, increasing genetic diagnoses in patients with common variable immunodeficiency or hyper-IgM syndrome, whose etiology remains unknown, can potentially lead to therapeutic changes 15. Some inborn errors of immunity have specific treatments, such as leniolisib in APDS1 and APD2, in phase three clinical trials. Leniolisib, a ΡΙ3Κδ inhibitor, can mitigate long-term complications associated with lymphoproliferation and positively impact immune dysregulation in APDS patients 16. This treatment helps to decrease lymphadenopathy and splenomegaly, increase B lymphocyte counts, address cytopenias, and improve overall symptoms 17. This underscores the need for greater awareness and genetic testing in the Latin American context to improve patient care and management 18.
Conclusion
Initially, the patient’s clinical immunoglobulin profile suggested hyper-IgM syndrome. However, inborn errors of immunity represent a dynamic field characterized by an ever-expanding array of newly described gene disorders. This evolving landscape underscores the importance of considering these novel gene disorders for patients without a molecular diagnosis.
Given the inherent heterogeneity within the spectrum of inborn errors of immunity, the identification of gene mutations becomes an invaluable tool in accurately diagnosing patients with clinical manifestations resembling autoimmune disorders, lymphoproliferative conditions, and antibody deficiencies. As our understanding of these conditions continues to evolve, early genetic diagnoses can significantly enhance patient care and management.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nguyen T Lau A Bier J Cooke KC Lenthall H Ruiz-Diaz S Human PIK 3R 1 mutations disrupt lymphocyte differentiation to cause activated PI 3Kδ syndrome 2J Exp Med 2023220 e 2022102010.1084/jem.2022102036943234 PMC 10037341 · doi ↗ · pubmed ↗
- 2Lucas CL Kuehn HS Zhao F Niemela JE Deenick EK Palendira U Dominantactivating germline mutations in the gene encoding the PI(3)K catalytic subunit p 110δ result in T cell senescence and human immunodeficiency Nat Immunol 201415889710.1038/ni.277124165795 PMC 4209962 · doi ↗ · pubmed ↗
- 3Singh A Joshi V Jindal AK Mathew B Rawat A An updated review on activated PI 3 kinase delta syndrome (APDS)Genes Dis 20207677410.1016/j.gendis.2019.09.01532181277 PMC 7063426 · doi ↗ · pubmed ↗
- 4Okkenhaug K Vanhaesebroeck B PI 3K in lymphocyte development, differentiation and activation Nat Rev Immunol 2003331733010.1038/nri 105612669022 · doi ↗ · pubmed ↗
- 5Redenbaugh V Coulter T Disorders related to PI 3Kδ hyperactivation: Characterizing the clinical and immunological features of activated PI 3-kinase delta syndromes Front Pediatr 2021910.3389/fped.2021.702872 PMC 837443534422726 · doi ↗ · pubmed ↗
- 6Yazdani R Hamidi Z Babaha F Azizi G Fekrvand S Abolhassani H PIK 3R 1 mutation associated with hyper Ig M (APDS 2 syndrome): A case report and review of the literature Endocr Metab Immune Disord Drug Targets 20191994195810.2174/187153031966619022511473930799802 · doi ↗ · pubmed ↗
- 7Condliffe AM Chandra A respiratory manifestations of the activated phosphoinositide 3-kinase delta syndrome Front Immunol 2018910.3389/fimmu.2018.00338 PMC 584494029556229 · doi ↗ · pubmed ↗
- 8Durandy A Kracker S Increased activation of PI 3 kinase-δ predisposes to B-cell lymphoma Blood 202013563864310.1182/blood.201900207231942637 · doi ↗ · pubmed ↗