Characterization of the complete mitochondrial genome of Hynobius bambusicolus Wang, Othman, Qiu and Borzée, 2023 (Amphibia, Caudata, Hynobiidae) and its phylogenetic implications
Yanpin Huang, Helin Wang, Haoran Luo, Honghui Zhong, Qingxian Lin, Xiaoping Zhou

TL;DR
This paper reports the full mitochondrial genome of a newly discovered salamander species and its evolutionary placement.
Contribution
The study provides the first complete mitochondrial genome sequence for Hynobius bambusicolus and its phylogenetic analysis.
Findings
The mitogenome of H. bambusicolus is 16,406 bp long with typical mitochondrial gene content.
Phylogenetic analysis shows H. bambusicolus is the basal branch in the Southern Chinese Hynobius clade.
Abstract
Hynobius bambusicolus (Caudata, Hynobiidae) is a recently described species, identified in 2022, and is thus not widely known. In this study, we sequenced and annotated the complete mitogenome of H. bambusicolus. The resulting mitochondrial genome is 16,406 bp in length and comprises 13 protein-coding genes (PCGs), two ribosomal RNA genes (rRNA), 22 transfer RNA genes, and a non-coding region. The base composition of the mitogenome is 33.3% A, 32.1% T, 20.8% C, and 13.8% G. The phylogenetic trees indicated that H. bambusicolus is the basal branch within the Southern Chinese Hynobius clade.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1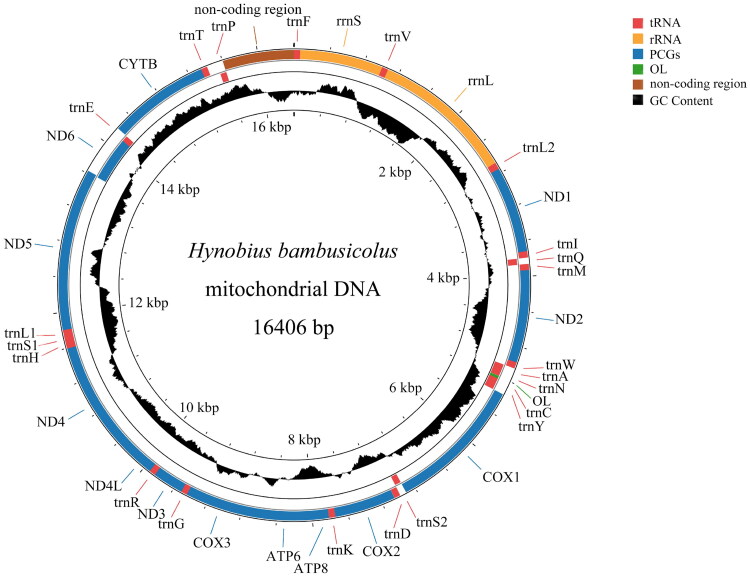 Figure 2
Figure 2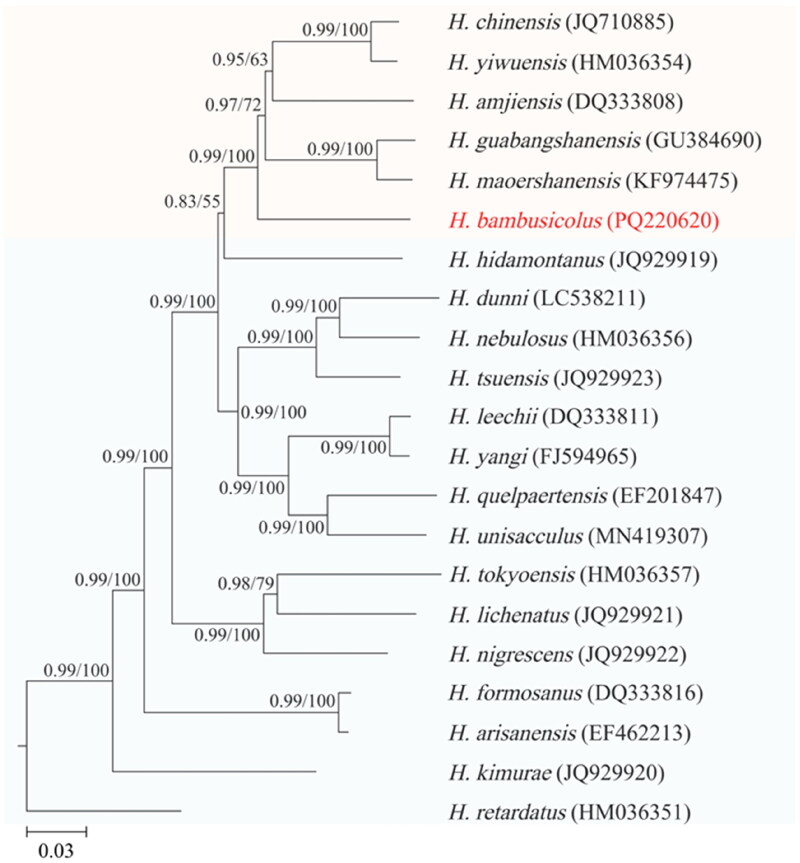 Figure 3
Figure 3| Species | Accession number | Source | Localities |
|---|---|---|---|
|
| This study | Fujian, China | |
|
| Zhang et al. ( | Zhejiang, China | |
|
| Unpublished | Southern China | |
|
| Unpublished | Guabang Mountain, China | |
|
| Zheng et al. ( | Zheijiang, China | |
|
| Huang et al. ( | Guangzi, China | |
|
| Zhang et al. ( | Northeastern China | |
|
| Unpublished | Central Taiwanese Island | |
|
| Zhang et al. ( | Northerncentral Taiwanese Island | |
|
| Unpublished | R. Korea | |
|
| Unpublished | R. Korea | |
|
| Unpublished | R. Korea | |
|
| Zheng et al. ( | Japan | |
|
| Unpublished | Japan | |
|
| Unpublished | Japan | |
|
| Igawa et al. ( | Japan | |
|
| Unpublished | Japan | |
|
| Unpublished | Japan | |
|
| Zheng et al. ( | Japan | |
|
| Unpublished | Japan | |
|
| Zheng et al. ( | Japan | |
|
| Pan et al. ( | Dabie Mountain, China |
- —Meihua Mountain National Nature Reserve Administration
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Amphibian and Reptile Biology · Microbial infections and disease research
Introduction
Hynobius is the most diverse genus in Hynobiidae family, which includes 66 species (Frost 2024). To date, the complete mitochondrial genomes have only been sequenced for approximately 20 Hynobius species. Hynobius bambusicolus Wang, Othman, Qiu and Borzée, 2023 was first discovered by Wang et al. (2023) in January 2022 in Quxi village, Liancheng County, Fujian, China (Figure 1). Currently, this species is known from a single locality, with an estimated population size likely to be fewer than 200 breeding individuals, aligning with the Critically Endangered (CR) status recommended by the IUCN Red List of Threatened Species (Wang et al. 2023). The colonies of H. bambusicolus inhabit artificial bamboo forests, which are significantly threatened by anthropogenic activities (Wang et al. 2023). To date, the molecular data available for H. bambusicolus is limited to segments of a single mtDNA gene, including rrnL, COX1, and CYTB (Wang et al. 2023). Phylogenetic trees constructed using these single or concatenated mtDNA gene segments have produced incongruent results, leaving the phylogenetic position of this species unresolved. In this study, we present the first complete mitochondrial genome of H. bambusicolus, and infer its phylogenetic position within the genus Hynobius based on the concatenation of two ribosomal RNA genes (rRNA) genes and 13 protein-coding genes (PCGs).
Reference image of the Hynobius bambusicolus (photographer: Zhenqi Wang, email: [email protected], used with permission). This photograph was captured in April 2024. The individual in the figure is an adult with a total length of 17 cm.
Materials and methods
Sample collection, DNA extraction, and sequencing
A dead larva of H. bambusicolus was collected in April 2024 from Quxi village, Liancheng County, Fujian, China (25.566° N, 116.938° E) and was deposited at −80 °C in the laboratory at College of the Environment and Ecology, Xiamen University, Xiamen, China (https://cee.xmu.edu.cn/, Xiaoping zhou, [email protected]) under voucher number CEE2024Anura-Hb-01. The larva specimen was identified as H. bambusicolus based on its brown body and blue speckles on dorsum, as well as its sequences of COX1 fragment 100.00% match to that reported for H. bambusicolus (GenBank accession number: OQ107447, Wang et al. 2023). A tail of the sample was sent to BGI Genomics Co., Ltd for DNA extraction and sequencing. DNA material was extracted using the Sodium Dodecyl Sulfate method (BGI Genomics Company Limited 2024). Genomic library was constructed using a BGI Optimal DNA Library Prep Kit (BGI-Shenzhen, China). The insert size was 300–400 bp. Sequencing was performed on the DNBSEQ platform using a pair-end sequencing protocol with a read length of 150 bp (PE150), and a total of 83,522,957 raw reads were yielded. Raw reads were filtered using SOAPnuke (Chen et al. 2018) to remove adapter sequences, contamination, and low-quality reads, which resulted in a total of 80,144,121 clean reads with the mean length of 150 bp.
Mitogenome assembly and annotation
The mitochondrial genome was assembled using NOVOPlasty 4.3.5 software (Dierckxsens et al. 2017) with the first 150 bp of COX1 (OQ107447) of H. bambusicolus (Wang et al. 2023) as a seed sequence. The coverage depth map, generated using the method described by Ni et al. (2023), showed an average depth of 220.31× (Supplementary Figure S1). The mitogenome was annotated using the MITOS (Bernt et al. 2013). The origin of L-strand replication (OL) and a non-coding region (putative D-loop) were identified by homology alignments with the corresponding known sequences of H. dunni Tago 1931 (GenBank accession number: LC538211, Igawa et al. 2020). The circular genome map was drawn using the CGView (Stothard and Wishart 2005).
Phylogenetic analysis
Twenty mitogenome sequences of other Hynobius species were downloaded from GenBank to reconstruct phylogenetic trees alongside H. bambusicolus, with Pachyhynobius shangchengensis Fei et al. 1983 used as the outgroup (Table 1). The fifteen genes (rrnS, rrnL, and 13 PCGs) were aligned individually using ClustalW (Thompson et al. 1994) with default parameters and then concatenated into a single alignment for phylogenetic analysis. Maximum likelihood (ML) and Bayesian inference (BI) were used to construct phylogenetic trees, and the concatenated alignment dataset was partitioned by gene. The nucleotide substitution model of each locus (Supplementary Table S1) employed in the both ML and BI analysis was selected based on the Bayesian Information Criterion (BIC) via MEGA11 (Tamura et al. 2021). The ML analysis was conducted utilizing RAxML-NG (Kozlov et al. 2019) with 1000 bootstraps. The BI analysis was performed with MrBayes 3.2.7 (Ronquist et al. 2012) under the following parameters: 10,000,000 generations with a sampling frequency every 1000 generations, four MCMC chains, and a burn-in of 25%. The convergence was evaluated by a split frequency value lower than 0.01 and the value of the Estimated Sample Size (ESS) for each parameter higher than 200 calculated using tracer v1.7.2 (Rambaut et al. 2018). The phylogenetic trees were visualized using TreeViewer (Bianchini and Sánchez‐Baracaldo 2024).
Results
The complete mitogenome sequence of H. bambusicolus is 16,406 bp in length and comprises 13 PCGs, 22 transfer RNA genes (tRNA) genes, 2 rRNA genes (rrnS and rrnL), an origin of L-strand replication (OL), and a non-coding region (putative D-loop). With the exception of one PCG (ND6) and eight tRNAs (trnQ, trnA, trnN, trnC, trnY, trnS2, trnE, and trnP), the majority of genes are encoded by the heavy (H) strand (Figure 2). Furthermore, the nucleotide composition of H. bambusicolus mitogenome consists of 33.3% A, 32.1% T, 20.8% C, and 13.8% G, exhibiting a slight bias toward A + T (65.4%). COX1 initiats with GTG, while the remaining 12 PCGs commence with ATG. The termination codons for the PCGs are as follows: TAA for COX1, COX2, ATP8, ATP6, ND4L, and ND5, AGA for ND6, incomplete stop codons TA for ND1, and incomplete stop codons T for COX3, ND2, ND3, ND4, and CYTB. The total length of tRNA genes is 1539 bp. The rrnS and rrnL in H. bambusicolus mitogenome are 935 bp and 1604 bp in length, respectively.
Circular map of the mitochondrial genome of Hynobius bambusicolus. Protein coding and ribosomal genes are shown with standard abbreviations. The genes in the inner are encoded on the L-strand. GC content plots are generated by calculating the GC content for each sliding window using the formula:G + C/Window Size, where values range between 0 and 1. The window moves by the step size, and the calculation repeats. The window size is set to 500 bp, and the step size is 1 bp. If the calculated value is higher than the GC content of the entire genome (0.346), it is displayed above the baseline; conversely, if it is lower, it is displayed below the baseline.
The BI and ML phylogenetic trees had identical topologies and most nodes were supported by high posterior probabilities (PP) and bootstrap percentages (BP) (Figure 3). The results revealed that the southern Chinese Hynobius formed a monophyletic group, and the H. bambusicolus was the basal lineage sister to other species within this group.
Phylogenetic tree for 21 Hynobius species distributed across East Asia based on the concatenated nucleotide sequences of rrnS, rrnL, and 13 PCGs. The outgroup taxa are not shown.The numbers on each node represent the Bayesian posterior probabilities (left) and ML bootstrap percentages (right). The number after the species name is the GenBank accession number. Orange background color represents Southern Chinese Hynobius, blue background color represents other Hynobius. Scale bar refers to a phylogenetic distance of 0.03 nucleotide substitutions per site.
Discussion and conclusions
The gene composition and organization of the mitochondrial genome of H. bambusicolus were identical to those observed in other Hynobius species. The sequence length of complete mitochondrial genome of Hynobius bambusicolus was also comparable to those reported for other Hynobius species, which ranged from 16,394 bp in Hynobius formosanus Maki 1922 (Zhang et al. 2006) to 16,495 bp in Hynobius chinensis Günther 1889. The primary factor contributing to length variations in the mitochondrial genome of Hynobius species was the variation in the intergenic spacer between the trnT and trnP, which ranged from 113 bp in Hynobius guabangshanensis Shen et al., 2004 to 203 bp in Hynobius yiwuensis Cai 1985 and H. chinensis.
The results of phylogenetic analysis in this study supported the monophyly of the southern Chinese Hynobius clade and the basal position of H. bambusicolus within this clade (Wang et al. 2023). The internal topology of the southern Chinese Hynobius clade was consistent with the previous findings of Igawa et al. (2020), but not with the findings of Li et al. (2011). In our results, Hynobius maoershanensis Zhou et al. 2006 was sister to H. guabangshanensis, and H. chinensis was sister to H. yiwuensis. In the study of Li et al. (2011), H. chinensis formed a sister group with H. maoershanensis before they shared a common ancestor with H. guabangshanensis. Future studies aimed at elucidating these phylogenetic issues should incorporate more extensive taxon sampling and the addition of nuclear data, which may enhance phylogenetic accuracy.
In conclusion, this study presented the complete mitogenome of H. bambusicolus and inferred its phylogenetic position based on the concatenated nucleotide sequences of two rRNAs and 13 PCGs. The results of this study enrich the available genomic databases for Hynobius genus, which can be useful for future studies focused on evolution and conservation.
Supplementary Material
Supplementary material revision.docx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319. doi:10.1016/j.ympev.2012.08.023.22982435 · doi ↗ · pubmed ↗
- 2Bianchini G, Sánchez‐Baracaldo P. 2024. Tree Viewer: flexible, modular software to visualise and manipulate phylogenetic trees. Ecol Evol. 14(2):e 10873. doi:10.1002/ece 3.10873.38314311 PMC 10834882 · doi ↗ · pubmed ↗
- 3BGI Genomics Company Limited. 2024. DNA extraction from animal tissues. [accessed 2024 Dec 24]. https://www.yuque.com/yangyulan-ayaeq/oupzan/ob 01swdvg 2al 7mfx.
- 4Cai C. 1985. A survey of tailed amphibians for Zhejiang, with description of a new species of Hynobius. Acta Herpetol Sin. 4(2):109–114.
- 5Chen Y, Chen Y, Shi C, Huang Z, Zhang Y, Li S, Li Y, Ye J, Yu C, Li Z, et al. 2018. SOA Pnuke: a Map Reduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 7(1):1–6. doi:10.1093/gigascience/gix 120.PMC 578806829220494 · doi ↗ · pubmed ↗
- 6Dierckxsens N, Mardulyn P, Smits G. 2017. NOVO Plasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e 18. doi:10.1093/nar/gkw 955.28204566 PMC 5389512 · doi ↗ · pubmed ↗
- 7Fei L, Qu W, Wu S. 1983. A new genus and species of Hynobiidae from Henan, China. Amphib Res Kunming. 1:1.
- 8Frost DR. 2024. Amphibian species of the world: an online reference. Version 6.2. New York (NY): American Museum of Natural History. [accessed 2024 Aug 12].https://amphibiansoftheworld.amnh.org/index.php.