Surgical treatment of subependymal giant cell astrocytoma in patients with tuberous sclerosis complex—an institutional experience and results
Mia Tuft, Ylva Østby Berger, Pål Bache Marthinsen, Bernt Johan Due-Tønnessen, Radek Frič

TL;DR
This study examines the outcomes of surgically removing brain tumors in children with a genetic condition called tuberous sclerosis complex.
Contribution
The paper provides a retrospective analysis of SEGA surgery outcomes in TSC patients, emphasizing safety and lack of cognitive impact.
Findings
Surgery for SEGA in TSC patients had low complication rates and no mortality.
There was no significant impact on cognitive function or epilepsy severity after surgery.
Three out of 11 patients required repeated surgeries during follow-up.
Abstract
Subependymal giant cell astrocytomas (SEGA) are present in patients with tuberous sclerosis complex (TSC), occasionally requiring surgical removal. The study aimed to analyze the results from our series of children undergoing surgery for SEGA. We retrospectively identified children with TSC undergoing resection of SEGA at Oslo University Hospital between 1982 and 2016. Patient charts, radiological images, epilepsy, and neuropsychological reports were reviewed. Out of 208 patients with TSC, 18 (9%) underwent resection of SEGA. Due to missing data, we could only analyze results from 14 surgeries in 11 children (median age 6 years, range 0–19; male/female ratio 2.7:1). The tumours were bilateral in four (36%) patients. The tumour diameter was a median of 19 mm (10–104 mm). The surgical approach was transcortical in eight (57%) and transcallosal in six surgeries (43%). Gross total…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
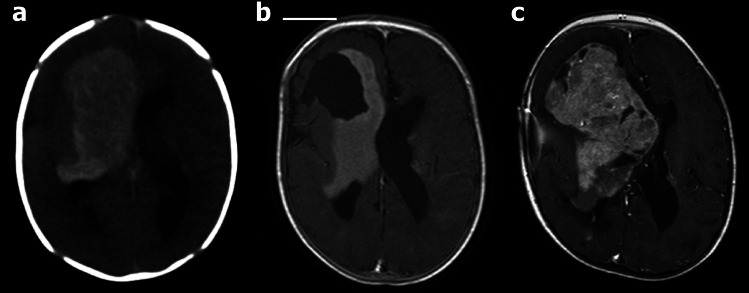 Figure 1
Figure 1- —University of Oslo (incl Oslo University Hospital)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTuberous Sclerosis Complex Research · Genetic and Kidney Cyst Diseases · Renal and related cancers
Introduction
Subependymal giant cell astrocytomas (SEGA) are defined as a tumour typically located in the caudothalamic groove, with a size of more than 10 mm in any direction, or a subependymal lesion at any location that has shown serial growth on consecutive imaging regardless of size [1]. SEGA usually grow slowly, but their progression may ultimately lead to the occlusion of the foramen of Monro and the development of obstructive hydrocephalus. They occur in 10–25% of patients with tuberous sclerosis complex (TSC) [2, 3], but are extremely rare in the general population [4]. SEGA usually do not present in patients older than 20–25 years [3], but up to 57 years has been reported [2], with an average age of 10–11 years and a range of 3–20 years [5, 6].
Tuberous sclerosis complex (TSC), in which SEGA most often occur, is a genetic neurodevelopmental disorder that affects about 1 in 6.000–10.000 individuals and about 2 million people worldwide [7]. It is characterized by a tendency to develop tubers in different organs, most often skin, kidneys, brain, and heart. About 80–90% of individuals with TSC present with typical radiological changes in the brain and epilepsy [8].
Treatment of SEGA is surgical, with the removal of the tumour, or medical, with the use of mammalian target of rapamycin (mTOR) inhibitors [9–11]. Furthermore, apart from traditional open surgical techniques for the removal of SEGA using transcallosal or transcortical approaches as in our sample of patients, both the use of endoscopic surgical technique and laser interstitial thermal therapy (LITT) has been increasingly reported in recent years [12–16], although currently applicable only in tumours smaller than 3 cm.
Data on outcomes from surgical treatment of SEGA is essential for counselling the patients and their families, as well as health care workers. More information is needed in terms of cognitive outcomes that may have a significant impact on patients’ educational and occupational potential. Clinical experience with patients with TSC in Norway shows that these patients often are not adequately assessed regarding their cognitive level of functioning and neuropsychiatric issues. Such assessments are necessary to facilitate appropriate educational, occupational, and family care adjustments for a better level of functioning and quality of life. Similar observations have been reported from other countries [17]. International consensus recommendations for the identification and treatment of neuropsychiatric disorders in TSC state the importance of such assessment [17, 18]. A special checklist to ensure that each patient gets the right diagnosis and treatment is developed (The TAND Consortium) [19] and is available in Norwegian translation.
Epilepsy in TSC is mainly related to the genetic condition itself and tubers in the cerebral cortex. The grade of epilepsy before and after surgery of SEGA should be assessed with EEG and epilepsy diary. Only a few studies, including the present study and the previous French study [20], address this particular issue, as well as the neuropsychiatric aspects.
Surgical outcomes after surgical resection of SEGA in patients with TSC have been reported in previous studies including from two to 64 patients [5, 6, 16, 20–37]. These studies showed excellent outcomes with low morbidity and mortality in asymptomatic SEGA, whereas resection is associated with higher surgical morbidity and mortality when diagnosed at a later stage and with bigger tumours that affect surrounding brain structures such as the basal ganglia, hypothalamus, fornix, and genu of internal capsule [37]. Yet, there is a need for more knowledge, due to the small samples in all present studies.
Our aim was primarily to better understand the possible side effects of surgical resection of SEGA in the Norwegian patient population with TSC. We also wanted to investigate whether the surgical treatment of SEGA had an impact on patients’ cognitive and neuropsychiatric functioning and epilepsy.
Material and methods
The study was approved by the regional ethical committee and Oslo University Hospital as a quality control study (reference number 2016/1187). We retrospectively analyzed medical records at Oslo University Hospital and The National Center for Rare Epilepsy-Related Disorders from 1982 to 2016 and identified all patients who underwent surgical resection of SEGA during this period.
Patient records were reviewed and the following information was retrieved: diagnosis of TSC; genetic test results; surgical notes; information about epilepsy, including EEG reports, seizure type, and frequency; pathology reports (to verify tumour classification); neuropsychology reports or other relevant information that could retrospectively describe the cognitive level of functioning; magnetic resonance imaging (MRI) or computed tomography (CT) scans. Surgical notes and pathology reports were reviewed by a neurosurgeon (R.F.), MRI and CT scans were reviewed by a neuroradiologist (P.B.M.), and information regarding epilepsy and cognitive functioning was reviewed by a neuropsychologist (M.T.).
Cognitive and behavioural functioning was assessed by analyzing all relevant information on the cognitive level of functioning, including test results from neuropsychological examinations (Wechsler Preschool and Primary Scale of Intelligence 3rd and 4th ed, The Beery-Buctencia Developmental Test of Visual_Motor Integration, 5th ed, BRIEF and PedsQL in the two presented cases), as well as information about diagnoses; reports on intellectual disability, ADHD, autism spectrum disorder, and behaviour; pedagogical reports; and other relevant clinical descriptions.
Epilepsy was classified into five grades: 0 (no epilepsy), 1 (no current epilepsy, no drugs), 2 (seizures controlled with drugs), 3 (weekly seizures), and 4 (daily seizures). We also noted whether there were focal seizures, Lennox Gastaut syndrome, or non-epileptic seizures.
Results
Patient sample
We identified 208 patients with TSC: 205 from the register at The National Center for Rare Epilepsy-Related Disorders and three from a journal search on the diagnosis (Q85.1 Tuberous Sclerosis Complex) at Oslo University Hospital. Out of these, we found 34 (17%) patients harbouring SEGA, of whom 18 patients (9%) underwent surgical resection.
Six (33%) of the 18 patients had a mutation of TSC1 and eight (44%) of TSC2, while four (22%) were not genetically specified. Pathology records confirming SEGA were available in only 11 patients; therefore, the remaining cases were excluded from further analysis. The male/female ratio was 2.7:1; the median age at the time of the first surgery was 6 years (range 0–19) (Table 1). Table 1. Characteristics of the patient sample in the present studyPatientSexAge^1^SurgeryepiGeneLocationSideSize (max, mm)HCSurgical approachResection gradeHistology verifiedFollow-up (years)RecurrenceComplications1M192001Yes?FHR15NoTranscorticalGTRYes12NoNone2M102001YesTSC1FHR34YesTranscorticalGTRYes11^†^ NoShunt infection, Shunt failure3M52002YesTSC1FHL25NoTranscorticalGTRYes21NoNone4M02006YesTSC2FTR57YesTranscorticalSTRYes10^†^ YesShunt failure32009FTR104YesTranscorticalSTRYes7NoNone5F62008YesTSC2FHBilat12L, 24RYesTranscorticalGTRYes15NoNone6F52008YesTSC1FHL20NoTranscorticalGTRYes12YesNone162019NoFHL15NoTranscorticalGTRYes1NoNone7F92009YesTSC1FHL28NoTranscallosalGTRYes1NoNone8M62012YesTSC1FHBilat16L, 10RNoTranscallosalGTRYes11YesNone9M62013YesTSC2FHBilat20L, 17RNoTranscallosalGTRYes9NoNone10M102016YesTSC2FHBilat19L, 13RYesTranscallosalGTRYes7YesChronic SDH162022YesFHBilat22L, 14RYesTranscallosalGTRYes1NoNone11M82016TesTSC2FHL19YesTranscallosalGTRYes7NoNone^1^At the time of the first surgery. GTR gross total resection, FH frontal horn, FT frontotemporal, HC hydrocephalus, L left, R right, SDH subdural hematoma, STR subtotal resection. ^†^Deceased
Radiology
Most of the SEGAs were in the typical location in the frontal ventricular horns, except for one case of a large frontotemporal (hemispheric) tumour (Fig. 1). The lesions were located on the right side in three (27%), on the left side in four (36%), and bilateral in four (36%) patients. The tumour diameter was median 19 mm (range 10–104 mm). Five patients (46%) had hydrocephalus (Table 1), although radiologically only mild in three of these cases.Fig. 1A case of a large SEGA in the right cerebral hemisphere, detected at birth (patient no. 4 in Tables 1 and 2). a Only CT was performed after birth and before the first emergency surgery when the tumour was only partially removed to achieve decompression and obtain material for histological diagnosis. b Axial MRI T1-weighted post-contrast image (T1 CE) acquired after the first surgery and biopsy showed partial removal of the tumour. c MRI scan obtained 3 years later showed tumour growth, resulting in the second surgical resection
Surgery
Fourteen tumour resections were performed in 11 patients. The surgical approach was transcortical in eight (57%) and transcallosal in six cases (43%). Gross total resection (GTR) was achieved in 12 out of 14 (86%) of surgeries. Two patients (18%) received a ventriculoperitoneal shunt (VPS), in one case before the surgical resection of SEGA. We experienced no mortality or major morbidity related to surgery of SEGA other than a chronic subdural hematoma in one patient (7% of all 14 resections), but one patient with VPS (9%) developed shunt infection and both patients with VPS experienced shunt failures during the follow-up.
During the follow-up of the median 11 years (range 1–21) following the first surgery, three patients (27%) underwent repeated surgery for SEGA due to radiologically documented recurrences. Two patients died from complications to their diagnosis of TSC, but not related to surgery.
Epilepsy
All patients had epileptic seizures before surgery, and the grade of epilepsy remained unchanged after surgery (Table 2). Table 2. Functional characteristics (epilepsy, cognitive skills, autism, and behaviour) of the patients included in the studyPatientEpilepsy gradeCognitive skills^1^ASD and autistic traits^2^Behaviour difficultiesBefore surgeryAfter surgery144Moderate IDYesNot verified244Deep IDYesYes322NormalNoNo44 (LGS)4 (LGS)Deep IDYesNo544Unspecified IDYesYes60–1^3^0^3^NormalNoNo722IQ scores lower normalNoNo84 (FS)4 (FS)Moderate-severe IDTraitsYes94 (FS)4 (FS)Reduced (equally to 4–5 ys at 6 ys of age), ID (unspecified)TraitsYes + ADHD104 (LGS)4 (LGS)Severe-deep ID (IQ < 20)YesYes1122Mild IDTraitsDescribed behavioural problemsEpilepsy grade: Grade 0 = no epilepsy; Grade 1 = no current epilepsy, no drugs; Grade 2 = seizures controlled with drugs; Grade 3 = weekly seizures; Grade 4 = daily seizures. FS focal seizures, ID intellectual disability, LGS Lennox Gastaut syndrome, na not available. Before and after surgery = the last EEG before surgery and the EEGs the following years after surgery^1^Mild ID, IQ 50–69; moderate ID, IQ 35–49; severe ID, IQ 20–34; profound ID, IQ < 20; unspecified ID, below IQ 70. Normal = normal IQ results or no special education or normal full-time work as an adult^2^ASD (autism spectrum disorder) refers to the diagnosis given by a clinician, and autistic traits refer to clinical signs of autism spectrum disorder^3^Used medication prophylactically, but did not have epileptic seizures; stopped taking medication 6 months after surgery and remained seizure-free
Neuropsychology
Neuropsychological assessments were scarcely described in the records of most patients, but in the majority of cases, it was possible to gather broad estimates based on journal descriptions and cognitive disability diagnoses, as seen in Table 2. Based on these descriptions of cognitive and behavioural functioning, no patients experienced any noticeable changes after surgery.
Only two patients (nos. 6 and 7 in Table 2) were neuropsychologically assessed both before and after surgery and may be described briefly here:
A case report (patient no. 10)
The testing of patient no. 10 (Tables 1 and 2), a 10-year-old boy with autism and severe to deep intellectual disability, was performed the day before the SEGA surgery and adjusted to the individual cognitive level of functioning. As the hospital had still not established routines for testing this group of patients before and after surgery, the conditions for testing were suboptimal. The boy was easily distracted, and appeared motorically restless, but cooperated when rewarded with iPad puzzles.
The investigation was based on the Bayley Scales of Infant and Toddler Development, Third Edition (cognitive and language scale) [38], Behaviour Rating Inventory of Executive Function (BRIEF), and PEDS QL Life quality in children version 4.0 [39], translated into Norwegian. Additional information was given from his mother and father, who were there during the first test sessions.
The postsurgical assessment was performed by the same neuropsychologist and took place at the boy’s school, with his special education teacher present throughout the session. The Bayley Scales (cognitive and language scale) was used for the post-surgery assessment, as well as observation of activities at his school teaching session with his special education teacher, and a clinical interview with the parents. His special education teacher supplied information about his level of words, and which specific words he knew and could pronounce.
The boy had a slightly better performance when tested 1 year after surgery. Before surgery, his test results were equivalent with children of 19–20 months on the cognitive scale and equivalent with 12–14 months old children on the scale for expressive communication. One year later, at the age of 11, his results had increased slightly, to the average of 21–22 months and 18–19 months on the same subscales, respectively. His mother reported that he had learned some more words than a year ago. The questionnaires BRIEF and PedsQL before surgery verified a need for comprehensive care. As expected, the answers verified severe problems with impulse control, attention and shifting attention from one activity to another, emotional control, social skills and communication, working memory, planning, and organizing. The same questionnaires were not filled out after surgery, but the clinical interview with the mother revealed a slight general improvement a year after surgery. He was more often in a good mood than he was before surgery, and he had shown a slight cognitive improvement and no stagnation in any area. He had learned some new skills, and the mother reported that a normal day in general was better than before surgery, in terms of life satisfaction. The information was supported by his special education teacher.
A case report (patient no. 11)
The patient, a 7.5-year-old boy, was assessed by the same psychologist (M.T.) the day before the surgery for SEGA and 1 year after surgery. Wechsler Preschool and Primary Scale of Intelligence, 3rd and 4th editition (WPPSI-III/IV) [40, 41]; the Beery-Buctencia Developmental Test of Visual-Motor Integration, 5th edition (Beery VMI) [42]; and Grooved Pegboard Test [43] were used in the first assessment. The patient’s mother filled out the questionnaires BRIEF and Paediatric Quality of Life Inventory (PedsQL). At the second assessment, the same assessment tools and questionnaires were administered, except that the third version of WPPSI had to be used, as the 4th version was unavailable at the time of testing. In both instances, test sessions were cut short because of a lack of compliance and/or endurance or because of difficulty understanding instructions. Additional anamnestic information was provided by the patient’s mother and teacher.
Before surgery, at the age of 7.5 years, the test Vocabulary from WPPSI-IV [44] scores were equivalent to the mean for 2.5-year-old children, and the test Picture Naming from the same test battery gave scores equal to mean scores for a 4-year-old child. He did not understand the part test Block Design. At a test of fine motor skills and speed (Grooved Pegboard), the scores were only slightly below mean scores for his age and there were no significant differences between his left and right hand. On a drawing task, he managed to copy simple geometrical figures (circles, simple lines, triangles, squares), but not any advanced figures. His results were equivalent to 5-year-old children. Results from the BRIEF questionnaire revealed problems within several areas: behavioural inhibition, transitions between activities, working memory, planning, organizing, and self-monitoring in social relations. Answers from the questionnaire PedsQL revealed some difficulties within social functioning and school functioning.
After surgery, at the age of 8 years, scores were equivalent to the mean scores for 4-year-olds on the subtests Information and Vocabulary and scores equivalent to mean scores for 3-year-olds on the subtest Block Design. It was not possible to calculate a full IQ score, because he lost patience and motivation in the test situation. On a test for fine motoric speed (Grooved Pegboard) [43], his scores were somewhat below the mean scores for his age, but like before the surgery, he mastered this skillfully, and there were no differences between his left and right hands. On a drawing task, copying geometrical figures, his results were equivalent to 6-year-old children.
The BRIEF questionnaire indicated that he had improved in several areas. He still had problems with transitions and had problems with self-monitoring (but it was a lot better than at the last assessment), planning, organizing, and working memory. However, his inhibition skills were improved, as were his behavioural regulation and social skills. In general, the mean scores were higher than before surgery.
Results from the questionnaire PedsQL were almost the same as the previous assessment, but there was an improvement in social function and school functioning.
Discussion
The rationale for the treatment of SEGA is primarily to alleviate the mass effect and secondary hydrocephalus caused by the tumour. In the most recent literature review of 18 studies published between 1980 and 2021 counting a total of 263 patients [36], the surgical morbidity was 4.9% and morbidity 33.6%, while ventriculoperitoneal was needed in 30.8% of cases. Although the comparison is difficult due to the low number of cases in the present series, our results are more in line with own data from the same study, showing very few complications and negligible impact of surgical removal of SEGA on the grade of epilepsy in the identical number of patients [36].
While surgical treatment of symptomatic or growing SEGA remains a valid treatment strategy, the risk of significant neurological morbidity reported in older publications (5–50%) is still considered its main limitation [15, 31, 36]. The risk of complications appears particularly high in large SEGA [37], in which the use of alternative treatment methods—such as laser interstitial thermal therapy (LITT) and mTOR inhibitors—is limited. It is therefore important to regularly screen TSC patients for SEGA and initiate early treatment when necessary.
Importantly, the present case series includes also patients treated already in the late 1990s and early 2000s, before the implementation of the new recommendations regarding the management of SEGA including the use of mTOR inhibitors [7]. Medical treatment with mTOR inhibitors was not available as an alternative to surgery for SEGA until around 2010 [45]. The advantageous effect of mTOR inhibitors is the shrinking of SEGA when tumours are large and infiltrate surrounding brain tissue. It can be used as neoadjuvant treatment before surgery [46] and on a long-term basis. A study showed that more than 60% of patients receiving treatment for ≥ 5 years exhibited a clinically relevant (≥ 30%) reduction of their SEGA [45]. In a Norwegian-Danish study, five patients were treated with everolimus for the indication of SEGA, achieving volume reduction in three of them; this study also stressed the importance of frequent and close evaluation of side effects [47]. Side effects of mTOR inhibitors used over a long time are still unknown, because of still limited long-term experience with these drugs. Moreover, there may be differences in side effects seen in children and adults [47, 48]. A major disadvantage is the recurrence of SEGA growth when the medication is discontinued [49].
Besides the medical treatment of SEGA, mTOR inhibitors are also used for the treatment of other manifestations of TSC [50].
We gathered data from 11 consecutive patients treated during the last two decades. Another seven patients were treated during the previous two decades (before 2000); here, the data on pathohistological verification were lacking, and these cases were therefore excluded from further analysis. However, it is still reasonable to assume that they also had SEGA, as SEGA is almost exclusively related to TSC and these were growing tumours. The available clinical patterns from these excluded cases were very similar to those presented in Tables 1 and 2: the semiology of epilepsy was similar and there were no major changes in the grade of epilepsy after surgery (except that epilepsy increased in one case and decreased in another) and no cognitive or neuropsychiatric changes related to surgery, either.
The low number of cases available from our tertiary high-volume institution reflects the rarity of SEGA, which presents in only 10–25% of patients with TSC [2, 3], and is in line with numbers presented in most other surgical series [5, 13, 16, 20, 22, 24, 29, 32, 33, 36]. Studies from larger samples of patients are rare [31, 35, 37], particularly after the introduction of medical treatment with mTOR inhibitors [51]. In Norway, we found that 17% of patients with TSC had SEGA, according to patient records compared with a national registry.
The grade of epilepsy remained unchanged in all patients. In general, epilepsy is present in about 85% of all patients with TSC, and up to 75% have seizures that are difficult to treat [52, 53]. Surgical treatment of SEGA, regardless of the surgical approach, does not seem to worsen epilepsy. A literature review reported that only three patients had new seizures after surgery, some patients improved, and the rest remained unchanged [36]. In the present study, we used a grading system from 0 to 4 for better comparison with the study of Fohlen and colleagues [20], since epilepsy outcomes are scarcely described in the surgical literature.
One important finding of the present retrospective study was the lack of systematic, standardized examination of cognitive functions in our historical series of patients with SEGA. The impression that also cognitive and behavioural functioning was mostly unaffected by the surgery in our series is therefore based on a certain confidence in the clinical information stated in the medical records. Ideally, this information should have come from dedicated clinical assessments. For the patients, their families, and medical personnel, information about cognitive and behavioural functioning should be an integral part of a preoperative workup and postoperative follow-up. According to a national tumour follow-up program for children in Norway, all children undergoing resection of a brain tumour are to be assessed by a neuropsychologist before and after surgery [54]. It may be suspected that patients with SEGA associated with TSC, which is a rare diagnosis, were not primarily regarded as brain tumour patients and therefore slipped from this routine. Another explanation could be that many of the patients have autism and intellectual disability, and their neuropsychological assessment was not considered feasible by clinicians. TSC is a diagnosis with a considerable range in terms of level of functioning [17]: if the patients with SEGA have a normal intellectual capacity and no identifiable neuropsychiatric symptoms, they can be tested by standard test batteries for their age, but most children with TSC have one or more neuropsychiatric problems, including intellectual disabilities, autism, and ADHD. The neuropsychological assessments therefore require individual adjustment. Although SEGA presents mostly in children, there are a few exceptions and individual adjustment may be needed also in adult patients. The TAND consortium (tandconsortium.org) with dedicated checklists can be used as a tool for neuropsychiatric screening for all patients with TSC, and further assessments should be carried out according to the screening results [19].
Limitations
The data in this study are retrieved retrospectively and based mostly on available patient records with clinical descriptions of cognitive functioning to epilepsy and reported surgical complications. The number of patients is small and therefore does not allow for any recommendations or relevant statistical analysis.
Conclusions
Surgical resection of SEGA in children with TSC was associated with a low complication rate. Recurrences were mostly related to subtotal resection. We could not document any impact of surgery on patients’ cognitive function or grade of epilepsy. However, the main finding was that neuropsychological data were limited in the vast majority of the cases. Neuropsychological assessment should be performed before the surgery and be a part of follow-up after surgery.
The reference list from the paper itself. Each links out to its DOI / PubMed record.