Adrenal Oncocytic Pheochromocytoma: Insights From a Challenging Diagnostic Journey
Charusheela Gore, Dipti Singh, Sushama Gurwale, Arpana Dharwadkar

TL;DR
This paper discusses a rare adrenal tumor case, highlighting the challenges in diagnosing oncocytic pheochromocytoma and the need for detailed histological analysis.
Contribution
The paper presents a new case of oncocytic pheochromocytoma and emphasizes the diagnostic challenges associated with it.
Findings
Oncocytic pheochromocytomas are rare, with only 150 cases reported.
Comprehensive histological and immunohistochemical studies are crucial for accurate diagnosis.
The case highlights potential diagnostic pitfalls in identifying this rare tumor variant.
Abstract
Pheochromocytomas are rare adrenal medulla tumors originating from chromaffin cells, accounting for 10% of primary adrenal neoplasms. Oncocytic variants of pheochromocytomas are rare and have been reported in only 150 cases. This report describes the case of a 60-year-old female who arrived with a non-functional adrenal tumor. This case report emphasizes the importance of a comprehensive histological and immunohistochemical study for diagnosing this rare diagnostic entity and its potential diagnostic pitfalls.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
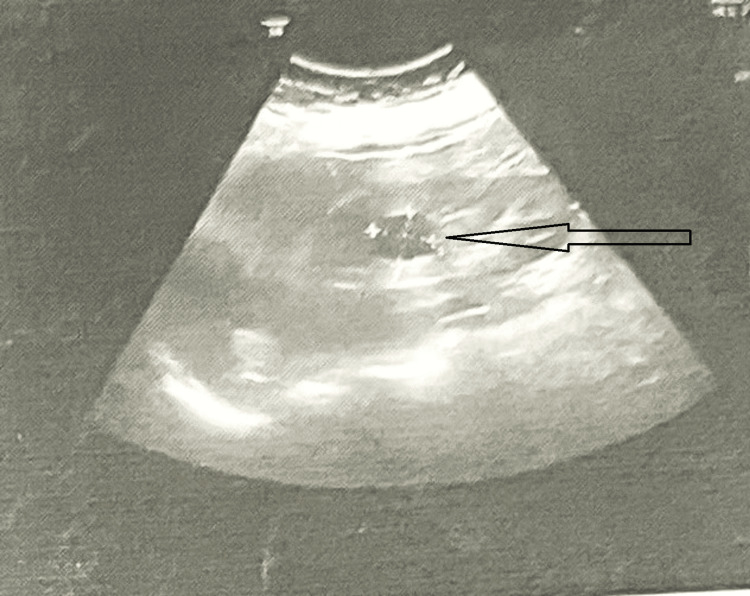 Figure 1
Figure 1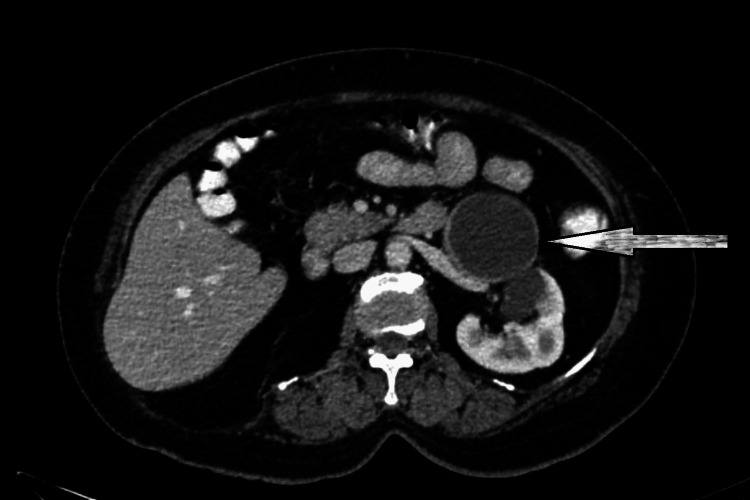 Figure 2
Figure 2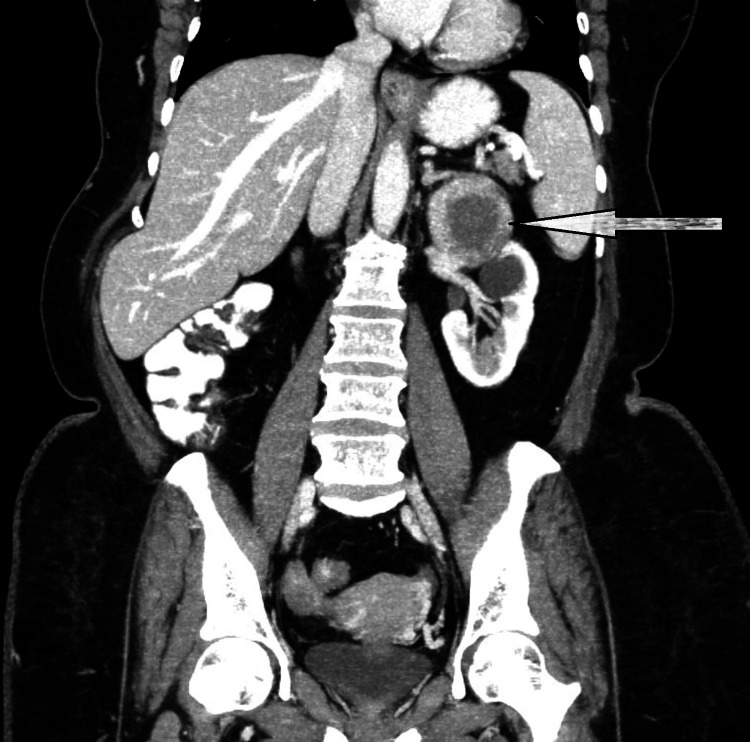 Figure 3
Figure 3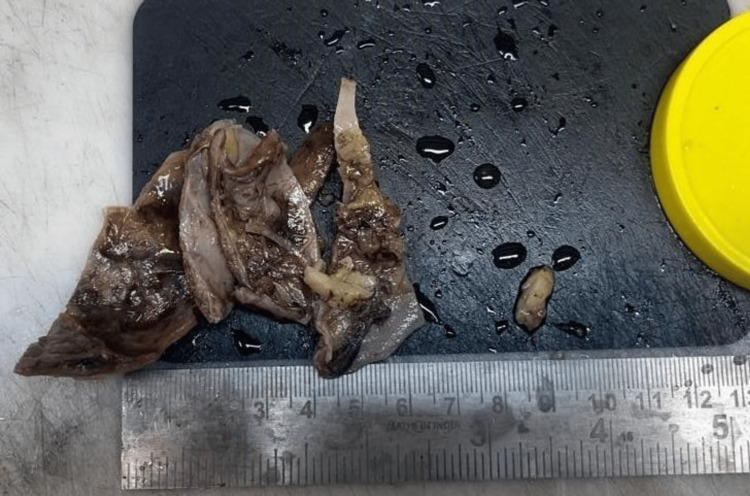 Figure 4
Figure 4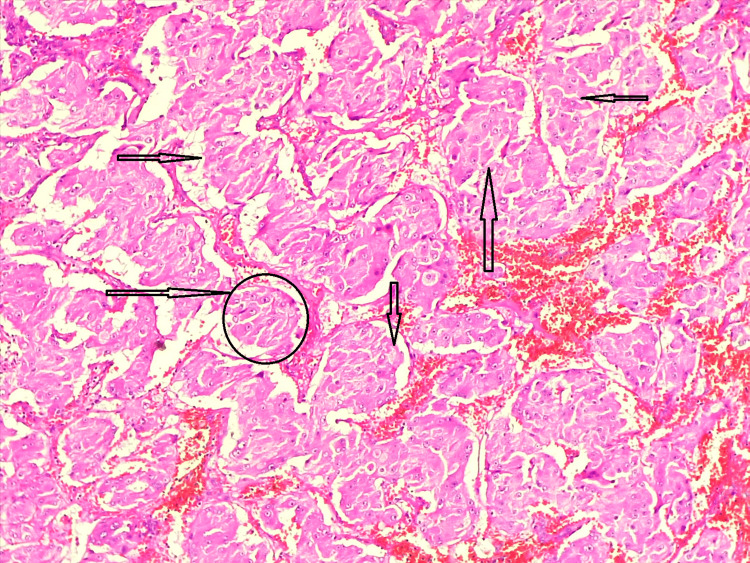 Figure 5
Figure 5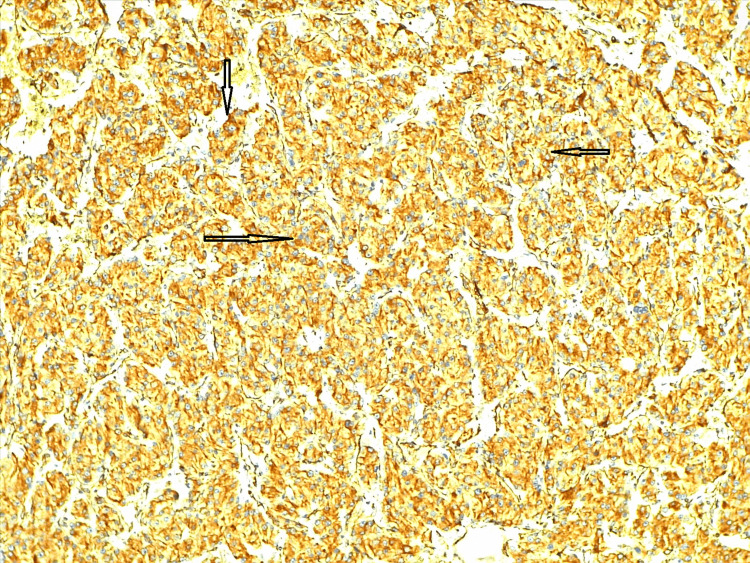 Figure 6
Figure 6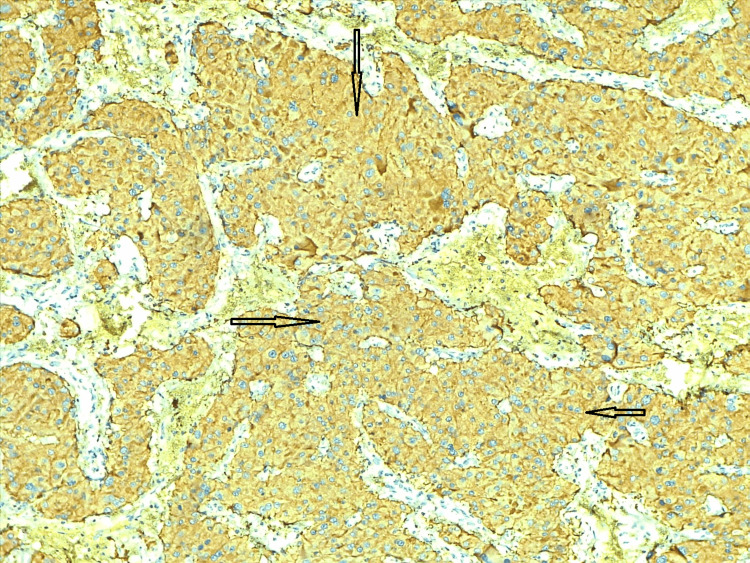 Figure 7
Figure 7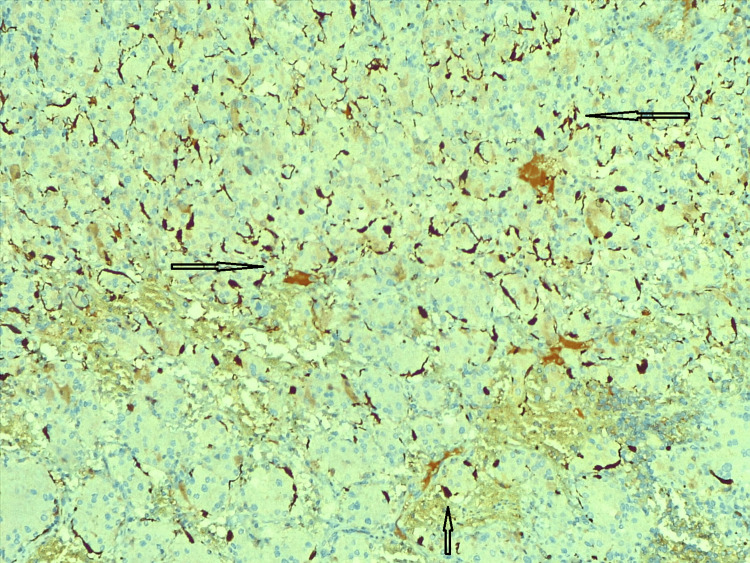 Figure 8
Figure 8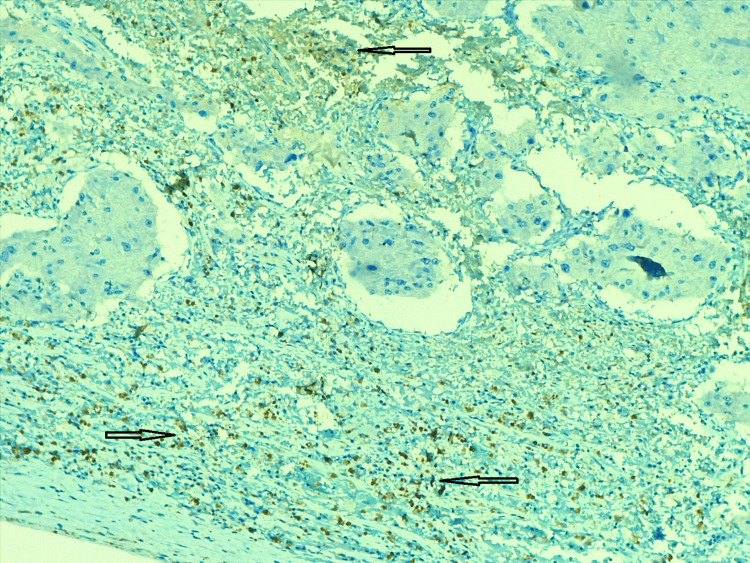 Figure 9
Figure 9| Serial No. | Test name | Observed values | Reference values |
| 1 | Vanillylmandelic acid (VMA) in urine | 2.8 mg/24 hour | 2-7 mg/24 hours |
| 2 | Adrenaline (epinephrine) in urine | 3 µg/24 hour | 0-20 µg/24 hours |
| 3 | Noradrenaline (norepinephrine) in urine | 20 µg/24 hour | 15-80 µg/24 hours |
| 4 | Metadrenalin in plasma | 0.42 nmol/L | <0.5 nmol/L |
| 5 | Normetadrenalin in plasma | 0.36 nmol/L | <0.9 nmol/L |
| 6 | Aldosterone in plasma | 85 pmol/L | 55-440 pmol/L |
| 7 | Cortisol in plasma (morning) | 9 µg/dL | 5-23 µg/dL |
| 8 | Adrenocorticotropic hormone (ACTH) in plasma | 5.3 pmol/L | 2.2-13.2 pmol/L |
| 9 | Renin in plasma | 0.21 ng/mL/hour | 0.2-2.3 ng/mL/hour |
| Demographic (author/year/country) | Sex/age (years) | Side | Clinical presentation | Weight | Size | Follow up | Remarks | Histopathological specific features |
| Wang et al./1997/USA [ | F/38 | L | Acute abdominal pain and hirsutism | 275 G | 34 mm | Nil | Areas of hemorrhage, cytokeratin+ | Fibrovascular stroma surrounding Nest of tumor cells, nuclear atypia |
| Li and Wenig/2000/USA [ | F/37 | L | Increase abdominal girth | 1150 G | 170 mm | 20 months | Pseudoglandular pattern | Psudoglandular pattern, a large nucleus with prominent nucleolus |
| Shibamori et al./2013/Japan [ | F/61 | R | Incidental finding | 260 G | 100 mm | 13 months | Alveolar configuration | Alveolar configuration, round nucleus |
| Kasem and Lam/2014/Australia [ | M/68 | L | Incidental finding | 36 G | 45 mm | 5 years | P53+ | Myelolipomatous metaplasia, capsular invasion |
| Nam et al./2014/USA [ | M/81 | L | Incidental finding | NA | NA | NA | CT guided fine needle aspiration | Sheets of cells, nuclear pleomorphism |
| Kenneth/2018/Wisconsin [ | M/54 | L | Hypertensive episode | 675 G | 135 mm | 3 months | Necrotic with capsular invasion | Nuclear pleomorphism with nuclear pseudo-inclusions |
| Present Case/2024/India | F/60 | L | Abdominal pain | 15G | 75 mm | 5 months | Hemorrhage with the cyst, nesting pattern | pleomorphic nuclei with nucleoli and abundant granular eosinophilic cytoplasm |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdrenal and Paraganglionic Tumors · Pituitary Gland Disorders and Treatments · Cancer, Hypoxia, and Metabolism
Introduction
Pheochromocytomas are rare adrenal medulla tumors that arise from chromaffin cells. These tumors originate from the embryonic neural crest and approximately 10% of primary adrenal neoplasms are pheochromocytomas [1]. Specific anatomic areas with histologic and ultrastructural features characteristic of their catecholamine-secreting tumor type include big polygonal cells, thick granular eosinophilic cytoplasm, and numerous mitochondria packed firmly into the cell. Since the first instance was identified in 1997 and given the name oncocytic pheochromocytoma, there have been few occurrences of the condition recorded [2].
Oncocytic tumors have been documented in several organs, including the kidney, thyroid, parathyroid, salivary glands, and pituitary [3]. Their histology demonstrates nesting, alveolar, and trabecular patterns [4]. Neuroendocrine markers such as chromogranin and synaptophysin are detected positively by immunohistochemistry [4,5].
Adrenal oncocytic tumors are extremely uncommon and only approximately 150 cases have been reported [6]. Of these, six cases were oncocytic tumors of the adrenal medulla: five pheochromocytomas and one adrenal medullary oncocytoma [7]. Since oncocytic forms of adrenal pheochromocytomas are exceedingly uncommon [8], they have not been included in recent research on adrenal oncocytic neoplasms [9].
Case presentation
A 60-year-old female experienced left-side flank discomfort, weight loss, and intermittent burning during micturition for the past three months. She had a right-side nephrectomy 10 years ago for a non-functioning kidney with staghorn calculus and mild hypertension. An ultrasonographic examination revealed a cystic lesion along the medial side of the left kidney with internal echoes, raising suspicion of either a renal parapelvic cyst or an adrenal cyst (Figure 1).
Ultrasound of the abdomen showing a left renal cystic lesionA cystic lesion measuring 23 x 23 mm without internal septations/ echoes was noted in the mid-pole region, suggesting a simple corticomedullary cyst (arrow).
A CT contrast revealed a 71 x 61 x 56 mm peripherally enhanced hypoattenuating cystic left adrenal mass in the retroperitoneum near the tail of the pancreas (Figure 2). The right kidney was not visualized, which was consistent with a history of nephrectomy.
CT with contrast, axial section abdomen showing left side pararenal and renal space occupying lesion (SOL)A CT scan showed a peripherally enhanced hypoattenuating large cystic left adrenal mass in the retroperitoneum and along the inferolateral aspect of the left adrenal gland, seen abutting the pancreatic tail (arrow). Metastatic deposits or masses were not found.
Left side simple renal cortical cyst with parapelvic cyst was identified. No other masses or metastatic deposits were found (Figure 3). The patient was diagnosed with hypertension (blood pressure near 140-160/80-90 mm Hg) for four years and has been effectively managed with medication. The pre-operative biochemical evaluation showed normal adrenalin and nor-adrenalin levels in urine and plasma (Table 1). With a clinical diagnosis of a non-functional adrenal cystic lesion, surgical excision was performed.
Coronal CT of abdomen and pelvisA large well-defined hypoattenuating, non-enhancing cystic lesion of size 71 x 61 x 56 mm, surrounded by enhancing adrenal tissue was noted arising from the inferolateral aspect of the left adrenal gland, seen abutting the tail of the pancreas and the small bowel loop (arrow). It showed no solid components/septations/calcifications.
A gross examination of the received specimen revealed a well-circumscribed, cystic, solid adrenal mass measuring 75 x 60 x 30 mm and weighing 15 grams. The external surface was grayish-white and covered with yellow fibrofatty tissue (Figure 4). The cut surface was cystic with a shiny thinned-out wall; a few thickened areas in the wall of the cyst were noted, which appeared yellow; and a few hemorrhagic regions were also noted.
Specimen (cystic adrenal lesion) received for histopathologyGross section showing greyish white external surface covered with fibro-fatty tissue having few hemorrhagic areas and measuring 75 x 60 x 30 mm.
Histopathological findings showed adrenal tissue and a cystic tumor. The wall of which comprised of nests separated by thin fibrovascular stroma (Figure 5). The cells were polygonal and showed mildly pleomorphic nuclei with small nucleoli and abundant granular eosinophilic cytoplasm. There was no evidence of necrosis or increase in mitotic activity (Figure 5). In this case, adrenal cortical adenoma, adrenal cortical cancer, medullary oncocytoma, adrenal cortical cyst, pheochromocytoma, and oncocytic pheochromocytoma were the differential diagnoses for adrenal oncocytic pheochromocytoma. A provisional diagnosis of pheochromocytoma/oncocytic pheochromocytoma was offered by correlating clinicopathological and immunohistochemistry findings.
Microscopic examination of the adrenal cystic tumor (hematoxylin & eosin stain, 10x)The polygonal cells had a lot of granular eosinophilic cytoplasm and partly pleomorphic nuclei with nucleoli. Arrows are placed in the surrounding septa around a bunch of cells. Necrosis or an increase in mitotic activity was not visible. There was a zellballen pattern (circle), which is indicative of pheochromocytoma.
Immunohistochemical markers like vimentin (Figure 6), synaptophysin (Figure 7), S100 (Figure 8), and pan CK (Figure 9) were positive in sustentacular cells. These findings confirmed the diagnosis of a cystic variant of oncocytic pheochromocytoma. There was no incident throughout the recovery time. The patient has not reported any new problems throughout the last five months of routine periodic follow-up. Her abdominal discomfort has lessened, and now she has resumed her normal activities.
Immunohistochemistry reveals vimentin-positive tumor cellsImmunohistochemistry staining of oncocytic pheochromocytoma showing positive vimentin expression (arrows). Vimentin, a mesenchymal marker, is detected in the tumor cells (brown staining). Areas with denser brown coloration indicate higher vimentin expression.
Immunohistochemistry - synaptophysinImmunohistochemistry shows synaptophysin identified the oncocytic component (arrows). Pheochromocytoma and paragangliomas are usually positive for synaptophysin.
Immunohistochemistry - S100The oncocytic pheochromocytoma adrenal gland component was identified by immunohistochemistry, with S100 shown in the figure (arrows). Sustentacular cells showed positivity for S100 IHC.IHC: immunohistochemistry
Immunohistochemistry - pan CKImmunohistochemistry revealing pan CK identified the oncocytic pheochromocytoma component (arrows). Some of the cells are showing (weak) positive (brown) for pan CK.
Discussion
Oncocytic pheochromocytoma is clinically nonfunctional and has an unclear cause. Oncocytic change is a degenerative phenomenon where mitochondria accumulate to compensate for oxidative phosphorylation uncoupling, leading to nonfunctional oncocytic tumors [10]. The literature provides six previous reports of oncocytic pheochromocytoma (Table 2) [2,4,5,10-12].
In this case, the differential diagnosis among adrenal cortical cyst, pheochromocytoma, and oncocytic pheochromocytoma was crucial. Initially, a diagnosis of cortical cystic pheochromocytoma was proposed; however, immunohistochemistry findings ruled out any cortical components [5]. An immunohistochemical study indicated that this tumor was pheochromocytoma because of the neuroendocrine markers, such as vimentin, synaptophysin, S100, pan CK, and CD-56. Adrenal cortical tumors frequently test positive for SF-1 and inhibin. However, SF-1 is not widely available or used, but inhibin-negative immunohistochemistry can be observed in some adrenocortical tumors [13]. Table 2 summarizes the characteristics of oncocytic pheochromocytoma from the literature.
Documented oncocytic pheochromocytoma patients do not show signs of excessive adrenaline, such as palpitations, flushing, diaphoresis, headaches, or hypertension. In three cases, hypertension was not caused by excess catecholamines. Chemical workups for pheochromocytomas showed no significant increase in catecholamines or their metabolites, indicating that oncocytic pheochromocytomas are "silent" and nonfunctional. These patients had an excellent prognosis and the tumors were clinically nonfunctional, according to the previously published research results (Table 2). Histologically, neither capsular nor lympho-vascular invasion was present, nor was there an increase in mitosis. It was difficult to identify the type of tumor - benign or malignant - until a thorough microscopic examination showed patches of hemorrhage, coagulation necrosis, and fibrotic degradation.
Oncocytic tumors share cytological characteristics such as rich eosinophilic granular cytoplasm and central or eccentric nuclei. The malignancies can be distinguished using immunohistochemical investigations. For instance, pheochromocytomas are specific for chromogranin A and CD56, while an adrenocortical origin would be suggested by positive for calretinin, MART-1, and inhibin A. Synaptophysin levels can be elevated in both pheochromocytomas and adrenal cortical tumors [14].
Pheochromocytoma's aggressive potential is difficult to predict. Two major scoring systems exist Thompson's PASS, based on histological criteria, and Kimura's Japanese scoring system, which includes Ki-67 immunoreactivity and catecholamine types, based on histological criteria and Ki-67 immunoreactivity [15]. PASS score includes histopathological characteristics like large nests, diffuse tumor necrosis, high cellularity, cell monotony, spindling, mitotic figures, extension into adipose tissue, vascular and capsular invasion, profound nuclear pleomorphism, and nuclear hyperchromasia for determining the aggressive nature of the lesion. A score of more than 4/20 is considered malignant [16]. The current case of pheochromocytoma is benign, showing large nests or diffuse growth exceeding 10% resulting in a PASS score of less than four.
Conclusions
Oncocytic pheochromocytomas are usually non-functional tumors with unique histological and ultrastructural features. They are often found incidentally and are differentiated from other adrenal gland tumors like adrenocortical oncocytomas and metastatic oncocytic tumors. Immunohistochemistry confirms the diagnosis by demonstrating neuroendocrine markers. This case underscores the need for a thorough diagnostic strategy for detecting and managing rare adrenal tumors, including accurate differentiation from other adrenal neoplasms and diligent follow-up.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adrenal tumours in Chinese Virchows Arch A Pathol Anat Histopathol Lam KY 13164211992163624510.1007/BF 01607133 · doi ↗ · pubmed ↗
- 2Oncocytic pheochromocytoma with cytokeratin reactivity: a case report with immunohistochemical and ultrastructural studies Int J Surg Pathol Wang BY Gabrilove L Pertsemlidis D 616711997
- 3Oncocytes, oncocytosis, and oncocytic tumors Pathol Annu Chang A Harawi SJ 263304271992 http://PMID: 17362461736246 · pubmed ↗
- 4Adrenal oncocytic pheochromocytoma Am J Surg Pathol Li M Wenig BM 155215572420001107585910.1097/00000478-200011000-00013 · doi ↗ · pubmed ↗
- 5Oncocytic pheochromocytoma of the adrenal gland with preoperative endocrine examination Int Cancer Conf J Shibamori K Kobayashi K Itoh N 165168220131
- 6Adrenal oncocytic neoplasm: a systematic review Urol Int Mearini L Del Sordo R Costantini E Nunzi E Porena M 1251339120132314719610.1159/000345141 · doi ↗ · pubmed ↗
- 7Oncocytoma of the adrenal gland medulla Ann Diagn Pathol ChistéM Poppiti RJ Bianco FJ 1231261720132205603810.1016/j.anndiagpath.2011.07.002 · doi ↗ · pubmed ↗
- 8Oncocytic lesions of the neuroendocrine system Adv Anat Pathol Canberk S Li Volsi VA Baloch Z 69822120142450869010.1097/PAP.0000000000000011 · doi ↗ · pubmed ↗