Metagenomes, metagenome-assembled genomes, and metatranscriptomes from a chlorinated ethene-dechlorinating culture amended with biochar pyrolyzed at different temperatures
Hongyu Dang, Weilun Zhao, Timothy E. Mattes

TL;DR
This study explores how biochar and its pyrolysis temperature affect a microbial community that breaks down chlorinated ethene.
Contribution
The novel contribution is the detailed metagenomic and metatranscriptomic analysis of a dechlorinating microbial consortium interacting with biochar.
Findings
Biochar amendment altered microbial community structure and function.
Pyrolysis temperature influenced microbial activity and gene expression.
Metagenome-assembled genomes revealed insights into dehalogenating pathways.
Abstract
We investigated the effects of biochar and pyrolysis temperature on a chlorinated ethene-dechlorinating anaerobic consortium. Sequencing of nucleic acids from suspended and biochar-attached cells yielded 9 metagenomes, 122 metagenome-assembled genomes, and 18 metatranscriptomes that provide insights into the structure, function, activity, and interactions of the dehalogenating consortium with biochar.
Click any figure to enlarge with its caption.
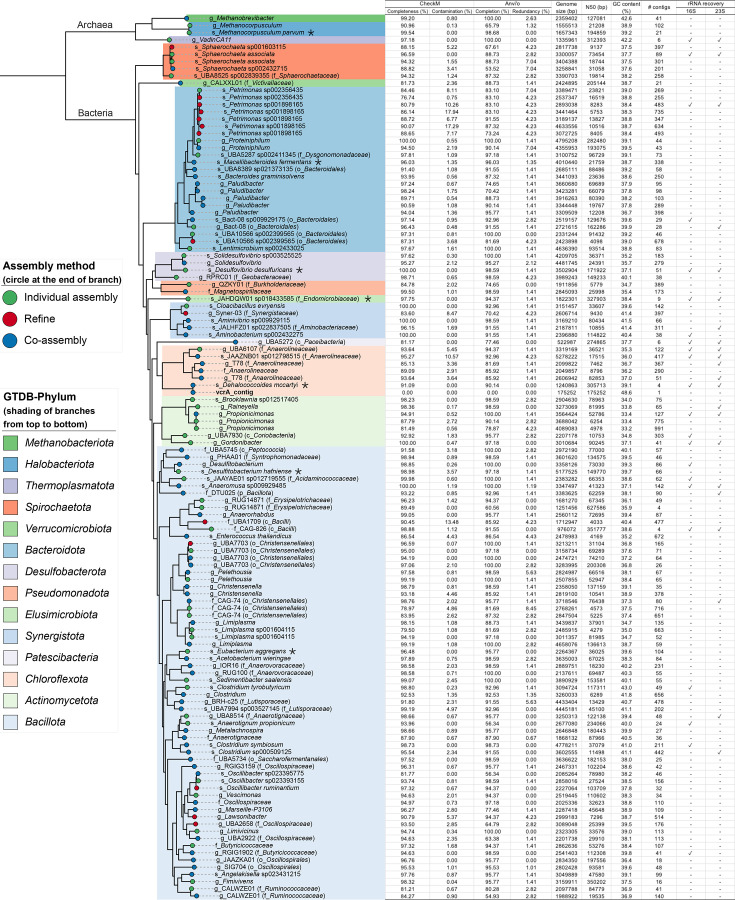 Fig 1
Fig 1| Sample name | SRA accession no. | BioSample no. | Pyrolysis temperature (°C) | Replicate | Sample type | Number of reads | |||
|---|---|---|---|---|---|---|---|---|---|
| Before trim | After trim-paired | After trim-unpaired R1 | After trim-unpaired R2 | ||||||
| Char350-A-liquid_DNA |
|
| 350 | – | Metagenome | 42,164,008 | 40,942,698 | 841,272 | 287,877 |
| Char350-B-attached_DNA |
|
| 350 | – | Metagenome | 53,558,356 | 52,003,807 | 1,030,067 | 386,197 |
| Char500-A-attached_DNA |
|
| 500 | – | Metagenome | 44,813,995 | 43,558,176 | 816,976 | 326,457 |
| Char500-B-liquid_DNA |
|
| 500 | – | Metagenome | 47,600,200 | 46,070,842 | 1,020,842 | 364,793 |
| Char700-A-attached_DNA |
|
| 700 | – | Metagenome | 49,407,025 | 48,054,961 | 848,396 | 379,281 |
| Char700-B-liquid_DNA |
|
| 700 | – | Metagenome | 50,301,937 | 48,829,200 | 952,342 | 385,050 |
| Char900-A-liquid_DNA |
|
| 900 | – | Metagenome | 48,444,322 | 47,201,060 | 794,642 | 338,191 |
| Char900-B-attached_DNA |
|
| 900 | – | Metagenome | 48,296,394 | 46,942,738 | 849,928 | 377,311 |
| NoChar-A-liquid_DNA |
|
| NA | – | Metagenome | 46,581,840 | 45,244,312 | 833,038 | 378,801 |
| Char350-A-attached_RNA |
|
| 350 | A | Metatranscriptome | 57,957,767 | 56,961,929 | 652,912 | 284,891 |
| Char350-A-liquid_RNA |
|
| 350 | A | Metatranscriptome | 48,115,301 | 47,220,372 | 698,649 | 145,630 |
| Char350-B-attached_RNA |
|
| 350 | B | Metatranscriptome | 48,588,053 | 47,579,440 | 796,578 | 143,771 |
| Char350-B-liquid_RNA |
|
| 350 | B | Metatranscriptome | 51,770,161 | 50,826,589 | 710,920 | 165,394 |
| Char500-A-attached_RNA |
|
| 500 | A | Metatranscriptome | 57,882,442 | 56,775,702 | 831,448 | 203,673 |
| Char500-A-liquid_RNA |
|
| 500 | A | Metatranscriptome | 57,447,734 | 55,879,132 | 1,329,767 | 158,883 |
| Char500-B-attached_RNA |
|
| 500 | B | Metatranscriptome | 54,685,915 | 53,406,752 | 1,054,073 | 147,996 |
| Char500-B-liquid_RNA |
|
| 500 | B | Metatranscriptome | 53,516,674 | 52,587,866 | 702,192 | 159,253 |
| Char700-A-attached_RNA |
|
| 700 | A | Metatranscriptome | 39,154,854 | 38,428,073 | 512,125 | 169,707 |
| Char700-A-liquid_RNA |
|
| 700 | A | Metatranscriptome | 60,394,555 | 59,237,767 | 918,897 | 169,161 |
| Char700-B-attached_RNA |
|
| 700 | B | Metatranscriptome | 66,681,063 | 65,239,485 | 1,150,431 | 196,916 |
| Char700-B-liquid_RNA |
|
| 700 | B | Metatranscriptome | 58,947,262 | 57,787,395 | 921,054 | 166,568 |
| Char900-A- attached_RNA |
|
| 900 | A | Metatranscriptome | 58,700,889 | 57,558,788 | 843,335 | 211,378 |
| Char900-A-liquid_RNA |
|
| 900 | A | Metatranscriptome | 66,675,677 | 65,344,965 | 1,061,992 | 181,614 |
| Char900-B-attached_RNA |
|
| 900 | B | Metatranscriptome | 62,066,578 | 60,842,217 | 969,950 | 178,548 |
| Char900-B-liquid_RNA |
|
| 900 | B | Metatranscriptome | 54,717,891 | 53,611,439 | 873,365 | 160,818 |
| NoChar-A-liquid_RNA |
|
| NA | A | Metatranscriptome | 51,288,938 | 50,335,534 | 743,633 | 146,313 |
| NoChar-B-liquid_RNA |
|
| NA | B | Metatranscriptome | 60,643,367 | 59,652,887 | 685,617 | 235,730 |
- —HHS | NIH | National Institute of Environmental Health Sciences (NIEHS)
- —HHS | NIH | National Institute of Environmental Health Sciences (NIEHS)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial bioremediation and biosurfactants · Microbial Community Ecology and Physiology · Microbial Metabolic Engineering and Bioproduction
ANNOUNCEMENT
Biochar has been used as a supplement to enhance anaerobic biodegradation of chlorinated ethenes, as it provides habitat, facilitates electron transfer, and stimulates microbial activity and microbe-microbe interactions (1, 2). However, specific biochar properties (e.g., pore size, electron donating/accepting, conductivity, etc.) that are beneficial to microbial ecology, growth, and activity are poorly understood. To fill these research gaps, biochars were synthesized at various temperatures (350°C, 500°C, 700°C, and 900°C) to provide a range of material properties. Each biochar type was added to duplicate microcosms constructed with 100 mL RAMM medium in 160 mL serum bottles and inoculated with an anaerobic tetrachloroethene-dechlorinating consortium (SDC-9) (3–5), together with duplicate live controls without biochar (10 bottles in total).
After 51–82 days of incubation, solid biochar was separated from the liquid culture in bottles by passing through 20 µm UV-sterilized filters (Just the Basics, Woonsocket, RI, USA) and washed with RAMM medium. RNA and DNA were extracted from both liquid and biochar samples with the RNeasy PowerSoil total RNA kit plus DNA Elution kit (Qiagen, Germantown, MD, USA). Residual DNA in RNA extracts was removed with the TURBO DNA-free kit (Thermo Fisher Scientific) and the Direct-zol RNA MiniPrep Plus kit (Zymo Research Corp., Irvine, CA, USA). High-quality RNA (i.e., with clear 16S and 23S rRNA peaks and RNA integrity number > 8), as confirmed with a 2100 Bioanalyzer RNA Pico assay (Agilent Technologies, Santa Clara, CA, USA), was submitted for sequencing.
RNA and DNA were sequenced at the Iowa Institute of Human Genetics (Iowa City, IA, USA). Sheared DNA (average size 550 bp) was used to prepare indexed DNA libraries with the KAPA HyperPrep Kit (Roche Sequencing and Life Science, Indianapolis, IN, USA). RNA libraries were indexed and rRNA-depleted with the stranded total RNA preparation with Ribo-Zero Plus Kit (Illumina Inc., San Diego, CA, USA). Supplemental rRNA probes (149) for dominant taxa in the culture, developed from eight 16S rRNA sequences retrieved from Silva 138.1 (6), were included to improve rRNA depletion and mRNA sequencing efficiency. The DNA and RNA libraries were pooled and sequenced with NovaSeq 6000 on SP (150-bp paired-end) and S1 flow cells (100-bp paired-end), respectively.
Raw sequencing reads were quality controlled by trimming adapters and low-quality sequences (average quality score per base < 15 or sequence length < 36 bases) with Trimmomatic (version 0.39) (7). Trimmed DNA and RNA data sets ranged from 38 to 60 million reads (Table 1), which provided sufficient sequencing depth for metagenomic and metatranscriptomic analysis (8). Reads were assembled into contigs using both individual assembly and co-assembly approaches with Megahit (version 1.2.9) (9). Trimmed short reads were mapped to the contigs with bowtie2 (version 2.2.5) (10). Contigs and their corresponding BAM files were used for binning with Metabat2 (version 2.15) (11). Bin completion and contamination were quantified with CheckM (version 1.2.2) (12) and Anvi’o (version 7.1) (13). Bins with a completion 80% and contamination 5% were designated as metagenome-assembled genomes (MAGs). Bins with a completion 80% and contamination > 5% were further refined with Anvi’o. All MAGs were dereplicated with dRep (version 3.4.2) (14). MAG marker genes, determined by checkM, were aligned with Clustal Omega (version 1.2.3) (15) and used to generate a phylogenetic tree with iqtree (version 2.0.3) (16). MAG taxonomy was classified using GTDB-tk (version 2.1.1; database release R214) and visualized in iTOL (version 5) (17) (Fig. 1). MAGs were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (version 6.0) (18).
Phylogenetic tree of 122 MAGs after dereplication, quality statistics determined by CheckM and Anvi’o, genome size, N50, GC content, number of contigs, and rRNA gene recovery (i.e., 16S and 23S, “✓” represents a recovery and “-” represents no recovery). Anvi’o was used to refine bins with completion ≥ 80% and contamination > 5%. Redundant sequences were manually removed based on their read coverage according to bin refinement instructions (https://merenlab.org/2015/05/11/anvi-refine/). The tree was generated using marker genes from each MAG as determined by checkM. The contig containing vcrA (vcrA_contig) was manually added next to the Dehalococcoides MAG on the tree as it has a 99% nucleotide identity with Dehalococcoides mccartyi determined by BLAST (19). Because the vcrA_contig and Dehalococcoides MAG had different tetranucleotide frequencies, they were binned separately. The vcrA_contig GenBank accession number is PP061217.1. The 16S rRNA gene sequences of dominant taxa (marked with “∗”) were collected from the Silva database to design probes for rRNA depletion. Silva taxa (and accession numbers) are Eubacterium (QTVG01000004); Dehalococcides mccartyi, (CP011127); Desulfitobacterium hafniense (AP008230); Desulfovibrio biadhensis (LM999902); Methanocorpusculum aggregans (LMVO01000026); Endomicrobium proavitum (CP009498); and Macellibacteroides fermentans, a(HQ020488).
A total of 122 MAGs belonging to 14 phyla were obtained, including one classified as Dehalococcoides mccartyi (Fig. 1). At least 87% of the metagenomic short reads were mapped to these MAGs, which indicated they represent the microbial composition in SDC-9. These data will advance knowledge of chlorinated ethene-dechlorinating microbial communities and their interactions with biochar.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wu Z, Man Q, Niu H, Lyu H, Song H, Li R, Ren G, Zhu F, Peng C, Li B, Ma X. 2022. Recent advances and trends of trichloroethylene biodegradation: a critical review. Front Microbiol 13:1053169. doi:10.3389/fmicb.2022.105316936620007 PMC 9813602 · doi ↗ · pubmed ↗
- 2Bolan S, Hou D, Wang L, Hale L, Egamberdieva D, Tammeorg P, Li R, Wang B, Xu J, Wang T, Sun H, Padhye LP, Wang H, Siddique KHM, Rinklebe J, Kirkham MB, Bolan N. 2023. The potential of biochar as a microbial carrier for agricultural and environmental applications. Sci Total Environ 886:163968. doi:10.1016/j.scitotenv.2023.16396837164068 · doi ↗ · pubmed ↗
- 3Steffan RJ, Vainberg S. 2013. Production and handling of dehalococcoides bioaugmentation cultures, p 89–115. In Stroo HF, Leeson A, Ward CH (ed), Bioaugmentation for groundwater remediation. Springer New York, New York, NY.
- 4Vainberg S, Condee CW, Steffan RJ. 2009. Large-scale production of bacterial consortia for remediation of chlorinated solvent-contaminated groundwater. J Ind Microbiol Biotechnol 36:1189–1197. doi:10.1007/s 10295-009-0600-519521729 · doi ↗ · pubmed ↗
- 5Shelton DR, Tiedje JM. 1984. General method for determining anaerobic biodegradation potential. Appl Environ Microbiol 47:850–857. doi:10.1128/aem.47.4.850-857.19846721493 PMC 239775 · doi ↗ · pubmed ↗
- 6Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D 590–6. doi:10.1093/nar/gks 121923193283 PMC 3531112 · doi ↗ · pubmed ↗
- 7Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi:10.1093/bioinformatics/btu 17024695404 PMC 4103590 · doi ↗ · pubmed ↗
- 8Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. 2014. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 15:121–132. doi:10.1038/nrg 364224434847 · doi ↗ · pubmed ↗