D-Hexopyranosides with Vicinal Nitrogen-Containing Functionalities
Jana Pospíšilová, Daniel Toman, Tomáš Ručil, Petr Cankař

TL;DR
This review summarizes the synthesis of D-hexopyranosides with nitrogen-containing groups, which are found in many natural and pharmaceutical compounds.
Contribution
The paper provides a comprehensive overview of synthetic methods for D-hexopyranosides with vicinal nitrogen-containing functionalities since the 1960s.
Findings
D-hexopyranosides with nitrogen-containing groups are important in natural and pharmaceutical compounds.
The review organizes syntheses based on substitution positions and methodologies.
These compounds exhibit diverse biological functions due to their structural variety.
Abstract
Various substituted D-hexopypyranosides units with nitrogen-containing functionalities are present in many important natural compounds and pharmaceutical substances. Since their complex structural diversity contributes to a broad spectrum of biological functions and activities, these derivatives are frequently studied. This review covers syntheses of D-hexopyranosides with vicinal nitrogen-containing functionalities since the 1960s, when the first articles emerged. The syntheses are arranged according to the positions of substitutions, to form a relative configuration of vicinal functionalities, and synthetic methodologies.
Click any figure to enlarge with its caption.
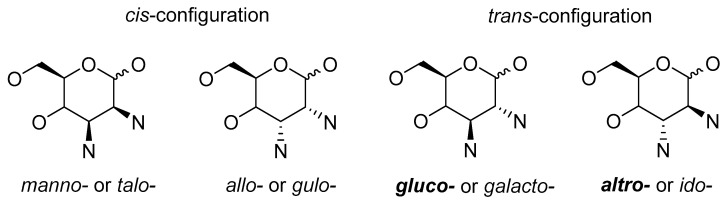 Figure 1
Figure 1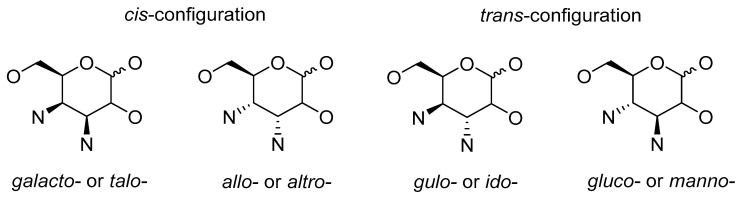 Figure 2
Figure 2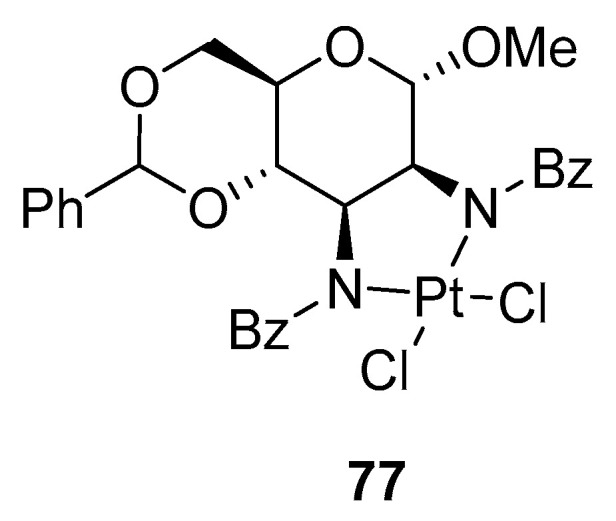 Figure 3
Figure 3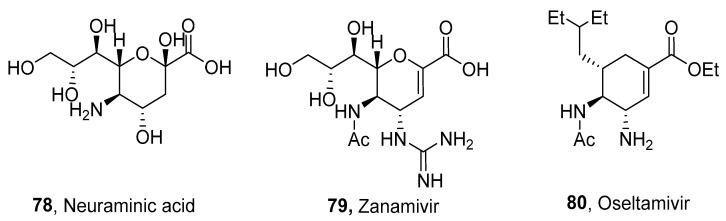 Figure 4
Figure 4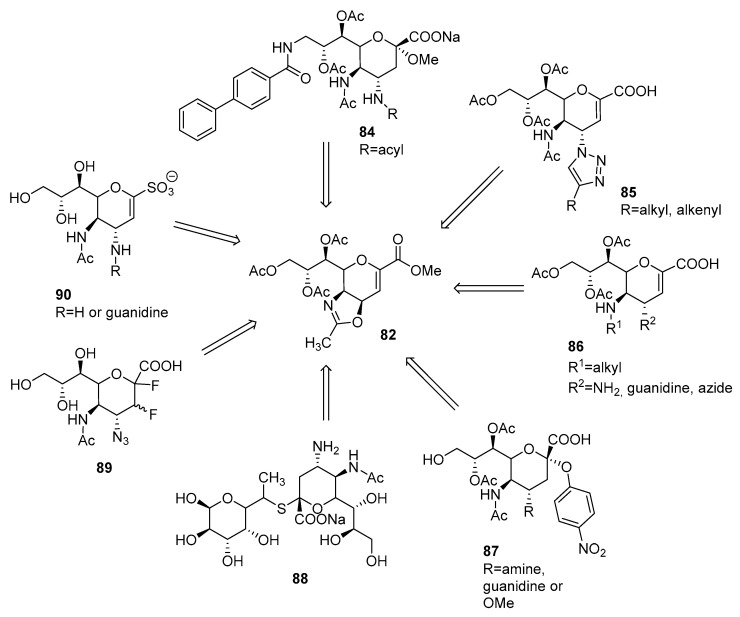 Figure 5
Figure 5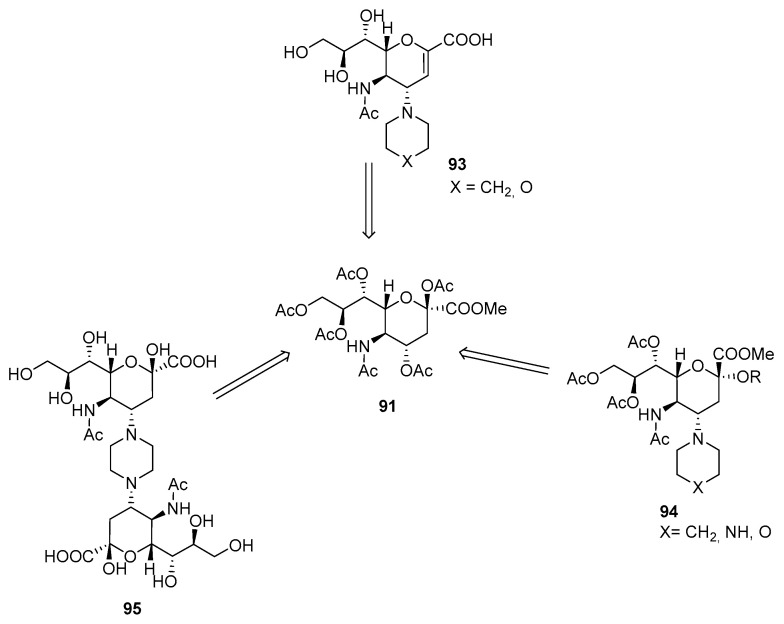 Figure 6
Figure 6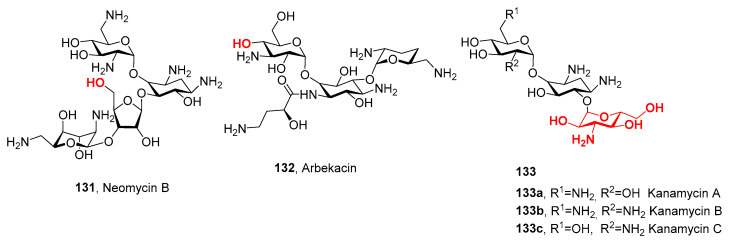 Figure 7
Figure 7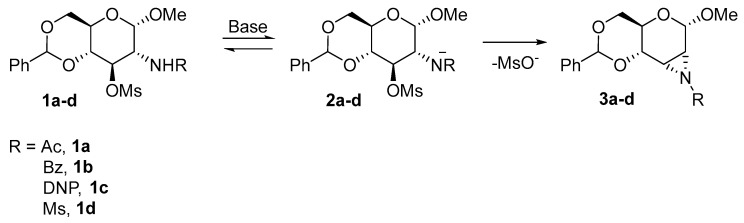 Figure 8
Figure 8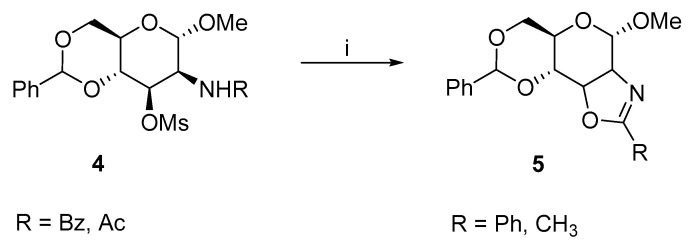 Figure 9
Figure 9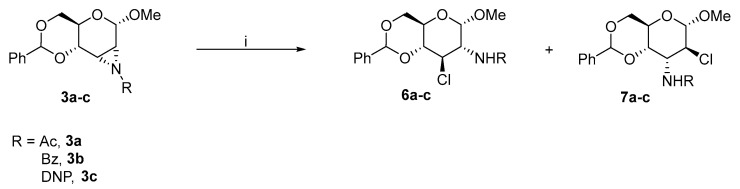 Figure 10
Figure 10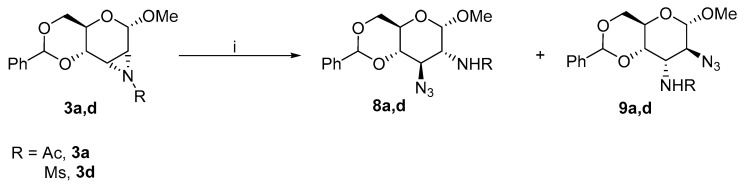 Figure 11
Figure 11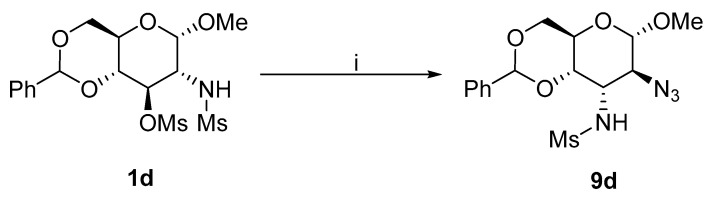 Figure 12
Figure 12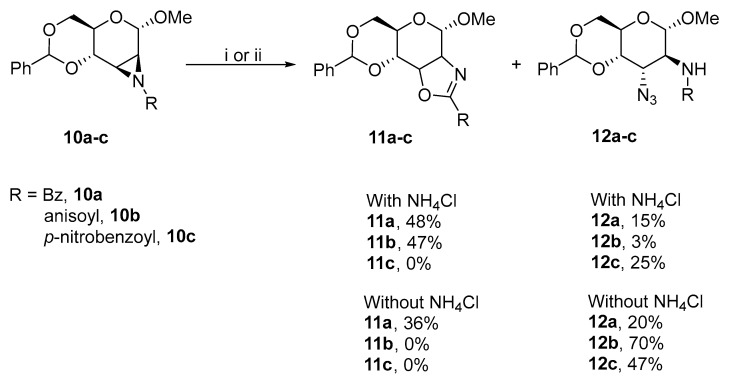 Figure 13
Figure 13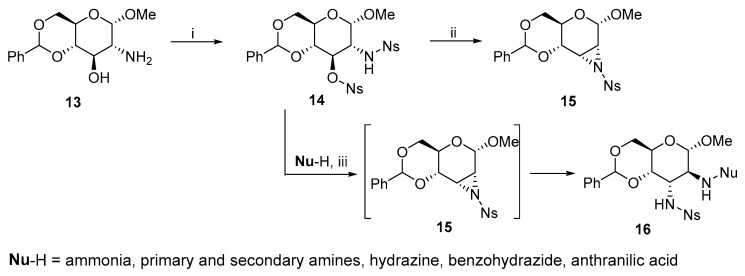 Figure 14
Figure 14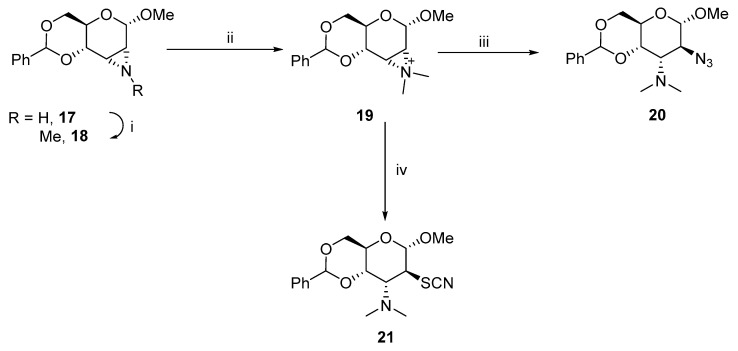 Figure 15
Figure 15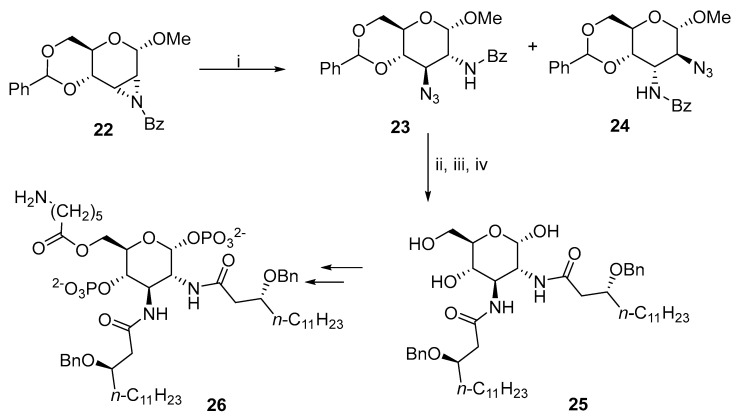 Figure 16
Figure 16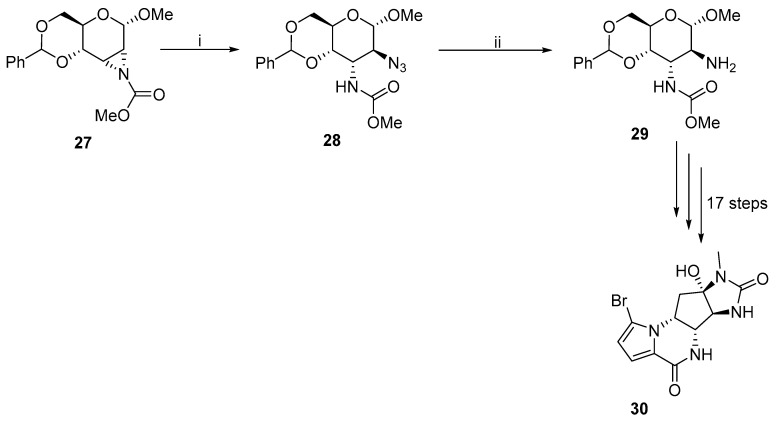 Figure 17
Figure 17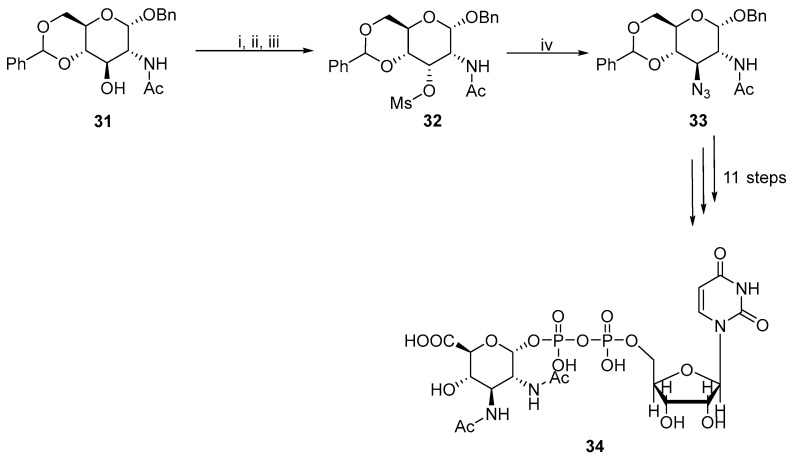 Figure 18
Figure 18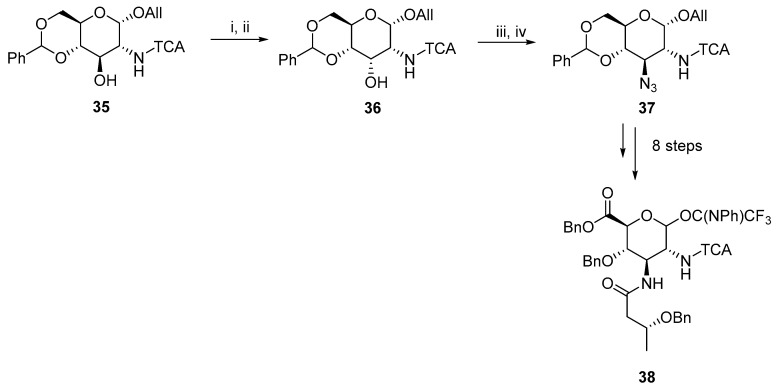 Figure 19
Figure 19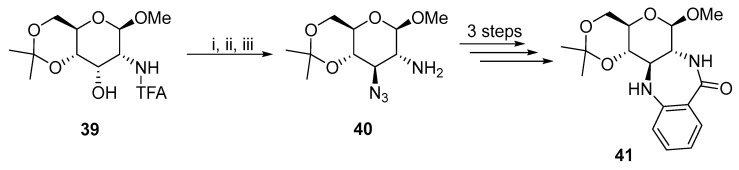 Figure 20
Figure 20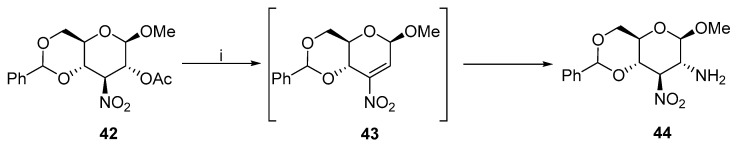 Figure 21
Figure 21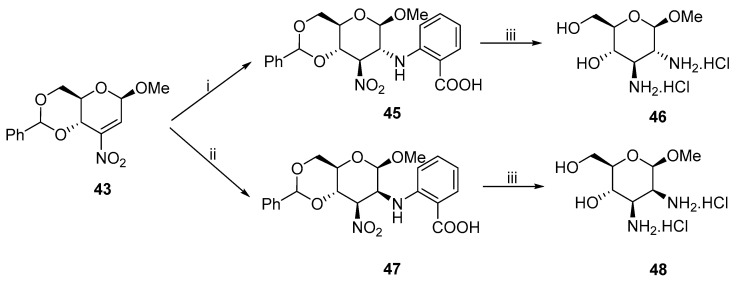 Figure 22
Figure 22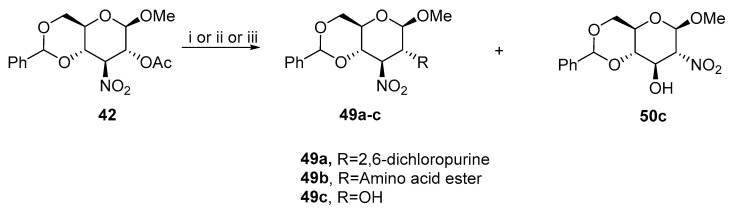 Figure 23
Figure 23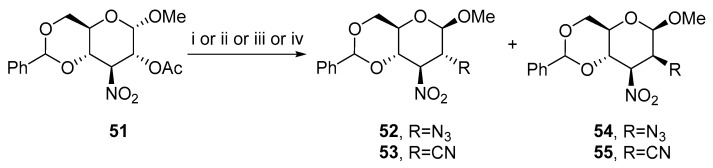 Figure 24
Figure 24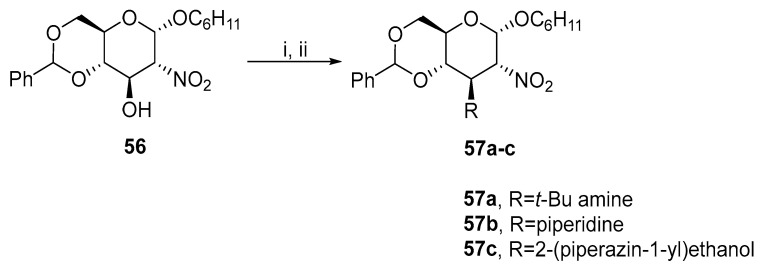 Figure 25
Figure 25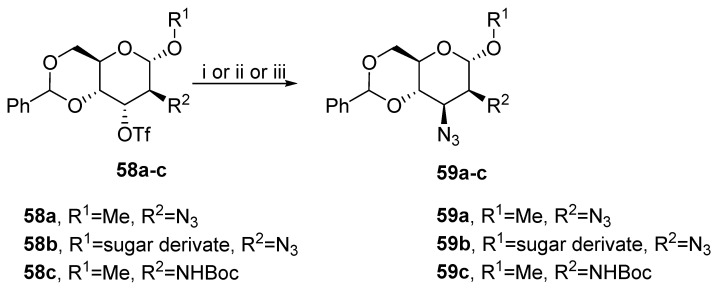 Figure 26
Figure 26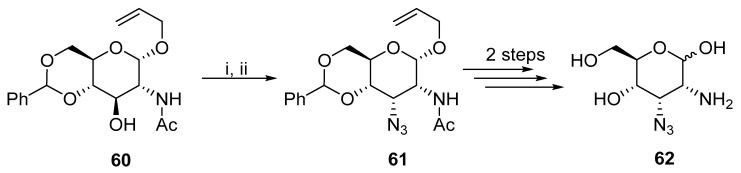 Figure 27
Figure 27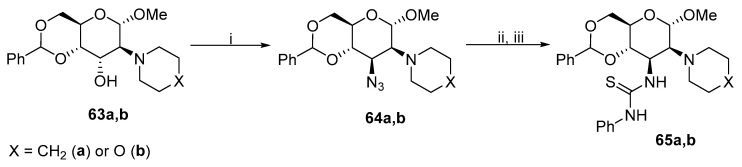 Figure 28
Figure 28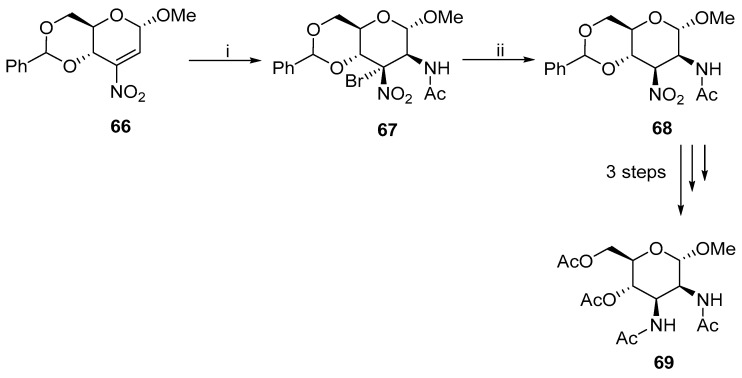 Figure 29
Figure 29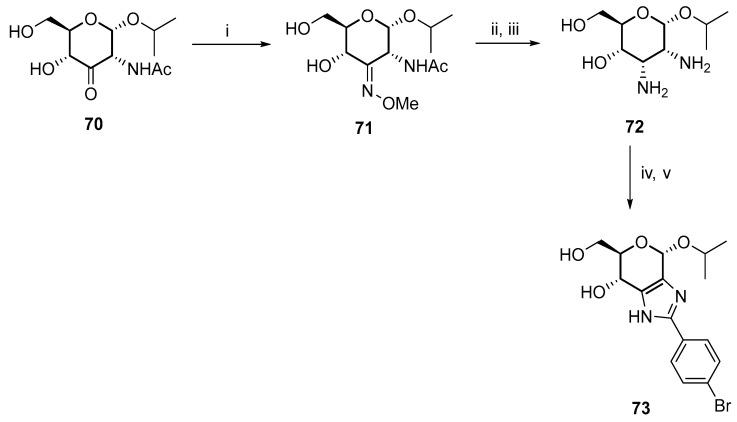 Figure 30
Figure 30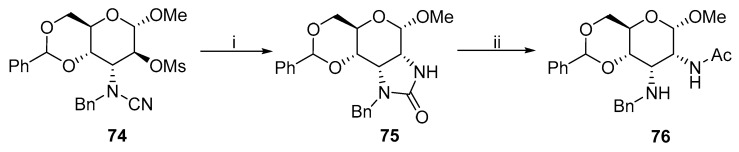 Figure 31
Figure 31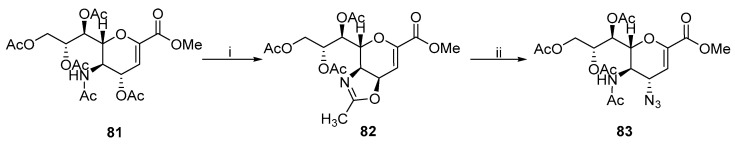 Figure 32
Figure 32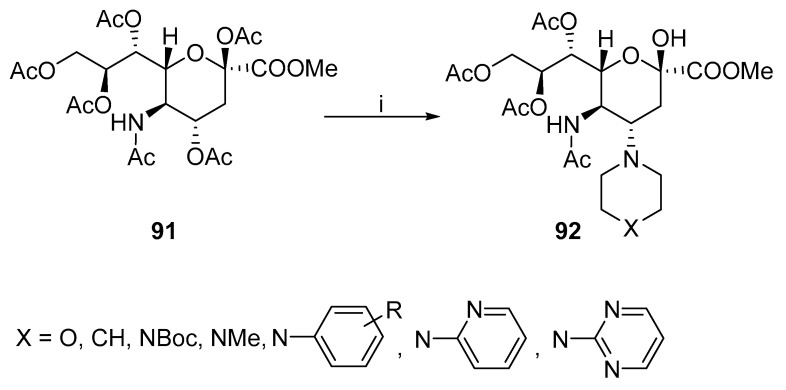 Figure 33
Figure 33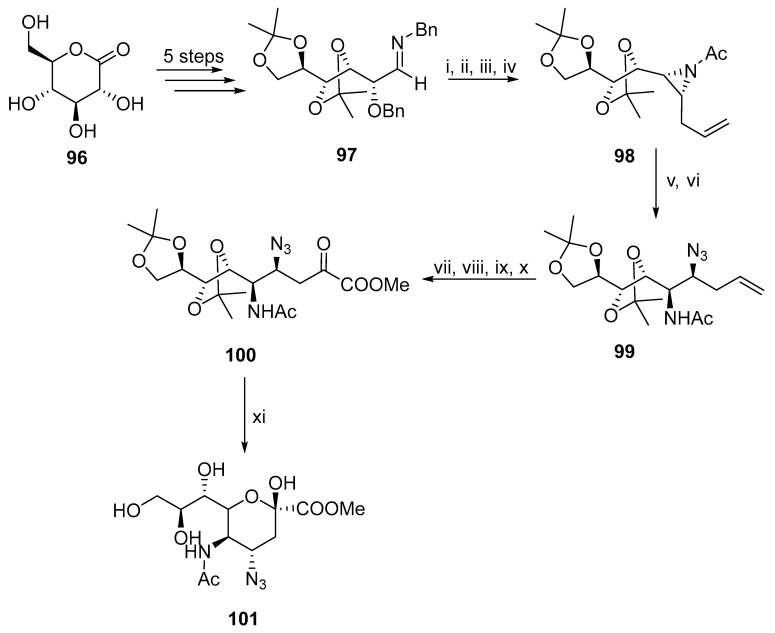 Figure 34
Figure 34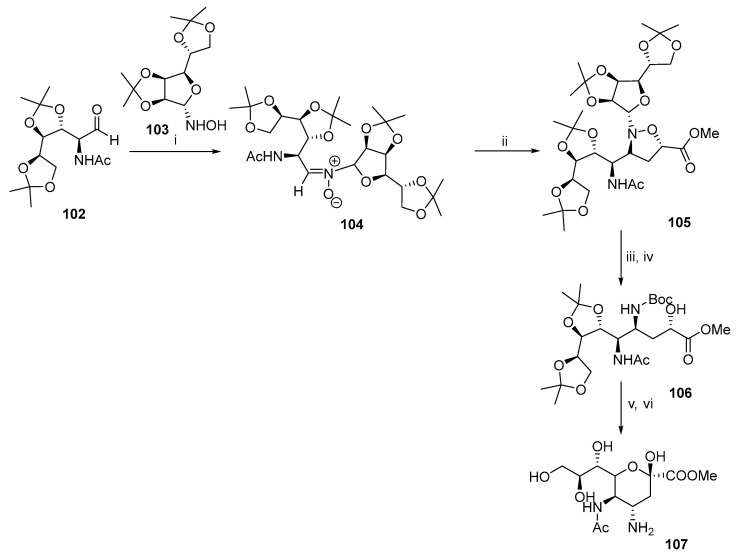 Figure 35
Figure 35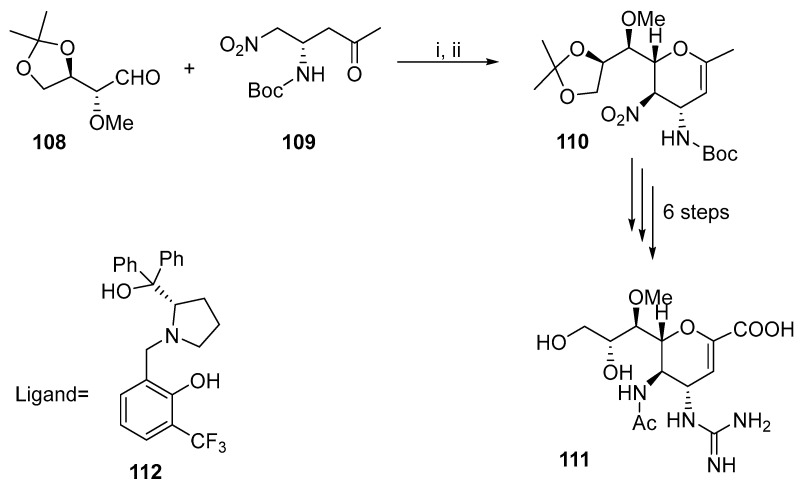 Figure 36
Figure 36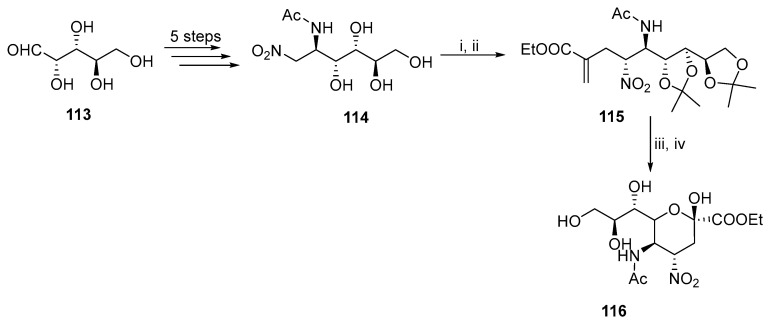 Figure 37
Figure 37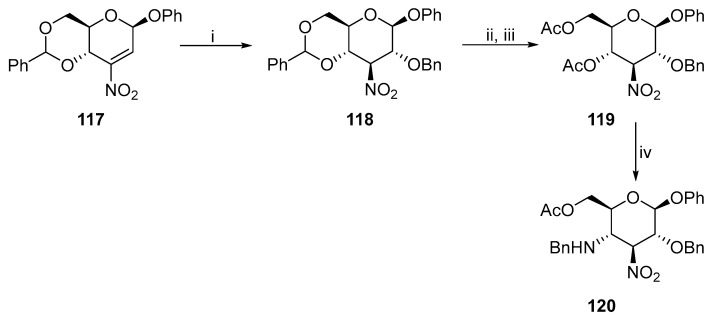 Figure 38
Figure 38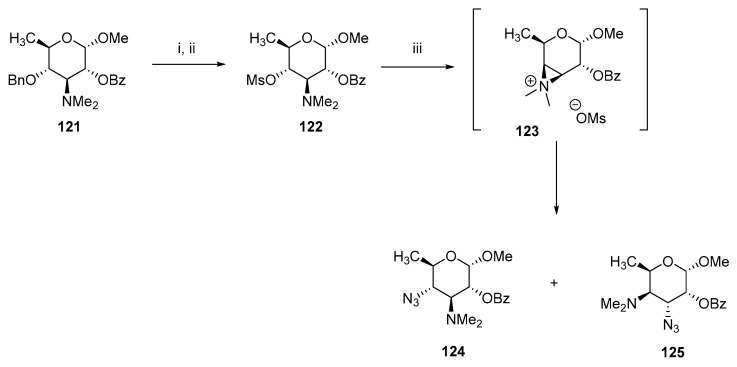 Figure 39
Figure 39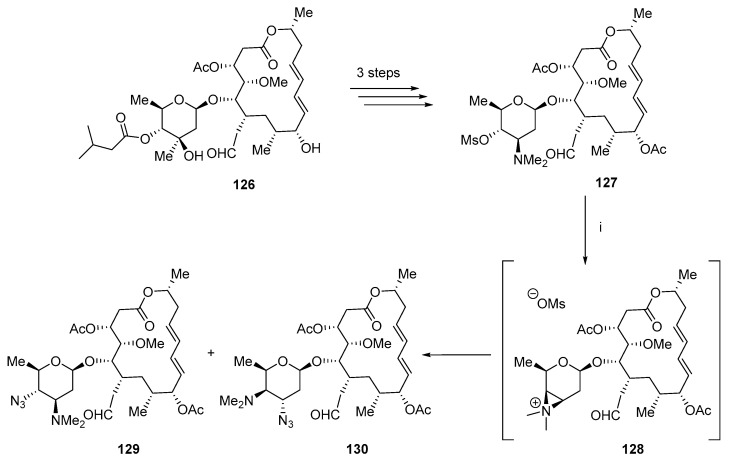 Figure 40
Figure 40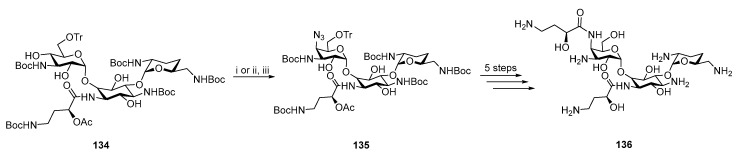 Figure 41
Figure 41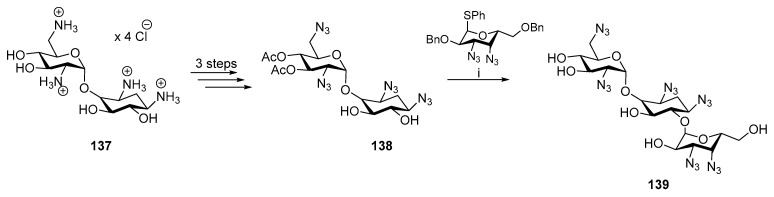 Figure 42
Figure 42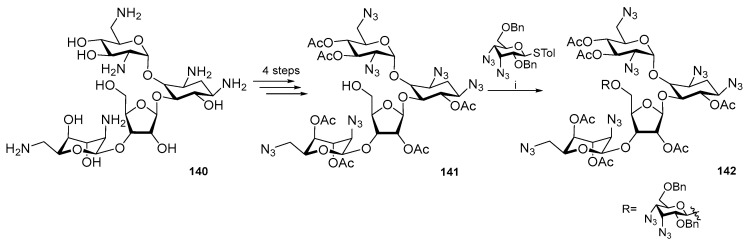 Figure 43
Figure 43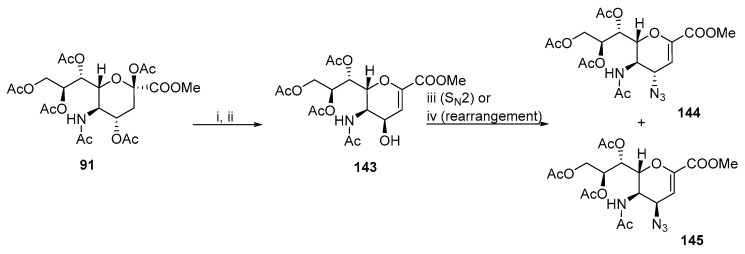 Figure 44
Figure 44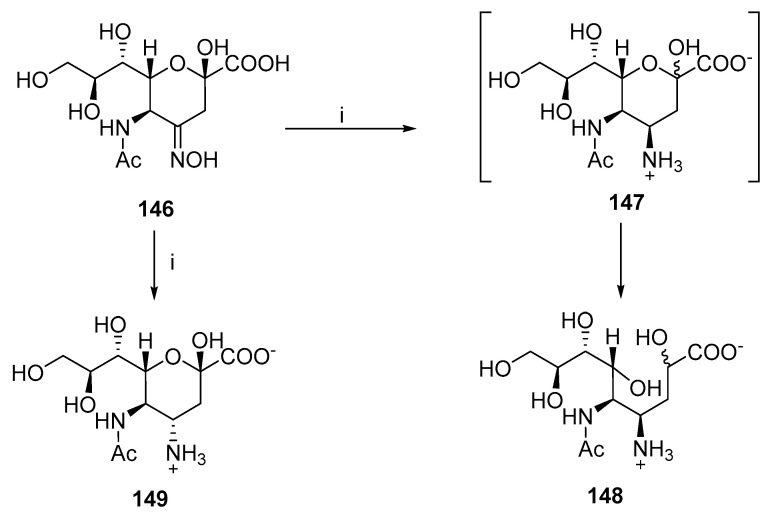 Figure 45
Figure 45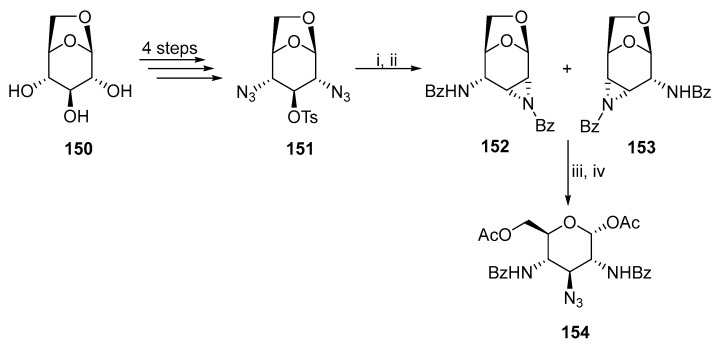 Figure 46
Figure 46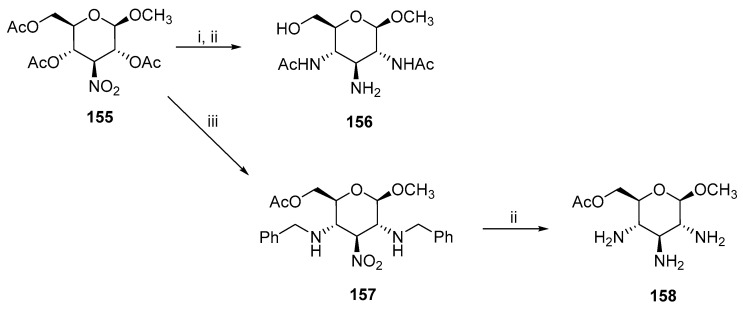 Figure 47
Figure 47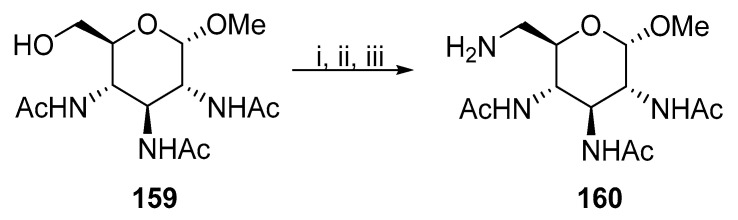 Figure 48
Figure 48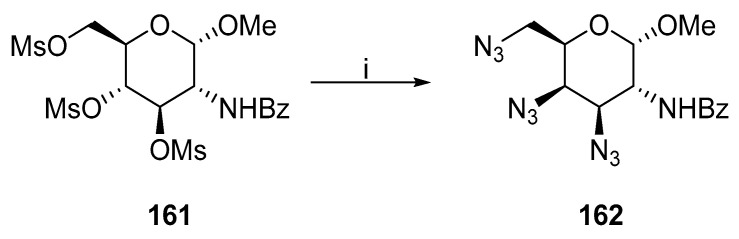 Figure 49
Figure 49- —Internal Grant Agency of Palacký University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCarbohydrate Chemistry and Synthesis · Synthesis of Organic Compounds · Synthetic Organic Chemistry Methods
1. Introduction
The presence of nitrogen functionalities in saccharides increases their molecular complexity and diversity. The evolution of living organisms uses amino sugars for various functions, e.g., as structural components, signaling molecules, transporting molecules, and post-translational modified proteins.
The most commonly known N-acetylglucosamine is present in chitin as a monomer unit that forms the polysaccharide chain. Deacetylation of chitin leads to chitosan, which has practical applications in medicine, agriculture, and industry. Chitin is also processed to obtain D-glucosamine, which is frequently used as a dietary supplement and intermediate for biologically relevant molecules. Furthermore, the N-acetylglucosamine units are essential for the biosynthesis of peptidoglycans and hyaluronic acid.
Epimeric D-galactosamine units are present in glycoprotein hormones such as luteinizing and follicle-stimulating hormones [1]. D-Mannosamine, which is another epimer, has been mostly revealed in glycoproteins and gangliosides. The N-acetylated form is a precursor for the biosynthesis of N-acetylneuraminic acid, which is a predominant derivative of sialic acid in human cells [2]. N-Acetylneuraminic acid is also involved in the development of influenza virus infections and the biology of pathogenic and symbiotic bacteria [3,4].
The structure of N-acetylneuraminic acid was a starting point for the rational design of neuraminidase inhibitors. Thus, Zanamivir, which was the first commercially developed neuraminidase inhibitor, was approved for the treatment and prevention of influenza A and B. Oseltamivir, which is another commercial inhibitor with a more simplified structure, preserves two nitrogen functionalities. Recent ongoing development of these inhibitors relies on the bio-isosteric substitution to replace the carboxylate with a phosphonate or sulfonate group to increase the total binding energy [5].
The presence of nitrogen functional groups also plays an important role in the binding of aminoglycoside antibiotics such as Arbekacin, Kanamycin B, or Neomycin B, which bind to the bacterial ribosomal subunits. The amino saccharides were also used as starting materials to synthesize compounds with various biological activities. For example, the alkaloid (−)-Agelastatin A with anticancer activity and the glycophospholipid ligand of a lipopolysaccharide receptor were synthesized from D-glucosamine.
Nitrogen-containing functionalities in D-hexopyranosides have an irreplaceable role in living organisms. Thus, research teams continue to develop synthetic methodologies to introduce nitrogen-containing functionalities into D-hexopyranosides. This review focuses on syntheses that lead to D-hexopyranosides with vicinal nitrogen-containing functionalities.
Figure 1 defines the structures of interest of derivatives with nitrogen-containing functionalities at positions 2 and 3, and the special emphasis is on the most common molecules that contain gluco- and altro- configurations. These carbohydrates offer many biological activities and synthetic opportunities for further transformations. Several of them are discussed herein: (−)-agelastatin A, which has anti-tumor activity [6]; glycophospholipid PPDm2-B, which interacts with the liposaccharide receptor of macrophages [7]; ligands for the Mo-catalysed allylic alkylation [8]; half-sandwich metal complexes with comparable anti-tumor activity to cis-platina [9]; and hybrids of β-D-glucose with benzodiazepine scaffolds [10].
Figure 2 shows the main motifs of the 3,4-disubstituted D-hexopyranosides. The cis-configuration of vicinal nitrogen-containing functionalities is in the derivatives of Neomycin, Kanamycin, and related compounds. The trans-configuration is incorporated in the skeleton of Zanamivir and its analogues.
2. Nitrogen Functionalities at Positions 2 and 3
Several synthetic methodologies provide D-hexopyranosides with nitrogen-containing functionalities at position 2 and 3. Generally, synthetic routes that lead to cis- and trans-derivatives are predetermined by the starting material. Both trans- and cis-derivatives are separately discussed.
2.1. Trans-Configuration
The derivatives with trans-oriented nitrogen-containing functionalities are synthesized based on the (a) aziridine formation and subsequent ring-opening reaction, (b) addition of an activated double bond, and (c) S_N_2 substitution of an activated hydroxyl group.
2.1.1. Aziridine Formation
Richardson and coworkers described the formation of aziridine 3 with an allo-configuration from substituted glucopyranosides 1a–d in 1965 (Scheme 1). The crucial condition for aziridine formation is the trans-diaxial configuration of glucosamine 1. The cis-configuration (e.g., in mannopyranoside) results in hexopyranosides 5 with an oxazoline ring (Scheme 2) [11]. A base-catalyzed aziridine formation requires strong nucleophilic anion of the acylamido group at the C2 carbon, which attacks the C3 carbon with the activated hydroxyl group to yield the desired aziridines 3a–d. There are many suitable bases; the method of choice predominantly depends on the N-substitution.
Richardson inspired other research groups. Later, Richardson [12] extended the substitution at C2–amine to anisoyl, dinitrophenyl (DNP), and mesyl groups with the desired activating effect and described the possibilities for ring-opening reactions. The product of the ring-opening reaction strongly depends on the amine substitution at the C2 carbon and nucleophile. As mentioned (in Scheme 1), a stronger anion favours the aziridine formation. A weaker anion could not accomplish the substitution of the O-mesyl group, and oxazoline 5 was obtained as a major side product. The same publication described the ring-opening reactions of acetylated, benzoylated, or DNP-substituted aziridines 3a–c with ammonium chloride. Initially, the ring-opening reactions were accomplished with halogen nucleophiles. When aziridines 3a–c were refluxed in DMF, the aziridine underwent trans-di-axial and trans-di-equatorial ring-opening reactions and formed gluco- (6a–c) and altro- (7a–c) products (Scheme 3).
The treatment of 3a with ammonium chloride provided almost exclusively gluco- derivative 6a, whereas aziridines 3b and 3c yielded mixtures of 6b,c and 7b,c, respectively. However, when sodium azide, which is a stronger nucleophile, was added to a reaction mixture dissolved in DMF in the presence of ammonium chloride, the formation of chloro- derivatives was suppressed, and azido derivatives 8 and 9 were formed (Scheme 4).
Aziridines 3a and 3d provided unsatisfactory yields of products with a mixture of ammonium chloride and sodium azide. The formation of side products or degradation of a starting compound was observed. Therefore, 1d was reacted with sodium azide in the absence of ammonium chloride to yield more 9d (Scheme 5).
The reaction proceeded via in situ formation of aziridine and a trans-diaxial ring-opening reaction, which exclusively resulted in 9d with the altro-configuration. With Richardson and coworkers, the Guthrie group [13,14] investigated the ring-opening reactions of manno-aziridine (Scheme 6). The reaction of 10b with sodium azide provided the highest yields. A similar conclusion was reached also by Meyer zu Reckendorf [15,16,17], who described the formation of an additional gluco-derivative.
The scope of the substitution at positions 2 and 3 was later expanded by ring-opening reactions of N-4-nosyl Hough–Richardson aziridine with 19 nitrogen nucleophiles (Scheme 7) [18]. The electron-withdrawing nitro group provided practical advantages. Aziridine 15 was synthesized under mild conditions with high yields. This aziridine can also be generated without isolation to furnish product 16 with an altro-configuration due to the highly regioselective ring-opening reactions. The altro-configuration is preferred over the gluco-configuration at ratios above 90:10.
D-hexopyranosides with vicinal trans-oriented nitrogen-containing functionalities can also be synthesized via aziridinium salts (Scheme 8) [19]. The hydrolysis of 19 with sodium azide and potassium thiocyanate yielded 20 and 21, which exclusively had altro-configurations.
The works of Richardson, Guthrie, and Meyer zu Reckendorf laid the foundations for further applications in the ring-opening reactions. Charon and co-workers used an aziridine ring-opening reaction to synthesize glycophospholipid ligands of lipopolysaccharide receptor 26 (Scheme 9) [7]. The ring-opening reaction of 22 afforded gluco-23 as a minor and altro-24 as a major product.
Hale used the aziridine-opening reaction to synthesize Agelastatin A 30, which is an inhibitor of GSK-3ß (Scheme 10) [6]. Aziridine 27 was treated with sodium azide in the presence of ammonium chloride, and 28 was a major product. No minor product was separated. Azide 28 was subsequently reduced to give derivative 29 at a very good yield. The desired Agelastatin A 30 was synthesized in a 17-step process.
2.1.2. Substitution of the Activated Hydroxyl Group
Nucleophilic substitution, which is associated with the inversion of a configuration, was performed after the hydroxyl group with mesyl, tosyl, or triflate agents had been activated. Rejzek and coworkers prepared phospho-derived glucuronic acid 34 using a double inversion at the C3 carbon (Scheme 11) [20].
A new strategy to synthesize 2,3-diamino-D-glucuronate was published for the total synthesis of Plesiomonas shigelloides serotype 51 aminoglycoside trisaccharide [21]. An interesting part of this synthesis was the Lattrel-Dax inversion from gluco- to allo-derivative 36. The described double inversion at C3 was first used to assemble a complex aminoglycoside 38 with various substitutions (Scheme 12). A trichloroacetamido (TCA) group was selected to mask the acetamido group of 38 to stereoselectively form the β-glycosidic bond.
A similar approach was used to synthesize cinnamon derivatives [22], 2,3-trans–diamino–metal-complexes (Mn, Pt, Rh, Ru, Ir, Cu, and Pd) [23,24,25,26,27,28,29,30,31], muramyl dipeptide analogues [32], or ß-D-glucose benzodiazepine derivatives 41 [10] (Scheme 13).
2.1.3. Michael Addition: Using Addition to Activated Double Bond
Michael addition to nitroolefins is another method to obtain 2,3–trans-diaminohexopyranosides. Baer reported the formation of olefins followed by the addition of ammonia in THF (Scheme 14) [33]. In addition to 44, an impurity with a yield below 10% was isolated but not characterized.
Afterwards, the synthesis was extended to anthranilic acid [34] and aminosugars [35]. The configuration of the diamine strongly depends on the pH of the reaction (Scheme 15). When the reaction occurred under basic conditions, gluco-product 45, which is thermodynamically more stable, was almost exclusively formed. In contrast, without KOH, manno- product 47 was isolated as a major isomer. Both 45 and 47 underwent reduction in the presence of the Kuhn catalyst and yielded diamines 46 and 48.
Subsequent publications used nucleosides [36], esters of amino acids [37], or sodium nitrite [38]. The reaction with 2,6-dichloropurine and amino acids esters (Gly, Ala, Phe, Ser, Tyr, and Val) exclusively produced 49a (a C–N bond was formed between C2 carbon and nitrogen at position 9) and 49b, respectively (Scheme 16). However, treating 42 with sodium nitrite resulted in 3-nitro derivative 49c as a major product and 50c as a minor isomer. The ratio of 49c to 50c could be slightly increased by adding hexadecyltributylphosphonium bromide.
Sakakibara and Sudoh studied the influence of the solvent and reagent on the substitution with azide or cyanide nucleophiles (Scheme 17) [39]. When sodium azide was used, 52 was isolated in 60% yield. When hydrazoic acid was added to THF, epimeric 54 was obtained in 79% yield. Therefore, more solvents were examined. Solvents such as DMSO or THF in the presence of hydrazoic acid favor the formation of 54. Chloroform or acetonitrile produced a mixture of 52 and 54. The study with hydrogen cyanide and potassium cyanide obtained the same conclusion. The reaction in DMSO led to major product 55 with the manno- configuration and the substitution in acetonitrile provided a mixture of 53 and 55.
The nitro group was also used at the C2 carbon (Scheme 18). Starting compound 56 was readily obtained by oxidizing protected glucosamine with m-CPBA [40]. Product 57c was further tested as a drug carrier.
2.2. Cis-Configuration
The cis configuration is commonly introduced by substituting an activated hydroxyl group with the appropriate configuration. Other less frequently used synthetic methods include subchapter miscellaneous reactions.
2.2.1. Substitution of Activated Hydroxyl Group
The most common method to prepare vicinal cis-oriented nitrogen-containing derivatives is based on nucleophilic substitution. Due to the inevitable inversion of the configuration during substitution, the hydroxyl group in the starting compound must be in the trans- position to the amine functional group. Walvoort used altropyranoside (58a) to synthesize mannopyranoside uronates. The number of azide groups was reduced in 59a, and the resulting intermediate was transformed into more complex derivatives (Scheme 19) [41]. Baer prepared disaccharose of the trehalose type 59b [42]. Finally, 59c was synthetized as a substrate for N-acetylneuraminic acid aldolase [43].
In contrast, Posakony prepared allopyranoside carbohydrate 61 from 60, where the final product 62 could serve as a catalytic cofactor analogue for glmS Rybozime (Scheme 20) [44]. The desired change in the configuration was achieved by the reaction of sodium azide with the mesylated hydroxyl group.
Alternatively, Mitsunobu reaction was used instead of the classical nucleophilic substitution of the activated hydroxyl group. This synthetic tool was applied to synthesize carbohydrate-based organocatalysts, where the C3 hydroxyl group was azidated with DPPA under Mitsunobu conditions (Scheme 21) [45].
2.2.2. Miscellaneous Methods
In addition to the S_N_2 reactions, the cis-configuration was achieved by other reactions. For example, Rank synthesized 69, where the key step was the addition of N-bromoacetamide to 3-nitroolefin 66 (Scheme 22) [46]. The reduction of 67 afforded nitro derivative 68, which was converted to acetylated mannopyranoside 69 in a three-step process. Similar results were obtained when talopyranoside was used.
Reductive amination at the C3 carbonyl provided another effective route to 3-aminoglucose (Scheme 23) [47]. Methyl oxime was formed as a mixture of the E and Z isomers, and subsequent hydrogenation provided the axially-oriented 3-amino group due to the anomeric isopropyl substituent. Furthermore, 2,3-dideoxy-2,3-diaminoallose 72 was used as a building block to synthesize imidazole derivative 73.
Another alternative method includes the formation of an imidazoline ring and its subsequent basic hydrolysis (Scheme 24). Baker et al. prepared 2,3–diamino allo-pyranosides 76 from the corresponding imidazolines 75, where phenyl could be attached to the imidazoline nitrogen instead of benzyl [48,49,50,51]. The long reaction time (up to 5 days) in each step was the main disadvantage of these synthetic routes.
Then, Baker et al. suggested the use of tosyl instead of benzyl groups, but the long reaction time and low yields unfortunately remained. Recently, allo- or manno-pyranosides were applied in the chemistry of complexes, where carbohydrates were used as ligands 77 and exhibited comparable anti-tumor activity comparable to cis-platina (Figure 3) [29].
3. Nitrogen Functionalities at Positions 3 and 4
The first part focuses on the synthesis of trans-diaminohexopyranoses, which are mainly incorporated into the skeleton of Zanamivir and its analogues. The second section discusses cis-dinitrogen-containing D-hexopyranoses, particularly derivatives of Neomycin, Kanamycin and related compounds. The final part discusses the reactions that lead to cis- and trans- products, where the configuration depends on the reaction conditions.
3.1. Trans-Configuration
A trans configuration with nitrogen-containing functionalities at positions 3 and 4 was introduced, particularly in the compounds derived from Neuraminic acid 78 (Figure 4); however, the trans configuration was found in other structures. Some neuraminidase inhibitors, such as Zanamivir 79 or Oseltamivir 80 are commercially available. These derivatives exhibit antiviral properties; therefore, their substitution is a topic of many research studies. There are several synthetic approaches to obtain the trans configuration: (a) oxazoline ring formation, (b) cyclization of acyclic intermediates with vicinal dinitrogen-containing functionalities, (c) Michael addition, and (d) aziridine ring formation.
3.1.1. Oxazoline Ring Formation
The formation and ring-opening of the oxazoline ring constitute a proven route to synthesize trans-3,4–diamino carbohydrates. Von Itzstein et al. prepared oxazoline 82 from O-acetylated derivative 81 (Scheme 25) [52]. The oxazoline ring is vulnerable to nucleophilic attacks at the C-O bond. Treating 82 with lithium azide forms 83.
The ring-opening reaction of the oxazoline ring with an azidation reagent was used to synthesize many derivatives. The substitution of deprotected primary hydroxyl group and a subsequent oxazoline ring-opening reaction with TMSN_3_ and azide reduction afforded carbohydrate 84 (Figure 5) [53]. The click reaction of azide with various acetylenes yielded triazole derivatives 85 [54]. Moreover, azide was reduced and converted to guanidine 86 [54,55,56,57]. The substitution at the anomeric hydroxyl group afforded product 87 [56]. The second sugar unit could be connected with the thioether bond and produced 88 [58]. A protocol to synthesize fluoro diastereomers 89 was described [59]. The reduction of the azide moiety of 83 and further insertion of the sulfonic acid group at position 1 yielded sialosyl α-sulfonate derivatives 90, which significantly inhibited the influenza virus sialidase activity [60].
In addition to the most commonly used azide reagents, the oxazoline ring can be opened by other nucleophiles. Ye et al. published a protocol to introduce morpholine, piperazine, piperidine, pyrrolidine, or primary and secondary amino moieties to obtain derivative 92 (Scheme 26) [61,62,63]. The protocol included an in situ formation of an oxazoline ring, which immediately proceeded to nucleophilically attack morpholine. The acetoxy group at position 6 also participates in the ring-opening reaction and undergoes selective deacetylation. This methodology was expanded by Bozzola et al. with various substituted N-aryl and N-heteroaryl piperazine derivatives [64].
This method was later used to synthesize other Zanamivir derivatives. Rota et al. (Figure 6) prepared Zanamivir derivatives 93 in a four-step synthesis [65], Further substitution was performed at the anomeric carbon of 94 [66], where various alcohols were used as nucleophiles. Then, 95 was synthesized via double ring-opening reactions to connect two carbohydrate units through the piperazine linker [63]. The linker can be extended with further substitution at piperazine.
3.1.2. Cyclization of Acyclic Intermediates with Vicinal Dinitrogen-Containing Functionalities
An alternative to the oxazoline ring-opening reaction is the cyclization of 100 (Scheme 27). The reaction sequence begins with lactone 96, where a five-step synthesis results in imine 97 [67]. Then, imine 97 is converted to aziridine 98 in a four-step synthesis [68]. Intermediate 99 is synthesized through an aziridine ring-opening reaction. The acidic cyclization of ketoester 100 yields 101.
Yao and co-workers developed an alternative synthesis of the similar derivative 107. Air-stabilized nitrone 104 was prepared by adding hydroxylamine derivative 103 to aldehyde 102 (Scheme 28) [69,70]. Heating 104 with methyl acrylate yielded isoxazolidine 105, which was hydrolyzed, and the N-O bond was subsequently cleaved to obtain alcohol 106. Dess-Martin oxidation afforded the keto intermediate, which was cyclized under acidic conditions to obtain 107.
Another method to construct a reactive intermediate suitable for ring closure is the Henry reaction (Scheme 29) [71]. An anti-selective Henry reaction in the presence of ligand 112 yielded the cyclic intermediate, which is dehydrated by thionyl chloride and pyridine to produce 110. Nitro derivative 110 was used as the starting material for the six-step synthesis of Zanamivir derivative 111. This synthesis can be conducted on a large scale.
The final example of cyclization is based on the key alkylation step (Scheme 30) [72]. Mannitol derivative 114 was prepared from arabinose 113 in a five-step synthesis. Subsequent protection and alkylation with ethyl α-(bromomethyl)acrylate formed acyclic intermediate 115, which produced 116 after ozonolysis and subsequent reductive deprotection.
3.1.3. Michael Addition
A primary amine was introduced by Michael addition to a D-glucosamine derivative [73]. The key step to synthesize 120 (Scheme 31) involves eliminating acetic acid and subsequent Michael addition of benzylamine.
3.1.4. Aziridine Ring Formation
In several cases, trans 3,4-diaminocarbohydrates were synthesized through the aziridine salt intermediate. Chen et al. prepared a mixture of azido modified desosamines 124 and 125 (Scheme 32) [74]. The synthesis began with 4-O-benzyl mycaminose 121, which was converted into reactive 4-mesyl intermediate 122 in two steps. Treatment of 122 with NaN_3_ produced two isomers 124 and 125, which indicates the in situ formation of aziridinium intermediate 123 and inevitable nucleophilic ring opening.
Similar results were reported to synthesize unsaturated josamycin derivatives (Scheme 33) [75]. The key mesylated intermediate 127 was prepared in three steps from josamycin 126. Sodium azide reacts with 127 through azirium salt 128 to produce a mixture of isomers 129 and 130.
3.2. Cis-Configuration
Glycoside antibiotics were derivatized by introducing nitrogen functionalities in the cis-configuration. Arbekacin, kanamycin B, and neomycin B are the most important antibiotics. For example, Arbekacin strongly inhibits methicillin resistant Staphylococcus aureus [76], kanamycin shows activity against the gram-negative bacteria E. coli and Klebsiella pneumonia [77], and neomycin B can be used to cure liver encephalopathy [78]. Subsequent derivatization of these glycoside antibiotics is highlighted in red (Figure 7).
Sasaki et al. activated the hydroxyl group of Arbekacin by mesylation and subsequent substitution with sodium azide to obtain 135, which was further converted in five steps to the final product 136 (Scheme 34) [76]. 136 has lower biological activity than Arbekacin 132. This paper confirmed the results of Hiariwa et al., where substituting the hydroxyl group with a nitrogen-containing functional group resulted in a derivative with decreased biological activity [79].
An alternative strategy using O-glycosylation can be used for the aza analogue kanamycin B (Scheme 35). The cis- configuration was introduced by O-glycosylation with 3,4-dinitrogen-containing carbohydrate. However, 139 exhibited lower minimum inhibitory concentration (MIC) than the original Kanamycin B [80].
A similar approach was used to synthesize neomycin B derivatives, where tolyl was used instead of phenyl in the anomeric thioether group (Scheme 36) [81,82]. The furanose hydroxyl group was derivatized. Biological testing was performed after the azide reduction and deacetylation to show lower activity against bacterial strains.
3.3. Methods Resulting in Cis- and Trans-Configurations
Some methods provide a mixture of trans- and cis-stereoisomers with nitrogen functionalities at positions 3 and 4. The formation of a major product usually depends on the reaction conditions. Zbiral and coworkers incorporated the azide functionality into the Neu5Ac molecule [83,84,85]. Substituting the hydroxyl group with an azide in 143 under Mitsunobu conditions produces two isomers 144 and 145 (Scheme 37). The ratio of isomers depends on the solvent. Toluene facilitates the S_N_2 reaction and predominantly produces 144 (ratio of 144:145 = 3:1), whereas THF favors the 3,3-rearrangement and produces 145 (ratio of 144:145 = 2:3) as the major product. Both 144 and 145 were subsequently reduced by the Staudinger protocol to the corresponding amines.
Another example is the reduction of the oxime functionality (Scheme 38) [86]. The catalytic hydrogenation of 146 produces two isomers: 147 and 149. Stereoisomer 147 is unstable due to syn-diaxial interactions, which immediately undergoes a ring-opening reaction and subsequently reduces the keto group to afford 148.
4. Nitrogen Functionalities at Positions 2, 3, and 4
The study of glycoside antibiotics containing polyaminated pyranoses motivated the development to synthesize 2,3,4-tri- or 2,3,4,6-tetra-substituted D-hexopyranosides with nitrogen functionalities. Derivatives of D-hexopyranosides containing nitrogen functionality at positions 2, 3, and 4 are rare in comparison to disubstituted derivatives. The first synthesis is based on the aziridine ring-opening reaction. Bailliez and coworkers published a synthesis (Scheme 39), where levoglucosan 150 was converted to diazide 151 [87]. Subsequent reduction and benzoylation produced a mixture of isomers 152 and 153. The mixture was treated with lithium azide and TFA to produce hexopyranoside 154 with three nitrogen functionalities without further purification.
Another synthesis used sequential acetoxy group elimination with a subsequent Michael addition with ammonia or benzylamine (Scheme 40) [88,89,90]. The substitution at C2 and C4 carbons provided 156 and 157, which was transformed into the corresponding triamine derivative 158.
Tetra-substituted hexopyranosides with nitrogen functionalities were synthesized via trisubstituted precursors. The C-N bond at position 6 was introduced in the last step or simultaneously through substitution at other positions. The first procedure was published by Baer and coworkers [91]. The primary hydroxyl group of 159 was mesylated and substituted with the azide anion, and the azide was subsequently reduced to give 160 (Scheme 41). A similar protocol was used by Cleophax [87] and Meyer zu Reckendorf [92].
Ali and coworkers synthesized 162 from glucopyranoside 161, where the galacto-configuration arose from the in-situ formed oxazoline ring and its ring-opening reaction with the azide anion and a simultaneous two-fold mesylate substitution (Scheme 42) [93]. A similar procedure was used for galactopyranoside or idopyranoside [94,95].
5. Conclusions
The nitrogen functionalities of D-hexopyranosides increase their structural diversity, which results in numerous derivatives with interesting biological activities. With ongoing basic research on the use of these compounds for advanced biological studies and the identification of other nitrogen-containing hexopyranosides with improved biological effects and selectivity, synthetic methods that introduce various nitrogen functionalities were developed. The first methods to synthesize derivatives with vicinal nitrogen-containing functionalities emerged in the 1960s. These methods use the ring-opening reactions of aziridine and oxazoline intermediates. In addition to conventional substitution reactions of tosylated or mesylated hydroxyl groups, the Michael addition reaction is frequently studied with nitrogen nucleophiles to produce Michael adducts from nitro-olefins. Other methods such as oxime reduction and ring-opening reactions of imidazoline derivatives further supplement the synthetic methodology. Moreover, the developed synthetic methods introduce nitrogen functionalities in various oriented configurations. Finally, considering the substantial expansion of photoredox catalysis, we can expect the development of novel methods using highly reactive radical intermediates.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cahoreau C. Klett D. Combarnous Y. Structure-Function Relationships of Glycoprotein Hormones and Their Subunits’ Ancestors Front. Endocrinol.201562610.3389/fendo.2015.00026 PMC 434156625767463 · doi ↗ · pubmed ↗
- 2Wang B. Brand-Miller J. The Role and Potential of Sialic Acid in Human Nutrition Eur. J. Clin. Nutr.2003571351136910.1038/sj.ejcn.160170414576748 · doi ↗ · pubmed ↗
- 3Zhao M. Zhu Y. Wang H. Zhang W. Mu W. Recent Advances on N-Acetylneuraminic Acid: Physiological Roles, Applications, and Biosynthesis Synth. Syst. Biotechnol.2023850951910.1016/j.synbio.2023.06.00937502821 PMC 10369400 · doi ↗ · pubmed ↗
- 4Han Z. Thuy-Boun P.S. Pfeiffer W. Vartabedian V.F. Torkamani A. Teijaro J.R. Wolan D.W. Identification of an N-Acetylneuraminic Acid-Presenting Bacteria Isolated from a Human Microbiome Sci. Rep.202111476310.1038/s 41598-021-83875-w 33637779 PMC 7910532 · doi ↗ · pubmed ↗
- 5HadháziÁ. Pascolutti M. Bailly B. Dyason J.C. Borbás A. Thomson R.J. von Itzstein M. A Sialosyl Sulfonate as a Potent Inhibitor of Influenza Virus Replication Org. Biomol. Chem.2017155249525310.1039/C 7OB 00947 J 28540971 · doi ↗ · pubmed ↗
- 6Hale K.J. Domostoj M.M. Tocher D.A. Irving E. Scheinmann F. Enantiospecific Formal Total Synthesis of the Tumor and GSK-3β Inhibiting Alkaloid, (−)-Agelastatin A Org. Lett.200352927293010.1021/ol 035036 l 12889910 · doi ↗ · pubmed ↗
- 7Charon D. Mondange M. Pons J.-F. Le Blay K. Chaby R. Synthesis and in Vitro Activities of a Spacer-Containing Glycophospholipid Ligand of a Lipopolysaccharide Receptor Involved in Endotoxin Tolerance Bioorg. Med. Chem.1998675576510.1016/S 0968-0896(98)00027-39681141 · doi ↗ · pubmed ↗
- 8Del Litto R. Benessere V. Ruffo F. Moberg C. Carbohydrate-Based Pyridine-2-Carboxamides for Mo-Catalyzed Asymmetric Allylic Alkylations Eur. J. Org. Chem.200920091352135610.1002/ejoc.200801240 · doi ↗