Isocitrate Dehydrogenase (IDH)-Mutant High-Grade Glioma in the Cerebellum: A Case Report
Divya Dinesh, Thanka Johnson, Divyalakshmi P.K., Santhanam Rengarajan, Venkatraman Indiran

TL;DR
This case report describes a rare instance of IDH-mutant high-grade glioma in the cerebellum, highlighting its diagnostic features and molecular confirmation.
Contribution
The novelty lies in presenting a rare cerebellar case of IDH-mutant high-grade glioma with detailed diagnostic and molecular analysis.
Findings
IDH-mutant high-grade glioma was diagnosed in the cerebellum of an elderly patient.
Histopathology and immunohistochemistry confirmed the glioma's IDH-mutant status.
Molecular studies aided in the accurate classification of the tumor.
Abstract
High-grade glial cancers typically arise in the cerebral hemisphere and rarely in the cerebellum. Our objective was to highlight the diagnostic features of isocitrate dehydrogenase (IDH)-mutant high-grade gliomas in the cerebellum. We present a case of an elderly patient admitted with giddiness who was diagnosed with IDH-mutant high-grade glioma in the cerebellum, presenting as multiple lesions. We evaluated an open biopsy specimen to arrive at a diagnosis and used molecular studies to confirm the diagnosis and further categorize the specimens. Histopathology and immunohistochemistry confirmed the diagnosis of IDH-mutant high-grade glioma in the cerebellum.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
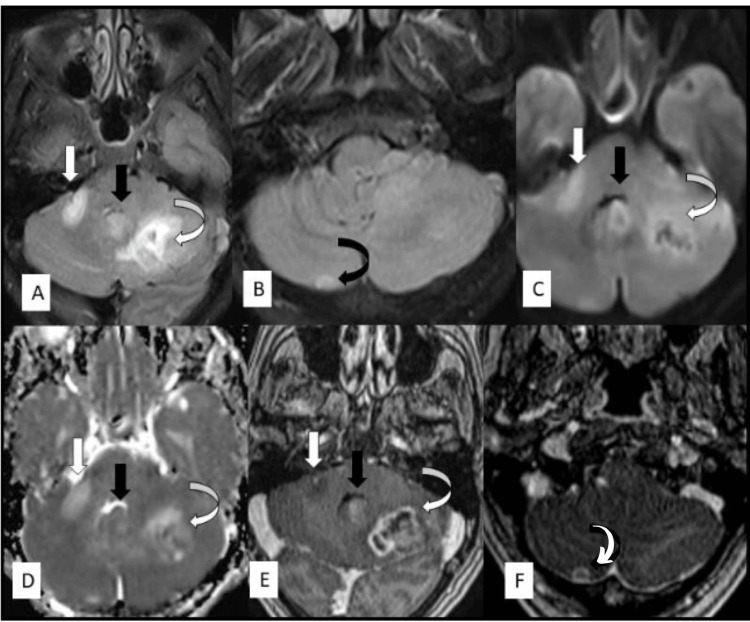 Figure 1
Figure 1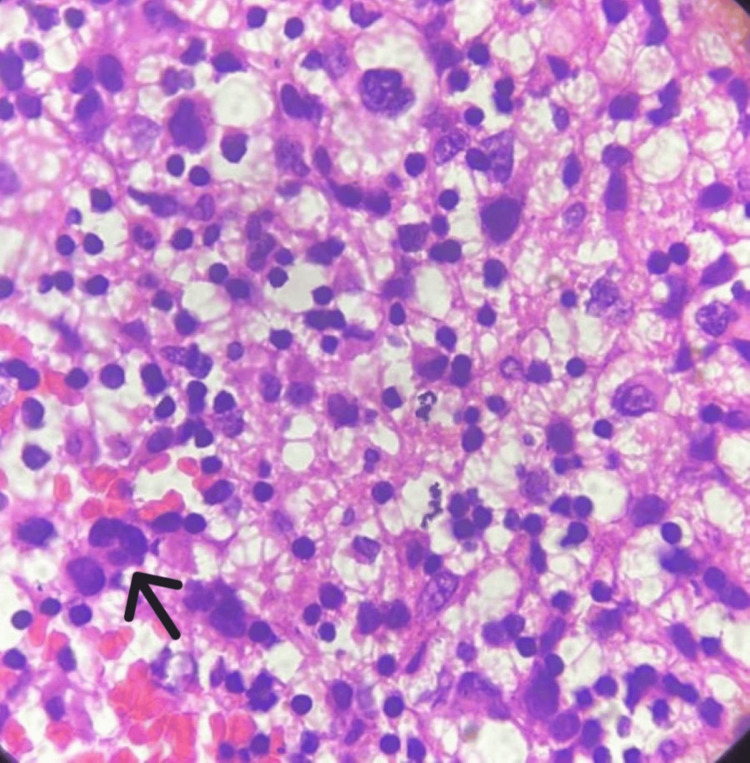 Figure 2
Figure 2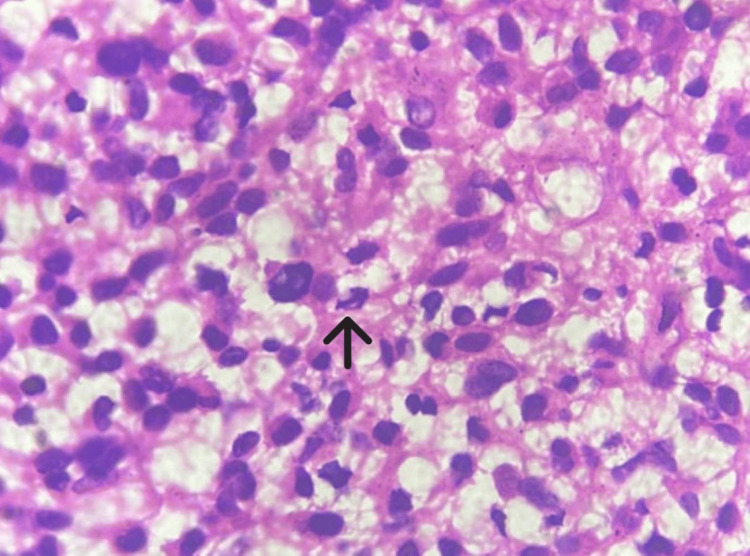 Figure 3
Figure 3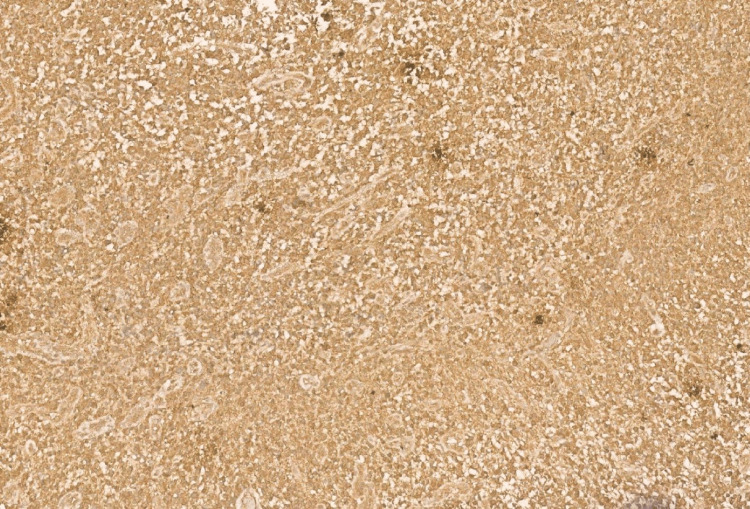 Figure 4
Figure 4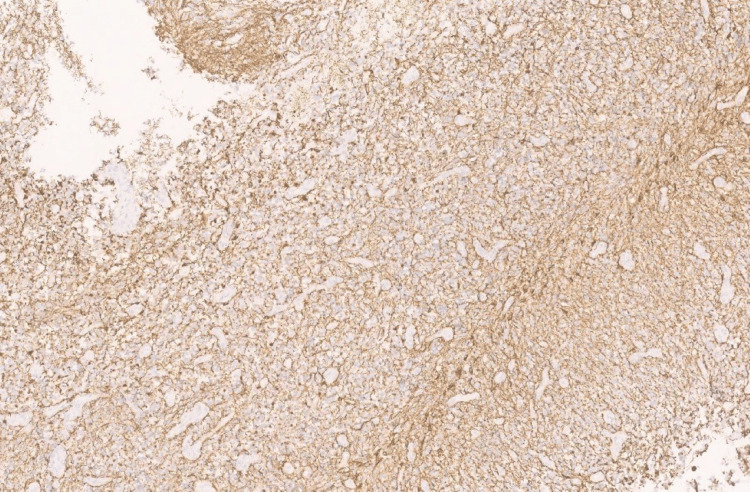 Figure 5
Figure 5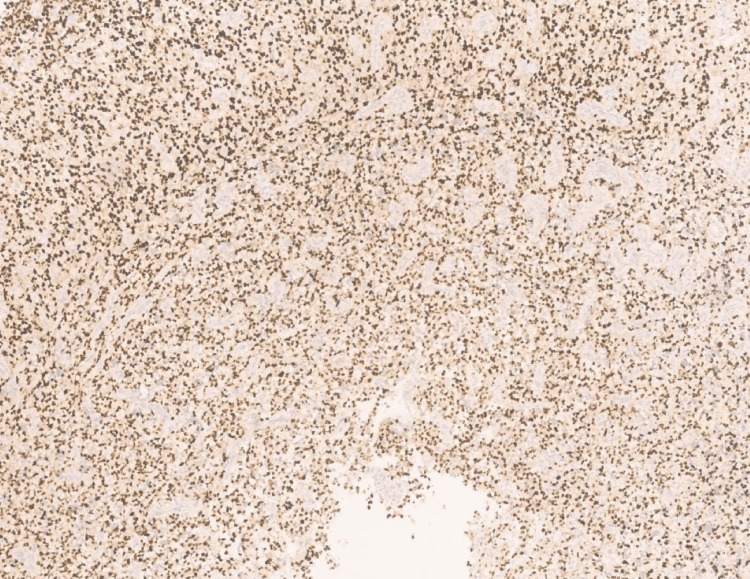 Figure 6
Figure 6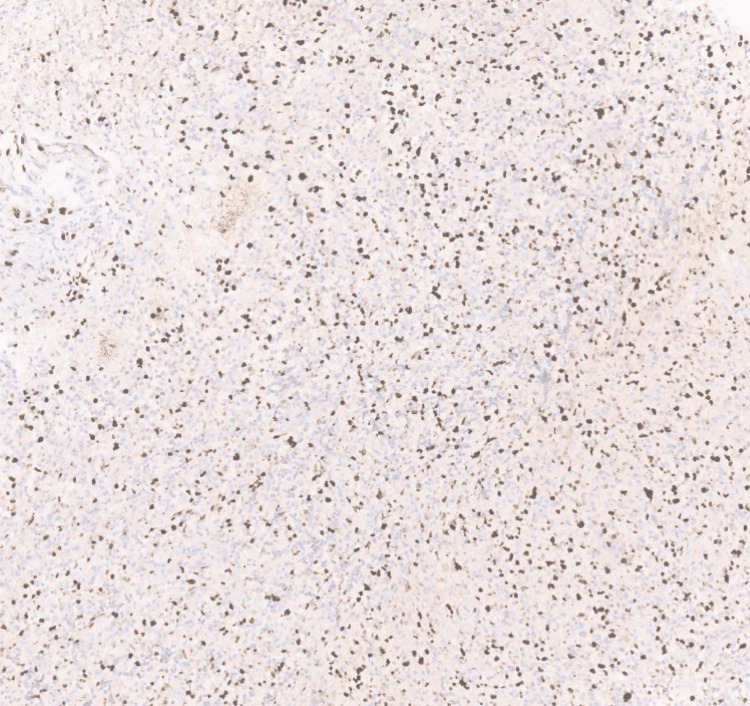 Figure 7
Figure 7 Figure 8
Figure 8 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlioma Diagnosis and Treatment · Cancer, Hypoxia, and Metabolism · Meningioma and schwannoma management
Introduction
Central nervous system (CNS) tumors account for only 2% of all malignant neoplasms. Nearly half of the tumors in the CNS are benign [1]. Gliomas are the most common primary malignant brain tumors in adults, presenting as supratentorial lesions. Cerebellar gliomas are rare and account for approximately 1% of CNS tumors. Cerebellar high-grade gliomas (cHGGs) have a poor prognosis and preferentially affect males and young adults [2].
Case presentation
A 62-year-old woman came to the neurology outpatient department with complaints of giddiness and vomiting of one-week duration. On evaluation, plain and contrast brain MRIs showed three T2 hyperintense intra-axial lesions in the posterior fossa (right middle cerebellar peduncle, vermis, and left cerebellar hemisphere). In addition, axial T1 postcontrast images showed heterogenous enhancement with a necrotic center in the left cerebellar lesion, solid enhancement in the cerebellar vermis, and absent enhancement in the right-middle cerebellar peduncle lesion (Figure 1).
MRI axial T2 image (A) and T2 FLAIR image (B) show three T2 hyperintense lesions in right middle cerebellar peduncle (white arrow), vermis (white arrow), left cerebellar hemisphere (curved white arrow), and right cerebellar hemisphere (curved black arrow). Axial DWI (C) and ADC map (D) show restricted diffusion in the left cerebellar and vermis lesions. Axial T1 post-contrast images (E,F) show heterogenous enhancement with necrotic center in the left cerebellar lesion, peripheral enhancement in right cerebellar lesion, solid enhancement in the cerebellar vermis, and absent enhancement in the right middle cerebellar peduncle lesion.FLAIR: fluid-attenuated inversion recovery; DWI: diffusion-weighted imaging; ADC: apparent diffusion coefficient
The patient underwent left-suboccipital decompression, and a biopsy was taken. Microscopy of the open biopsy specimen showed a glial tumor with pleomorphic cells having nuclear atypia, eccentrically placed nuclei, a moderate amount of glassy eosinophilic cytoplasm, and occasional binucleate and multinucleate cells (Figure 2), mitosis (Figure 3), and apoptosis. There was no necrosis, and microvascular proliferation was visible in the received specimen.
Photomicrograph of tumor cells showing eosinophilic cytoplasm with eccentrically placed nuclei. Black arrow shows a multinucleated cell (100x, H&E).
Photomicrograph showing mitosis (black arrow) (100x, H&E).
Immunohistochemistry showed diffuse cytoplasmic positivity for isocitrate dehydrogenase (IDH) 1 (Figure 4), diffuse, strong positivity for glial fibrillary acidic protein (GFAP) (Figure 5), and diffuse nuclear positivity for Olig2 (Figure 6).
Photomicrograph showing IDH1 with diffuse cytoplasmic positivity (40x, H&E).
Photomicrograph showing GFAP with diffuse strong positivity (40x, H&E). GFAP: glial fibrillary acidic protein
Photomicrograph showing Olig2 with diffuse nuclear positivity (40x, H&E).
Furthermore, Ki-67 showed 40% positivity (Figure 7), and p53 showed positivity in 10-15% of cells. There was scattered positivity for H3k27m (Figure 8), ATRX (alpha-thalassemia/mental retardation, X-linked) was retained in the cell (Figure 9), and epithelial membrane antigen (EMA) was negative.
Photomicrograph showing Ki-67 with 40% positivity (40x, H&E).
Photomicrograph showing H3K27m positivity in scattered cells (40x, H&E).
Photomicrograph showing ATRX retained in tumor cells (40x, H&E).ATRX: alpha-thalassemia/mental retardation, X-linked
After evaluating the histopathology and immunohistochemistry results, which showed highly pleomorphic cells with mitosis and IDH mutation respectively, we arrived at a diagnosis of IDH-mutant high-grade glioma in the cerebellum, and the patient was referred to another facility for further management. At present, the patient has completed radiotherapy and four cycles of chemotherapy with temozolomide.
Discussion
Approximately 1% of high-grade gliomas in the CNS are cHGGs (WHO Grade 4) [2]. Subclasses of cHGG include high-grade astrocytomas with piloid features (HGAP) (~31% of tumors), H3k27m diffuse midline gliomas (~8% of tumors), and IDH wildtype glioblastomas (~43% of tumors). These tumor types were included in a recent series of cHGGs. In comparison to their supratentorial counterparts, gliomas occur disproportionately less frequently in the cerebellar parenchyma [3].
The actual presence of cHGGs was questioned until the 20th century because of their low incidence [3] and the possibly inadequate CT visibility of the posterior fossa structures [4]. The depletion of Substance P in the adult cerebellum is one theory that has been offered to explain this relative resistance to gliomagenesis in the cerebellum [5]. Some researchers have even suggested the majority of these cancers are metastases originating from an unknown supratentorial or brainstem location rather than the cerebellum [6]. Nevertheless, this theory is not well supported by the available data. The lack of FOX1-telencephalic markers and other hallmark gene expression, as well as other recent molecular findings, amply demonstrate that cHGGs are actually cerebellar in origin rather than cerebral.
The fifth edition of the WHO Classification of Head and Neck Tumors classifies adult-type gliomas into three types: (a) IDH-mutant astrocytomas presenting as diffusely infiltrating gliomas with IDH1 or IDH2 mutations and loss of nuclear ATRX expression, (b) oligodendrogliomas presenting with IDH mutations, combined whole arm deletions of 1p and 19q, and retained nuclear expression of ATRX, and (c) IDH-wildtype glioblastomas presenting with any one feature such as microvascular proliferation, necrosis, TERT (telomerase reverse transcriptase)-promoter mutations, epidermal growth factor receptor (EGFR) amplification, or +7/-10 chromosome copy number alterations [7].
Features of cHGGs include uncommon IDH mutations, rare EGFR amplification, moderately common p53 mutations [8], more frequent RAS (Rat sarcoma) mutations, ATRX alterations, PDGFRA (platelet-derived growth factor receptor alpha) amplification, less frequent TERT promotor mutations, CDK2a/2b loss, CDK4 amplification, scant expression of telencephalic markers (FOX1), widespread expression of cerebellar markers (PAX3) and widespread expression of oligodendrocyte progenitor markers (CSPG 4). Treatment for glioma is maximal resection followed by radiotherapy and concurrent chemotherapy with TMZ. Adjuvant TMZ or carmustine chemotherapy regimens are also often employed. Methylation classes of high-grade gliomas include (a) HGAPs, (b) diffuse midline gliomas H3k27-altered (DMG H3k27), (c) GBM IDH-wildtype subclass RTK I, and (d) GBM IDH-wildtype subclass midline (GBM-MID) [2].
HGAPs
A distinct DNA-methylation signature characterizes the recently identified tumor entity known as HGAP. These tumors most often develop in the posterior fossa, and the cerebellum is the most common site. Piloid astrocytomas typically occur in older patients. IDH wildtype HGAPs frequently exhibit CDK2a/2b deletions, ATRX mutations, and changes to the mitogen-activated protein kinase (MAPK) pathway. Potential treatments target the frequent MAPK pathway abnormalities in HGAP [9].
DMG H3k27m
An uncommon glioma known as DMG, H3k27-mutant, was mentioned in the fourth updated edition of the WHO Classification of Head and Neck Tumor classification of CNS malignancies. It was later renamed as DMG, H3 k27-altered in the fifth edition to reflect the presence of additional modifications such as EZHIP (Enhancer of Zeste Homologs Inhibitory Protein) overexpression [1,3]. Because this tumor typically develops in deep midline tissues such as the brainstem or thalamus and spreads widely, surgical removal is frequently not viable, and in some instances, even a biopsy is not an option. Neurosurgeons are also less likely to undertake resection or biopsy because of the poor prognosis [10].
IDH wildtype glioblastoma
Cerebellar glioblastomas are rare. These lesions are smaller and more frequently seen in young individuals [11]. IDH wildtype glioblastoma is an aggressive primary brain cancer with a median survival time of 14-21 months after diagnosis under ideal conditions. With the extended longevity of glioblastoma patients, extra neural glioblastoma metastasis is becoming more common, albeit still extremely rare. It is now expected that 2% of patients with glioblastoma may advance to extra neural metastasis, but this usually affects a small number of extra neural sites. Metastatic glioblastoma cases have been observed in the neck, lymph nodes, liver, lungs, bones, and systemic vasculature [12].
Conclusions
Patients with high-grade glioma fall into two groups, those with a good prognosis and those with a poor prognosis. Younger patients often have a good prognosis when compared to older patients. The treatment regimen consists of maximum resection with adjuvant conventionally fractionated radiotherapy, with or without chemotherapy. However, the majority of high-grade glioma patients have a poor prognosis, and the most effective technique to treat them is currently unknown.
Given that cHGGs and supratentorial HGGs have distinct molecular characteristics, the cerebellum appears to be an area that is not prone to HGG formation. With the realization that a sizable fraction of these tumors may reflect HGAP or DMG H3k27m, the approach to treating cHGG is beginning to vary. In this report, the elderly female patient presenting with multiple lesions in the cerebellum had an atypical presentation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1The global prevalence of primary central nervous system tumors: a systematic review and meta-analysis Eur J Med Res Salari N Ghasemi H Fatahian R 392820233667046610.1186/s 40001-023-01011-y PMC 9854075 · doi ↗ · pubmed ↗
- 2Cerebellar high-grade glioma: a translationally oriented review of the literature Cancers (Basel) Raghu AL Chen JA Valdes PA 1741520223661216910.3390/cancers 15010174 PMC 9818238 · doi ↗ · pubmed ↗
- 3The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting J Comp Neurol von Bartheld CS Bahney J Herculano-Houzel S 3865389552420162718768210.1002/cne.24040 PMC 5063692 · doi ↗ · pubmed ↗
- 4Brain Tumors: Their Biology and Pathology Springer-Verlag Zülch KJ Berlin/Heidelberg Springer-Verlag 112013
- 5Why cerebellar glioblastoma is rare and how that indicates adjunctive use of the FDA-approved anti-emetic aprepitant might retard cerebral glioblastoma growth: a new hypothesis to an old question Clin Transl Oncol Kast RE 4084101120091957419810.1007/s 12094-009-0379-x · doi ↗ · pubmed ↗
- 6Cerebellar glioblastoma: a clinical series with contemporary molecular analysis Acta Neurochir (Wien) Hong B Banan R Christians A 2237224816020183020336210.1007/s 00701-018-3673-y · doi ↗ · pubmed ↗
- 7The 2021 WHO classification of tumors of the central nervous system: a summary Neuro Oncol Louis DN Perry A Wesseling P 123112512320213418507610.1093/neuonc/noab 106PMC 8328013 · doi ↗ · pubmed ↗
- 8The impact of TP 53 and RAS mutations on cerebellar glioblastomas Exp Mol Pathol Milinkovic VP Skender Gazibara MK Manojlovic Gacic EM Gazibara TM Tanic NT 2022079720142503640410.1016/j.yexmp.2014.07.009 · doi ↗ · pubmed ↗