Germline Sequencing of DNA Damage Repair Genes in Two Hereditary Prostate Cancer Cohorts Reveals New Disease Risk-Associated Gene Variants
Georgea R. Foley, James R. Marthick, Sionne E. Lucas, Kelsie Raspin, Annette Banks, Janet L. Stanford, Elaine A. Ostrander, Liesel M. FitzGerald, Joanne L. Dickinson

TL;DR
This study identifies new genetic risk variants in DNA repair genes linked to hereditary prostate cancer, which could help in developing targeted treatments.
Contribution
The study discovers novel rare germline variants in DNA damage repair genes associated with prostate cancer risk.
Findings
Rare ERCC3 and BRIP1 variants are significantly associated with prostate cancer risk.
PARP2 and MUTYH variants show risk associations in separate datasets.
No evidence of younger age or higher-grade disease in variant carriers.
Abstract
An urgent demand exists to identify inherited genetic risk variants for prostate cancer (PrCa), particularly in DNA damage repair genes targetable with precision medicine-based strategies. Though the most heritable common cancer, discovery of rare germline PrCa risk variants is hampered by their low frequency, even in sizeable population datasets. Through utilising two large, independent, familial PrCa resources and their likely enrichment of rare causative variants, we provide robust evidence for several novel risk variants in DNA damage repair genes. Rare, inherited variants in DNA damage repair (DDR) genes have a recognised role in prostate cancer (PrCa) susceptibility. In addition, these genes are therapeutically targetable. While rare variants are informing clinical management in other common cancers, defining the rare disease-associated variants in PrCa has been challenging.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
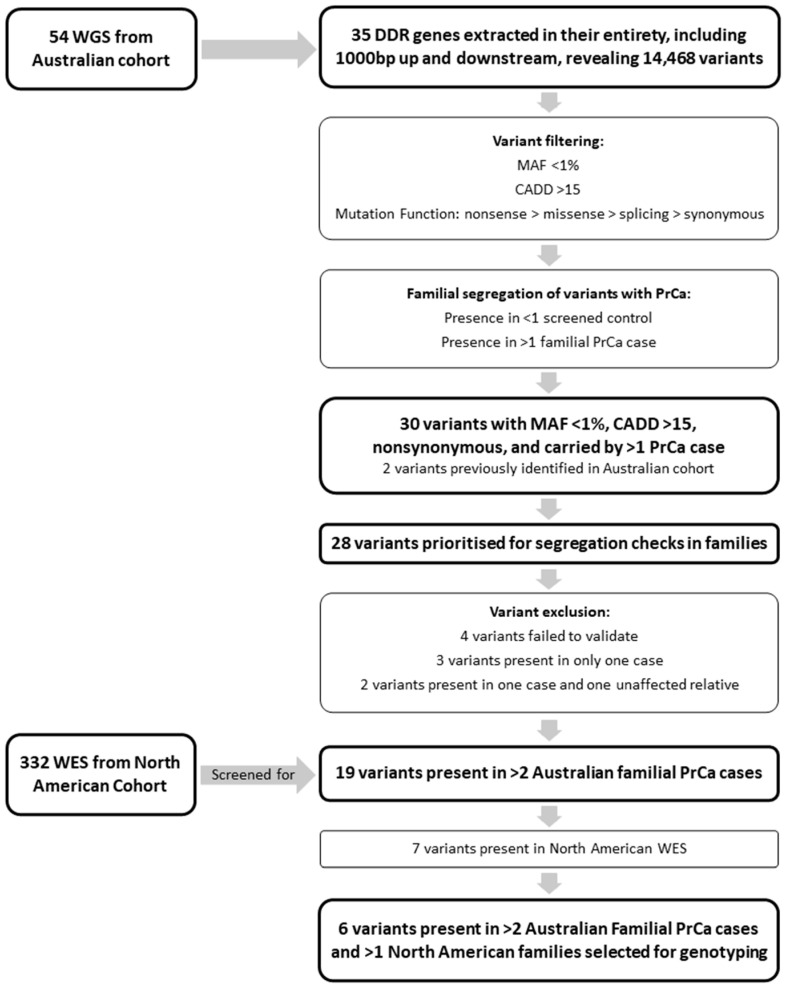 Figure 1
Figure 1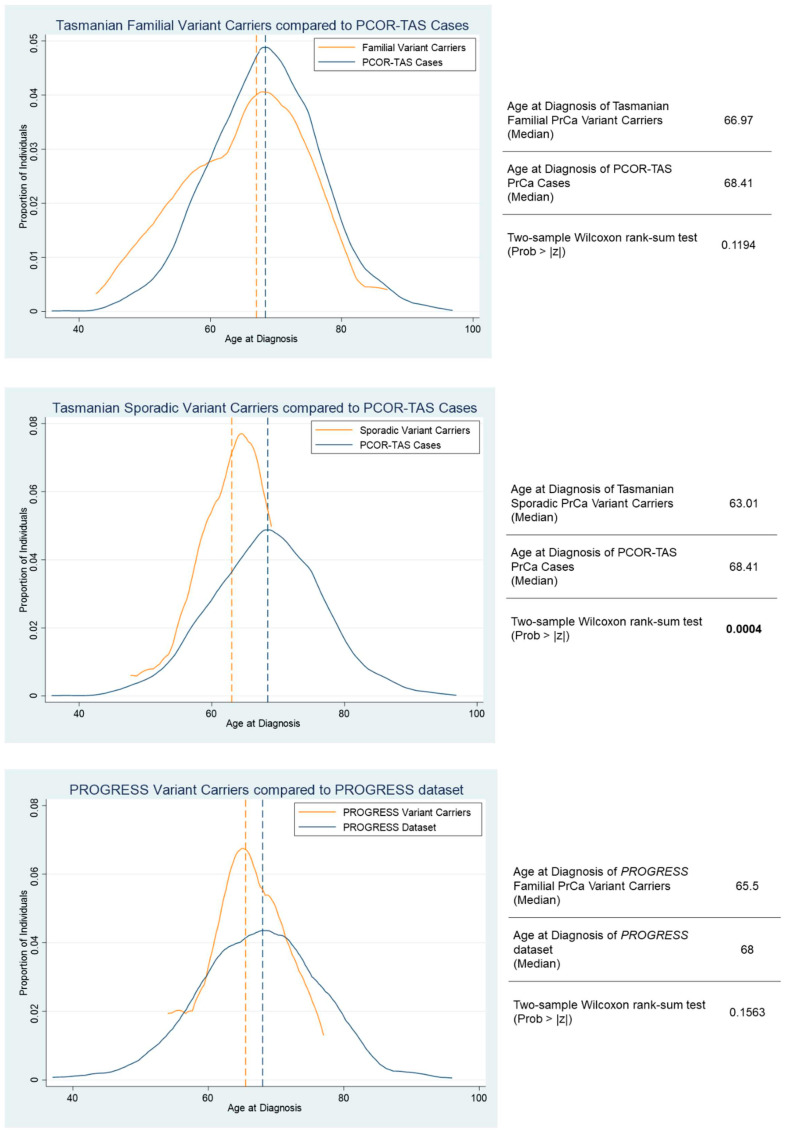 Figure 2
Figure 2- —Cancer Council Tasmania and IMPACT Perpetual Trustees
- —Royal Hobart Hospital Research Foundation (RHHRF)
- —Cancer Australia
- —The Mazda Foundation
- —Max Bruce Trust
- —The Estate of Dr RA Parker
- —Tasmanian Community Fund
- —Robert Malcom Familial Prostate Cancer Bequest
- —Fred Hutchinson Cancer Center
- —Institute for Prostate Cancer Research of the University of Washington Medicine and Fred Hutchinson Cancer Center
- —Cancer Council Tasmania/College of Health and Medicine Scholarship
- —National Cancer Institute of the National Institutes of Health
- —National Human Genome Research Institute of the National Institutes of Health and the Intramural Program of the National Human Genome Research Institute
- —Cancer Council Tasmania/College of Health and Medicine Senior Research Fellowship
- —Williams Oncology RHHRF
- —Gerald Harvey University of Tasmania Senior Research Fellowship
- —Australian Research Council Future Fellowship
- —Select Foundation Cancer Research Fellowship
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProstate Cancer Treatment and Research · Prostate Cancer Diagnosis and Treatment · PARP inhibition in cancer therapy
1. Introduction
Prostate cancer (PrCa) is responsible for a significant proportion of cancer-related deaths in men worldwide and presents a substantial health burden in terms of morbidity, mental health, and economic costs associated with treatment. Family history is one of the strongest risk factors for PrCa: heritability is estimated at ~57% [1], and both common and rare variants contribute to inherited risk. Notably, a considerable percentage of men with advanced disease harbour clinically actionable germline genetic variants, many of which are aberrations in DNA damage repair (DDR) genes [2,3,4,5,6,7]. Specifically, germline variants in these genes have been observed in 8–16% of metastatic PrCa patients [2,5,8].
Despite recognition of their potential to facilitate early diagnosis and assist in treatment selection, there remains a significant gap in our understanding of the full spectrum of DDR gene variants contributing to PrCa risk [8,9,10,11,12]. Several DDR genes, particularly BRCA1/2 and ATM, have been associated with a substantial increase in disease risk, poorer prognosis, differing responses to treatment, and more aggressive disease [13,14,15,16,17]. Notably, there are many other genes involved in the DDR pathways (>100), and their contribution to PrCa risk has only more recently been explored. For example, two recent studies have provided evidence for a role of CHEK2, PALB2, BRIP1, RAD51C, RAD51D, BARD1 and NBN in PrCa risk [18,19]. Importantly, tumours harbouring loss-of-function mutations in DDR genes exhibit a therapeutic response to poly (ADP-ribose) polymerase inhibitors (PARPi) [20] and platinum-based chemotherapy [21]. Thus, screening for clinically actionable germline variants in PrCa patients, particularly those with a significant family history together with advanced disease, represents an important strategy to improve PrCa outcomes. However, the rarity of these variants in population-based PrCa datasets, which represent the majority of PrCa DDR gene discovery studies to date, has hampered research advances. In addition, many of these studies have not differentiated between germline and acquired mutations, and those variants that have been identified remain largely of unknown clinical significance.
Curation of the full spectrum of DDR genetic variants contributing to PrCa risk has significant potential in the healthcare setting, where precision medicine can be implemented for both diagnosis and treatment. Moreover, the observation that germline and acquired mutations are frequently identified in the same DDR genes underscores the importance of these pathways in tumour development. Here, we interrogated whole-genome and -exome germline data from two high-risk familial PrCa datasets with the aim of identifying novel, rare DDR gene variants contributing to PrCa risk.
2. Methods
2.1. Study Resources
This study utilised clinicopathological and genetic data available from two independent PrCa resources: the Australian resource, consisting of the Tasmanian Familial Prostate Cancer Study and the population-based Tasmanian Prostate Cancer Case-Control Study, and the North American resource, consisting of the Prostate Cancer Genetic Research Study (PROGRESS) from the Fred Hutchinson Cancer Center (FHCC).
The Tasmanian Familial Prostate Cancer Study included 73 PrCa families comprising DNA from 379 affected men and 471 unaffected male and female relatives of Northern European heritage [22,23,24]. The study was initiated in Tasmania in the late 1990s, prior to the implementation of wide-spread prostate-specific antigen (PSA) testing as a PrCa screening tool. Families with more than two affected first-degree relatives spanning two or more generations were identified through interrogation of the Menzies Genealogical database and the Tasmanian Cancer Registry (TCR), in addition to collaboration with local clinicians.
The second, population-based Tasmanian Prostate Cancer Case-Control Study comprises 459 cases and 322 male controls of Northern European ancestry [22,23,24]. Cases were identified from the TCR. Controls were selected at random from the Tasmanian electoral roll, frequency matched by five-year age groups to the cases, and checked bi-annually against the TCR for PrCa diagnosis.
The FHCC resource comprises PROGRESS, which includes >300 PrCa families from across North America [25]. Whole exome sequencing (WES) data were available for 130 families, which included 11 older unaffected men and 321 affected men. Men prioritised for WES were those diagnosed with an early-onset/aggressive disease phenotype, uncle–nephew and/or cousin pairs from families with densely aggregated affected men.
Details of the clinical data available for these resources are presented in Table 1.
Additionally, age-at-diagnosis and Gleason score (GS) data from 2126 participants enrolled in the population-based Prostate Cancer Outcomes Registry—Tasmania (PCOR-TAS) were available for clinicopathological analyses. PCOR-TAS was established in 2015, with an aim to improve all aspects of the quality of care for men diagnosed with PrCa. The opt-out registry is an ongoing initiative that records data on the diagnosis, treatment, outcomes, and quality of life for Tasmanian men diagnosed with PrCa. Details of the registry, including data collection methods, have been described previously [26]. As of 15 July 2021, 2126 men had been recruited into PCOR-TAS, with ~3% having opted out of the registry.
2.2. Whole-Genome Sequencing and Bioinformatic Sequence Analysis
Whole-genome sequencing (WGS) data were generated from germline DNA (additional details: Supplementary Materials: Method S1) for 54 individuals from eight Australian families (Supplementary Materials: Table S1) and seven unaffected men from the Australian Case-Control Study.
Of the familial individuals, 43 had been diagnosed with PrCa, with the remaining individuals comprising a female relative with a self-reported breast cancer diagnosis (n = 1) and unaffected male relatives (n = 10). WGS (mean coverage = 38.7x; range = 29.2x–49.8x; the sequencing coverage and quality statistics for each sample are summarized in Supplementary Materials: Table S2) was completed in five instalments at the Australian Genome Research Facility (Melbourne, Australia), the Ramaciotti Centre for Genomics (Sydney, Australia), and the Texas Biomedical Research Institute (San Antonio, TX, USA). Sequence data were aligned to the hg19 reference genome with BWA-MEM [27], and variants were called with GATK [28], using bcbio-nextgen (https://github.com/bcbio/bcbio-nextgen, accessed on 27 June 2024).
2.3. Variant Filtering, Prioritisation, and Validation
A panel of 35 genes involved in DDR pathways was compiled (Supplementary Materials: Table S3), in addition to the established PrCa risk gene, HOXB13 [29]. Variants located in a genomic window 1000 bp up and downstream of the nominated candidate genes were extracted from WGS data using bcftools [30] and annotated using ANNOVAR [31]. Included genes and genomic positions can be found in Supplementary Materials: Table S4.
Variant filtering and prioritisation occurred according to a range of criteria (Figure 1). Variants were filtered to include those with a minor allele frequency (MAF) < 1% in gnomAD non-Finnish Europeans (NFE) and Combined Annotation-Dependent Depletion (CADD) score > 15, with further prioritisation informed by predicted mutation function (e.g., nonsense > missense > splicing > synonymous). Variants were excluded if present in >1 of the seven screened unaffected male control genomes, or if present only in PrCa unaffected familial individuals.
Short-listed variants (MAF < 1%, CADD > 15, nonsynonymous, and carried by >1 PrCa case; (Figure 1)), which had been validated by Sanger sequencing on the ABI 3500 Genetic Analyser (Applied Biosystems, Foster City, CA, USA), were genotyped in additional non-WGS relatives to determine segregation in the relevant discovery family. Primers were designed to amplify fragments approximately 300 bp in length for each of the selected variants. Primer sequences are presented in Supplementary Materials: Table S5, and PCR conditions are available upon request.
2.4. Additional Genotyping in Expanded Australian Resources
Six prioritised variants were genotyped in the full Australian familial and case–control resources, using TaqMan™ genotyping assays (ThermoFisher Scientific, Waltham, MA, USA; Supplementary Materials: Table S6) on the LightCycler^®^ 480 system (Roche, Basel, Switzerland). Existing whole exome data were interrogated for prioritised gene variants in the PROGRESS study individuals.
2.5. Statistical Analysis
The distribution of clinical disease features in the Australian and PROGRESS prostate cancer cases is reported as percentages, median and interquartile range. Association between genotyped variants and PrCa risk was tested for using Modified Quasi-Likelihood Score (M_QLS_) analysis [32] (additional details: Supplementary Materials: Method S2) [33,34]. Population prevalence of PrCa was set at one in seven, and the analyses were conducted in the Australian familial and case–control datasets alone, the FHCC PROGRESS cohort alone, and all datasets combined. For DDR variants significantly associated with PrCa risk, a two-sided Wilcoxon rank-sum test was used to compare median diagnosis age of Tasmanian Familial, Sporadic and PROGRESS variant carriers with population-based PCOR-TAS cases (TAS) or the full PROGRESS cohort (FHCC). The distribution of variant carriers across several age-at-diagnosis categories (<50, <55, <60, <65, <70) is reported as numbers and percentages.
3. Results
3.1. Clinical Characteristics of Australian and North American PrCa Resources
Clinical characteristics of the study resources are presented in Table 1. Age-at-diagnosis, time interval between diagnosis and death, and proportion of PrCa-specific deaths were similar across the datasets.
3.2. Identification of Candidate Rare DDR PrCa Risk Variants
WGS data were interrogated for rare, potentially pathogenic variants in 35 DDR genes (Supplementary Materials: Table S3). Initial filtering identified 30 variants in 20 genes, of which two in HOXB13 and RAD51C have previously been shown to be significantly associated with PrCa risk in our Australian cohort [22,23], providing proof-of-principle for our approach. Of the 28 remaining variants, four failed to validate via Sanger sequencing and were excluded from further investigation.
To determine segregation with disease, the remaining 24 variants underwent Sanger sequencing in additional non-WGS affected and unaffected relatives with DNA from each of the Australian discovery families (Table 2). Five variants were subsequently excluded: three variants that were each only present in a single affected man and two variants that were only present in a single affected man and one unaffected relative. The remaining 19 variants, ATM rs56128736, BARD1 rs3738888, BRCA1 rs28897673, BRCA2 rs28897727, BRCA2 rs55639415, BRCA2 rs786202915, BRIP1 rs4988345, ERCC2 rs142568756, ERCC3 rs145201970, MRE11 rs777373591, MSH6 rs142254875, MUTYH rs36053993, PARP2 rs200603922, PMS2 rs1554304601, POLE chr12: 133219216, POLE rs41561818, PTEN rs587779989, PTEN rs773513402, and RECQL4 rs780723602, were present in at least two affected relatives from the Australian discovery cohort.
For further prioritisation, we then determined whether any of the 19 variants were present in the North American PROGRESS families. Examination of exome data from 332 individuals revealed seven variants in 34 cases from 22 kindreds. Four variants, ATM rs56128736, BRCA2 rs28897727, ERCC3 rs145201970, and MUTYH rs36053993, were present in two or more PrCa cases in a single family (Table 2).
Six DDR variants, BARD1 rs3738888, BRCA2 rs28897727, BRIP1 rs4988345, ERCC3 rs145201970, MUTYH rs36053993, and PARP2 rs200603922, that partially segregated with disease in an Australian PrCa family and were present in two or more PROGRESS families, were selected for additional investigation (Table 3). These variants were genotyped in the extended Australian familial and case–control resources via TaqMan genotyping (Supplementary Materials: Table S6). All six variants were identified in additional individuals (n_range_ = 9 to 33; Supplementary Materials: Table S7) within the Australian datasets, and all except MUTYH rs36053993 were observed in additional familial PrCa cases. With the inclusion of the PROGRESS dataset, the BARD1 rs3738888 and BRIP1 rs4988345 variants were each observed in the most PrCa cases (n = 22), which included ten and nine sporadic cases, respectively. The predicted pathogenicity of these variants was determined using multiple bioinformatic tools (additional details: Supplementary Materials: Method S3) [35,36,37,38,39,40,41,42,43,44], and outputs are shown in Table 3.
3.3. Statistical Analysis
Genotypes were available for six variants in 1963 individuals, including 700 familial and 459 sporadic PrCa cases overall. M_QLS_ association analysis permitted the inclusion of related and unrelated individuals while also appropriately controlling for Type 1 error [32]. In the Australian dataset, a significant association was observed between PARP2 rs200603922 and PrCa risk (p = 0.028), whilst in the PROGRESS dataset, a significant association was observed between BRIP1 rs4988345 (p = 0.034), ERCC3 rs145201970 (p = 0.010), and MUTYH rs36053993 (p = 0.031) and PrCa risk (Table 4). In the combined Australian and PROGRESS datasets, a significant association with PrCa risk was observed between BRIP1 rs4988345 (p = 0.025) and ERCC3 rs145201970 (p = 2.57 × 10^−4^). The ERCC3 variant remained significant following Bonferroni correction for multiple testing. PrCa status of variant carriers is provided in Supplementary Materials: Table S7, and clinical characteristics of affected familial carriers are presented in Supplementary Materials: Table S8.
Age-at-diagnosis amongst variant carriers was compared to relevant population datasets (Figure 2), with a shift towards younger age-at-diagnosis observed, most evident in Australian sporadic rare variant carriers compared with the population-based PCOR-TAS cohort. While a slightly higher proportion of DDR variant carriers was observed in cases diagnosed before 55 years of age (9.4%; Supplementary Materials: Table S9), variant carriers were relatively consistent at ~6% across the remaining age-at-diagnosis categories. Population data from PCOR-TAS revealed that ~20% of Tasmanian men were diagnosed with a GS ≥ 8. Of men carrying a risk DDR variant, 23% (15/66) were diagnosed with GS ≥ 8, while 58% (38/66) of rare variant carriers were diagnosed with a GS ≤ 6.
4. Discussion
The discovery of rare, high-risk germline variants has long proven challenging due to their very low frequency, which substantially impacts power to detect significant statistical associations. However, there remains considerable impetus to characterise rare risk variants in DDR genes, especially considering the increasing availability of therapies targeting this pathway. In a candidate gene approach designed to take advantage of large familial PrCa resources, where rare risk variants are expected to be enriched, we examined massively parallel sequencing data from two independent datasets to identify rare, likely deleterious DDR variants. Subsequent analysis of 1963 individuals from the Australian and PROGRESS datasets revealed statistically significant associations between rare variants in ERCC3 and BRIP1 and PrCa risk, with ERCC3 surviving correction for multiple testing. In addition, a variant in PARP2 was significantly associated with PrCa risk in the Australian dataset alone, while a variant in MUTYH was significantly associated with PrCa risk only in the PROGRESS dataset.
ERCC3 encodes one of two ATP-dependent DNA helicases, which are core members of the nucleotide excision repair pathway. The ERCC3 rs145201970 variant (MAF 0.17%), located in exon 7, causes an amino acid change at position 283 (p.R283C), which is predicted to disrupt the arginine-aspartic acid salt bridge via the inclusion of a more hydrophobic residue. The variant is located within two domains listed by Interpro as likely required for ERCC3 protein function [45]. Topka et al. comprehensively examined germline mutations in the ERCC2, 3, 4, and 5 genes in 16,712 patients affected by multiple different cancers [46]. Numerous likely pathogenic/pathogenic loss of function (LoF) germline variants were observed in ERCC3, with rs145201970 (n = 42) representing the second most observed LoF variant in this gene in cancer patients after rs34295337 (n = 70) [46]. While there are no previous reports describing rs145201970 as a PrCa risk variant, other germline pathogenic/likely pathogenic ERCC3 variants in PrCa patients have been recently reported by Kohaar et al. [47], Carignan et al. [48] and Rantapero et al. [49]. Additionally, an intronic ERCC3 variant has been associated with increased risk of biochemical recurrence after low-dose-rate prostate brachytherapy, potentially due to reduced mRNA expression in variant carriers [48]. In breast cancer, a recurrent truncating mutation has been associated with familial disease [50,51]. In vitro studies have demonstrated that mutations in ERCC3 impair DNA repair capability and confer a selective sensitivity to Irofulven, a sesquiterpene that has demonstrated some efficacy in clinical trials for metastatic PrCa [46].
BRIP1 is a member of the Fanconi Anaemia gene family and functions in the double-strand break repair pathway, interacting closely with BRCA1. The rare rs4988345 variant (MAF 0.43%) is in exon 5, located within the nuclear localisation signal domain. As a result of the p.R173C amino acid change, there is a loss of positive charge and a more hydrophobic residue introduced within a helicase ATP-binding domain and a region annotated as a nuclear localization signal. BRIP1 rs4988345 has been previously identified in a study enriched for familial PrCa but was only observed in a single PrCa case (0.52%) [52]. Other rare BRIP1 variants were detected in five hereditary PrCa cases (MAF < 1%) [53]; however, no statistical analyses were performed due to their low frequency. BRIP1 has been included on screening panels for several clinical trials investigating the response of metastatic PrCa patients with DDR defects to Olaparib, a PARPi (ClinicalTrials.gov Identifier: NCT02987543) [54]. A cohort of that study comprised men harbouring mutations in 12 DDR genes, including BRIP1, however, only four individuals were identified as carriers of a variant in this gene, below the pre-set threshold for statistical analysis. Evaluation of BRIP1 has also been included in the Phase 2 TRITON2 trial (ClinicalTrials.gov Identifier: NCT02952534), where one patient with a BRIP1 variant responded to the PARPi, Rucaparib [55].
PARP2 is a poly (ADP-ribose) polymerase involved in the base excision repair pathway (BER), and rs200603922 is located in the first exon (p.R15G). This variant (MAF 0.12%) has previously been observed to partially segregate with PrCa in familial cases who tested negative for BRCA1 and BRCA2 mutations [56]. Although several bioinformatic tools predict the variant allele to be benign (Table 3), the R15G amino acid change introduces a more hydrophobic residue, which may impact protein interactions and the phosphorylation of distal residues. There is one other report of a PARP2 variant, rs3093926 (MAF 4.2%), segregating in a PrCa pedigree, but the contribution of this variant to PrCa risk remains undetermined [57], and though common, it was not observed in our Australian discovery families. PARP2 mutations have been associated with breast cancer risk [58], but similarly to PrCa, their functional impact remains unclear. However, PARP2 remains of interest given the ongoing development of PARPi. Though most primarily target PARP1, some, such as Niraparib [59], also affect PARP2, which may be relevant when assessing therapeutic PARPi in men with PARP2 mutations.
MUTYH encodes a DNA glycosylase involved in oxidative DDR and the BER pathway. The rs36053993 variant (MAF 0.47%) results in an amino acid change from a neutral residue to a negatively charged, less hydrophobic residue (p.G368D), with this change located in the highly conserved nudix hydrolase domain. The NCBI human variant database, ClinVar, lists this variant as pathogenic/likely pathogenic arising from its association with MUTYH-associated polyposis, an autosomal recessive hereditary condition typified by the development of colorectal carcinomas. Kohaar et al. (2022) previously reported the rs36053993 SNP in germline samples from PrCa patients [47]. Others have also reported several pathogenic/likely pathogenic variants in MUTYH, including a study reporting 1.8% of 1351 PrCa cases [60] and another reporting 2.4% of 3607 PrCa cases as carrying pathogenic variants in this gene [61]. Furthermore, reduced gene and protein expression of MUTYH in prostate tumours has been associated with an increase in total somatic mutations, which may result from impaired DDR capacity [62].
In this study, the strategy for filtering and prioritisation of variants was developed to detect moderate to highly penetrant, rare DDR gene variants that contribute to familial PrCa risk. It is notable that rare germline variants predicted to be deleterious have been previously observed in BRIP1 (n = 7), ERCC3 (n = 8), MUTYH (n = 10), and PARP2 (n = 5) in a cohort of 5545 non-aggressive and aggressive sporadic PrCa cases [12]. While no statistically significant association with aggressive disease risk was observed, due to the very low frequency of these variants, their association with PrCa risk in general was not explored in this case-only cohort (see Supplementary Tables, Darst et al. [12]). Notably, this study examined 5545 cases, and statistically significant associations with aggressive disease were only demonstrated for previously known PrCa risk genes, BRCA2 and PALB2, with a nominal association seen for ATM. This highlights the fact that the innate rarity of DDR gene variants presents a significant challenge for rare variant evaluation, even in larger sporadic case datasets. Our approach was designed to maximise power by seeking to identify rare variants enriched in two large familial PrCa cohorts. However, it is possible that additional rare, disease-associated variants were not detected due to not being present in the Australian WGS discovery cases. It was also necessary to restrict follow-up to only those candidate variants observed in more than one North American family, as the rareness of these variants impacts statistical power to detect associations. However, this may have resulted in risk variants associated with disease in the Australian cohort being missed, e.g., ATM rs56128736. Furthermore, instances where prioritised variants were subsequently not found to be associated with PrCa could be attributed to their rarity and, thus, lack of statistical power. Thus, to establish the necessary evidence base to inform clinical decision making, it is critical that both our significant DDR risk variants and all prioritised variants be validated/examined in additional familial and population-based datasets, including large publicly available resources such as the UK Biobank. Concurrently, it is worth considering the expansion of candidate gene screening strategies in current clinical trials of PARPi and in current germline testing guidelines for men with a family history of PrCa.
Examining rare variant association with clinicopathological variables presented significant challenges, again due to their rarity, but also due to biases introduced by recruitment strategies. For example, while the aim was to collect all known relatives with PrCa in the Australian and PROGRESS familial cohorts, population-based cases with an early age-at-diagnosis were targeted for recruitment to the Australian case–control study.
Carriers of putative pathogenic DDR variants were slightly more frequently observed in the earlier age-at-diagnosis group, and mildly increased in the GS ≥ 8-at-diagnosis group when compared with the population-based PCOR-TAS cohort. However, it is notable that the majority of variant carriers were diagnosed with a GS ≤ 6 (58%) and/or at age ≥65 years (53%), consistent with the findings of Darst et al. [12], where carriers of DNA repair mutations were, on average, diagnosed only ~1 year younger than non-carriers. Taken together, these findings raise the question as to whether limiting screening for putative genetic DDR variants to very early-onset (<50 years) or only high-grade disease patients is likely to result in a substantial proportion of DDR variant carriers being excluded from testing, and subsequently denied access to effective treatment modalities.
5. Conclusions
This study implicates several additional DDR genes as contributors to inherited genetic risk of PrCa. The existing evidence that rare DDR gene variants are associated with aggressive disease and the growing use of cancer therapies targeting this pathway highlights the potential significance of these findings. However, this study raises the concern that confining genetic screening to only those patients with early-onset and/or high-grade PrCa may result in a missed opportunity for some men to receive effective, targeted gene-based therapies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mucci L.A. Hjelmborg J.B. Harris J.R. Czene K. Havelick D.J. Scheike T. Graff R.E. Holst K. Möller S. Unger R.H. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries JAMA 2016315687610.1001/jama.2015.1770326746459 PMC 5498110 · doi ↗ · pubmed ↗
- 2Castro E. Romero-Laorden N. del Pozo A. Lozano R. Medina A. Puente J. Piulats J.M. Lorente D. Saez M.I. Morales-Barrera R. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer J. Clin. Oncol.20193749050310.1200/JCO.18.0035830625039 · doi ↗ · pubmed ↗
- 3Abida W. Armenia J. Gopalan A. Brennan R. Walsh M. Barron D. Danila D. Rathkopf D. Morris M. Slovin S. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making JCO Precis. Oncol.2017110.1200/PO.17.0002928825054 PMC 5558263 · doi ↗ · pubmed ↗
- 4Abida W. Cyrta J. Heller G. Prandi D. Armenia J. Coleman I. Cieslik M. Benelli M. Robinson D. Van Allen E.M. Genomic correlates of clinical outcome in advanced prostate cancer Proc. Natl. Acad. Sci. USA 2019116114281143610.1073/pnas.190265111631061129 PMC 6561293 · doi ↗ · pubmed ↗
- 5Robinson D. Van Allen E.M. Wu Y.-M. Schultz N. Lonigro R.J. Mosquera J.-M. Montgomery B. Taplin M.-E. Pritchard C.C. Attard G. Integrative Clinical Genomics of Advanced Prostate Cancer Cell 20151611215122810.1016/j.cell.2015.05.00126000489 PMC 4484602 · doi ↗ · pubmed ↗
- 6Holt C. Comprehensive Genetic Profiling Unravels Molecular Targets in Prostate Cancer Oncol. Times 201941910.1097/01.COT.0000577096.71826.2f · doi ↗
- 7Beltran H. Yelensky R. Frampton G.M. Park K. Downing S.R. Mac Donald T.Y. Jarosz M. Lipson D. Tagawa S.T. Nanus D.M. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity Eur. Urol.20136392092610.1016/j.eururo.2012.08.05322981675 PMC 3615043 · doi ↗ · pubmed ↗
- 8Pritchard C.C. Mateo J. Walsh M.F. De Sarkar N. Abida W. Beltran H. Garofalo A. Gulati R. Carreira S. Eeles R. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer N. Engl. J. Med.201637544345310.1056/NEJ Moa 160314427433846 PMC 4986616 · doi ↗ · pubmed ↗