Information Theoretic Study of COVID-19 Genome
Philippe Jacquet

TL;DR
This paper uses information theory to analyze the COVID-19 genome and compare it with other genomes to measure similarities.
Contribution
The paper introduces a low-complexity joint complexity tool for genome comparison, more efficient than the Smith–Waterman algorithm.
Findings
Joint complexity can effectively quantify genome similarities with lower computational cost.
The method allows for large-scale genome comparisons due to its efficiency.
The approach was applied to compare the COVID-19 genome with past and present genomes.
Abstract
In this paper, we analyse the genome sequence of COVID-19 on a information point of view, and we compare that with past and present genomes. We use the powerful tool of joint complexity in order to quantify the similarities measured between the various potential parent genomes. The tool has a computing complexity of several orders of magnitude below the classic Smith–Waterman algorithm and would allow it to be used on a larger scale.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
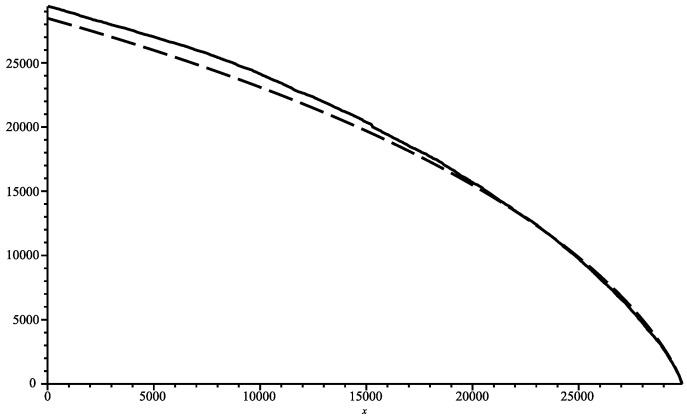 Figure 1
Figure 1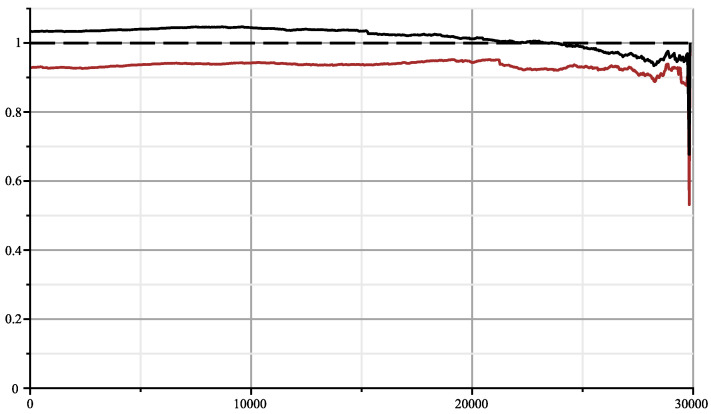 Figure 2
Figure 2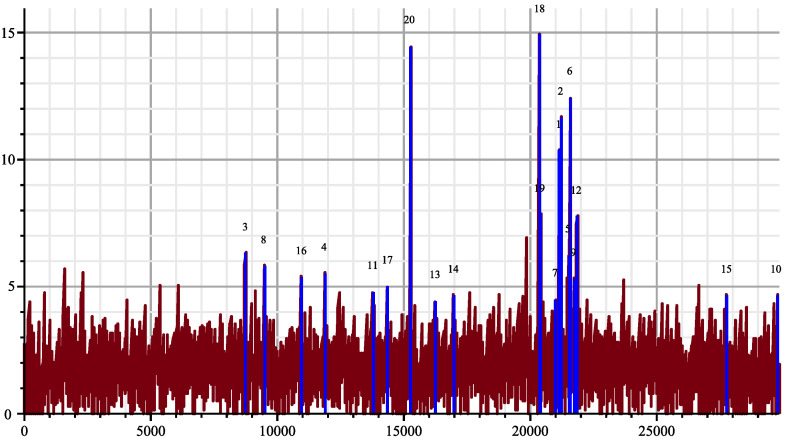 Figure 3
Figure 3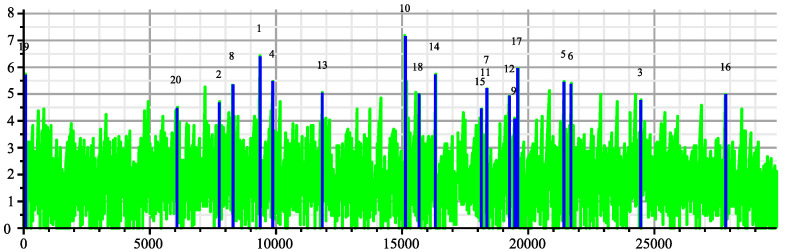 Figure 4
Figure 4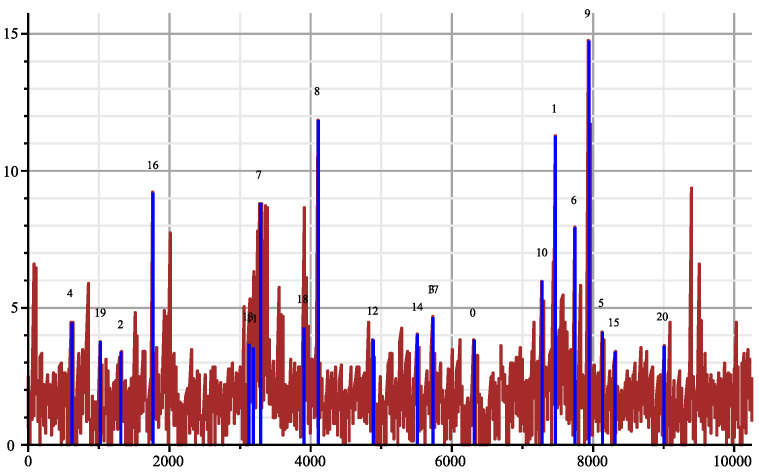 Figure 5
Figure 5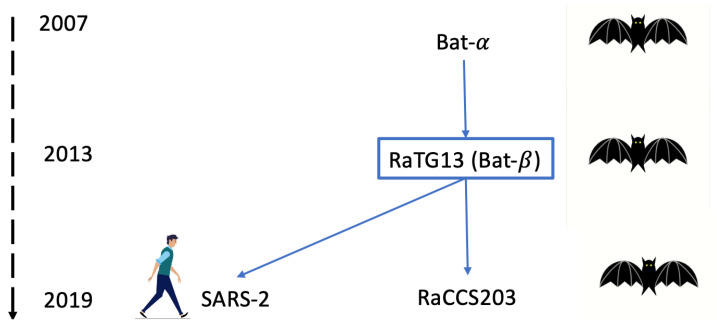 Figure 6
Figure 6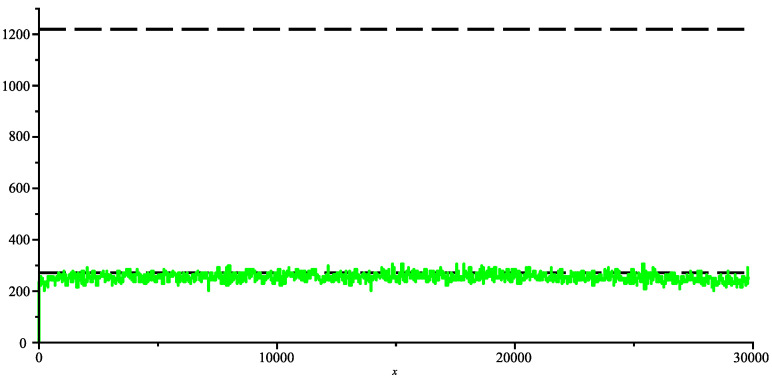 Figure 7
Figure 7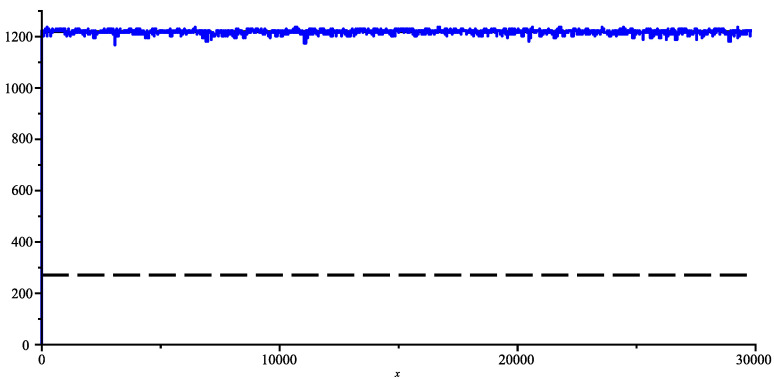 Figure 8
Figure 8 Figure 9
Figure 9 Figure 10
Figure 10 Figure 11
Figure 11 Figure 12
Figure 12 Figure 13
Figure 13Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFractal and DNA sequence analysis · Algorithms and Data Compression · Machine Learning in Bioinformatics