Magnetic structure of monatomic Fe chains on Re(0001): emergence of chiral multi-spin interactions
Andr\'as L\'aszl\'offy, Levente R\'ozsa, Kriszti\'an Palot\'as,, L\'aszl\'o Udvardi, L\'aszl\'o Szunyogh

TL;DR
This study uses first-principles calculations to explore how the magnetic structure of monatomic Fe chains on Re(0001) evolves with chain length, revealing the emergence of chiral multi-spin interactions influencing spin spiral states.
Contribution
The paper introduces a classical spin model incorporating chiral four-spin interactions derived from ab initio calculations, explaining the observed magnetic behaviors.
Findings
Transition from antiferromagnetic to spin spiral states with increasing chain length
Opposite chirality of spin spirals predicted by different methods
Chiral four-spin interactions are essential to accurately model the magnetic structure
Abstract
We present results of first-principles calculations of the magnetic properties of Fe chains deposited on the Re(0001) surface. By increasing the length of the chain, a transition is found from an almost collinear antiferromagnetic state for a 5-atom-long chain to a spin spiral state with the rotational plane slightly tilted from the surface of the substrate for the 15-atom-long chain. It is shown that a classical spin model derived from the ab initio calculations containing only two-spin interactions supports opposite chirality of the spin spiral compared to a direct optimization of the spin configuration within the ab initio method. The differences between the results of the two methods can be understood by introducing chiral four-spin interactions in the spin model.
Click any figure to enlarge with its caption.
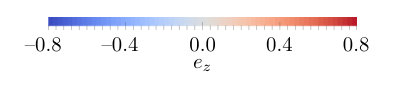 Figure 1
Figure 1 Figure 2
Figure 2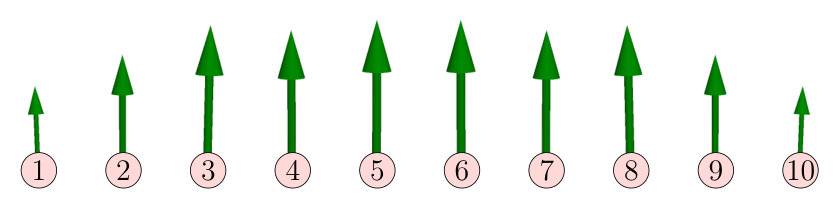 Figure 3
Figure 3 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6 Figure 7
Figure 7 Figure 8
Figure 8 Figure 9
Figure 9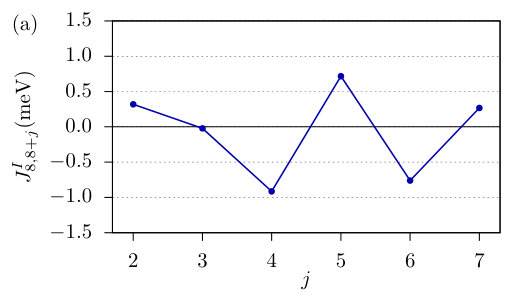 Figure 10
Figure 10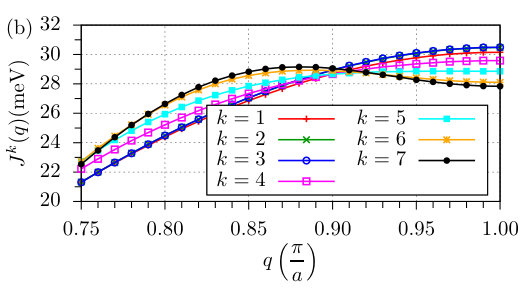 Figure 11
Figure 11 Figure 12
Figure 12 Figure 13
Figure 13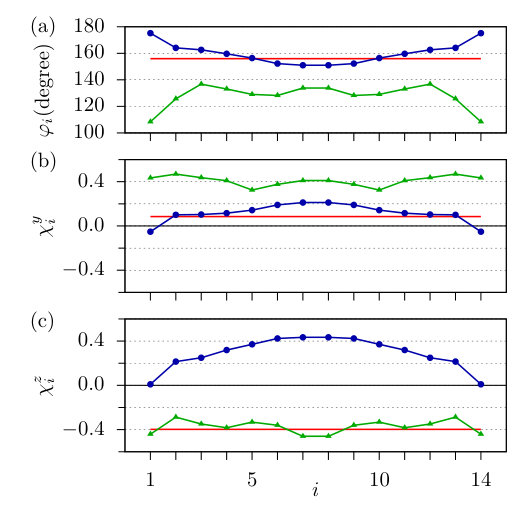 Figure 14
Figure 14 Figure 15
Figure 15 Figure 16
Figure 16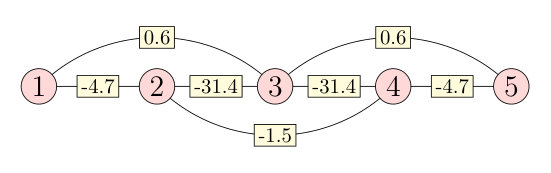 Figure 17
Figure 17 Figure 18
Figure 18 Figure 19
Figure 19 Figure 20
Figure 20 Figure 21
Figure 21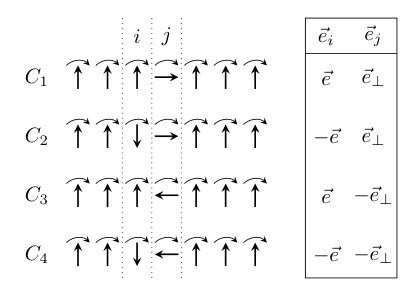 Figure 22
Figure 22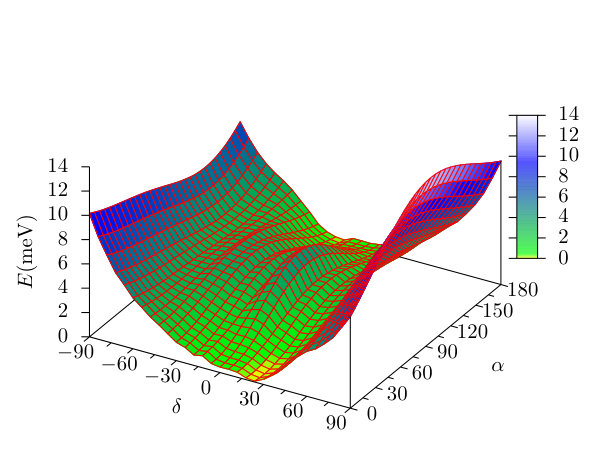 Figure 23
Figure 23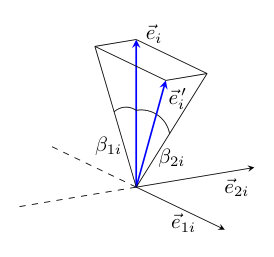 Figure 24
Figure 24| –0.60 | 0.64 | 0.23 | 0.42 | 0.18 | –0.03 | –0.03 | |
| –3.95 | –0.71 | –0.98 | –1.24 | –1.76 | –1.62 | –1.43 | |
| –1.24 | 2.96 | 1.61 | 1.80 | 1.34 | 1.48 | 1.49 | |
| 0.05 | –0.25 | –0.30 | –0.04 | –0.02 | –0.07 | 0.00 | |
| –4.06 | –4.47 | –5.12 | –5.13 | –4.92 | –4.96 | –5.03 | |
| 0.44 | 0.84 | 0.73 | 1.16 | 1.16 | 1.13 | 1.12 |
| 7 | 8 | –0.243 | –0.033 | |
| 7 | 8 | –3.107 | –1.428 | |
| 7 | 8 | –2.306 | 1.491 | |
| 7 | 9 | –0.376 | 0.000 | |
| 7 | 9 | –1.083 | –5.030 | |
| 7 | 9 | –1.319 | 1.117 | |
| 8 | 9 | –0.167 | 0.033 | |
| 8 | 9 | –3.154 | –1.428 | |
| 8 | 9 | –2.313 | 1.491 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Magnetic structure of monatomic Fe chains on Re(0001): emergence of chiral multi-spin interactions
A. Lászlóffy
Department of Theoretical Physics, Budapest University of Technology and Economics, Budafoki út 8., HU-1111 Budapest, Hungary
L. Rózsa
Department of Physics, University of Hamburg, D-20355 Hamburg, Germany
K. Palotás
Department of Theoretical Physics, Budapest University of Technology and Economics, Budafoki út 8., HU-1111 Budapest, Hungary
Institute for Solid State Physics and Optics, Wigner Research Center for Physics, Hungarian Academy of Sciences, P. O. Box 49, H-1525 Budapest, Hungary
MTA-SZTE Reaction Kinetics and Surface Chemistry Research Group, University of Szeged, H-6720 Szeged, Hungary
L. Udvardi
Department of Theoretical Physics, Budapest University of Technology and Economics, Budafoki út 8., HU-1111 Budapest, Hungary
MTA-BME Condensed Matter Research Group, Budapest University of Technology and Economics, Budafoki út 8., HU-1111 Budapest, Hungary
L. Szunyogh
Department of Theoretical Physics, Budapest University of Technology and Economics, Budafoki út 8., HU-1111 Budapest, Hungary
MTA-BME Condensed Matter Research Group, Budapest University of Technology and Economics, Budafoki út 8., HU-1111 Budapest, Hungary
Abstract
We present results of first-principles calculations of the magnetic properties of Fe chains deposited on the Re(0001) surface. By increasing the length of the chain, a transition is found from an almost collinear antiferromagnetic state for a five-atom-long chain to a spin spiral state with the rotational plane slightly tilted from the surface of the substrate for the 15-atom-long chain. It is shown that a classical spin model derived from the ab initio calculations containing only two-spin interactions supports opposite chirality of the spin spiral compared to a direct optimization of the spin configuration within the ab initio method. The differences between the results of the two methods can be understood by introducing chiral four-spin interactions in the spin model.
I Introduction
The investigation of clusters of magnetic atoms on nonmagnetic surfaces has recently opened several intriguing prospects for the storage and transfer of information on the nanometer scale. The reduced dimensionality of the clusters often leads to an enhancement of the magnetic anisotropy energy Gambardella et al. (2002), stabilizing the magnetic structure in one of two states connected by time-reversal symmetry. These two states can in turn be used for designing logic gates Khajetoorians et al. (2011). Besides the anisotropy, the interactions between the magnetic adatoms mediated by the substrate also crucially influence the magnetic state. One prominent type of these couplings is the Dzyaloshinskii–Moriya (DM) interaction Dzyaloshinsky (1958); Moriya (1960), the presence of which can be attributed to the spin–orbit coupling and the inversion-symmetry breaking caused by the surface. This interaction leads to the formation of noncollinear structures with a preferred chirality by which the information may be encoded Menzel et al. (2012). Linear chains of magnetic atoms on a superconducting surface also offer a possibility for realizing Majorana bound states Alicea (2012) as fundamental elements of topological quantum computing, the signatures of which have been investigated experimentally in chains both with collinear Nadj-Perge et al. (2014) and noncollinear Kim et al. (2018) magnetic ground states.
Determining the ground state for an interacting magnetic system based on ab initio electronic structure calculations remains a considerable challenge. Such computations can efficiently be performed by mapping the energy or grand potential of the system to a classical spin model. It was demonstrated in Ref. Liechtenstein et al. (1987) how Heisenberg exchange interactions between pairs of spins may be determined based on the derivatives of the energy, that is, the torque acting on the spin directions. The torque method has been generalized to tensorial two-spin interactions appearing in the presence of spin–orbit coupling Udvardi et al. (2003); Ebert and Mankovsky (2009), and it was validated for various systems over the last decade Antal et al. (2008); Vida et al. (2016); Simon et al. (2018). A fully real-space calculation of the interactions in a magnetic cluster is presented in Ref. Cardias et al. (2016). However, only considering two-spin interactions in the spin model is not sufficient for describing all types of magnetic order. It was demonstrated in various ultrathin film systems that isotropic four-spin interactions may stabilize up-up-down-down states Al-Zubi et al. (2011); Krönlein et al. (2018); Romming et al. (2018), conical spin spirals Yoshida et al. (2012); Zimmermann et al. (2014), or nanoskyrmion lattices Heinze et al. (2011).
The problem of finding a spin model which contains all types of magnetic interactions relevant in the system may be circumvented by updating the directions of the magnetic moments during the ab initio calculations. Because of the higher number of degrees of freedom the computational complexity increases dramatically, but such methods enable a more accurate determination of the magnetic ground state. It was proposed in Refs. Stocks et al. (1998); Újfalussy et al. (1999) that the constrained local moment method within density functional theory is applicable for performing first-principles spin dynamics simulations. Using this method, it was demonstrated in Ref. Újfalussy et al. (2004) that the reduction of the symmetry leads to a canted magnetic configuration in a finite Co chain along a step edge on the Pt(111) surface. An alternative procedure for updating the spin directions based on the Landau–Lifshitz–Gilbert equation Lan ; Gil was introduced in Ref. Rózsa et al. (2014), where the torques acting on the spins are determined directly from the electronic structure at each time step within a fixed electronic potential.
In the present paper the magnetic properties of monatomic Fe chains are investigated on the Re(0001) substrate. Spin-polarized scanning tunneling microscopy measurements performed for a 40-atom-long Fe chain on superconducting Re in Ref. Kim et al. (2018) revealed a spin spiral ground state with both in-plane and out-of-plane spin components and a period of approximately four lattice constants.
The paper is organized as follows. In Secs. II.1 and II.2 the details of the ab initio calculations are discussed, performed using the Vienna Ab-initio Simulation Package (vasp) Kresse and Furthmüller (1996) and the embedding technique within the KKR method Lazarovits et al. (2002), respectively. In Sec. II.3 the spin model including two-spin interactions is introduced. The results for the parameters entering the spin model are discussed in Sec. III.1. The possible magnetic ground states of the chains obtained from the spin model and from a direct optimization within the ab initio method are compared in Sec. III.2. The deviations between the different methods observed for the chirality of the spin structure for the 15-atom-long chain are resolved by taking into account four-spin chiral interactions introduced in Sec. III.3. Finally, the results are summarized in Sec. IV.
II Methods
II.1 VASP calculations
To model the geometries of Fe atomic chains on the Re(0001) surface, the equilibrium structure of an Fe adatom on Re(0001) has been first calculated by using the VASP method. The obtained structure corresponds to the total energy minimum after geometry optimization. In the calculation the generalized gradient approximation (GGA) within density functional theory (DFT) has been used with the exchange–correlation (XC) functional parametrized following the work of Perdew, Burke, and Ernzerhof (PBE) Perdew et al. (1996). The system has been modeled as a surface cell in a slab geometry consisting of four atomic layers of Re (in total Re atoms), and an Fe adatom in the hcp hollow position Kim et al. (2018). The chosen geometry ensures that the interactions between Fe atoms in repetitive supercells are negligible due to their large separation of Å that corresponds to , where Å is the in-plane lattice constant of Re. In the (0001) direction a 10-Å-thick vacuum region has been considered to avoid interaction between repetitive slabs. The Brillouin zone was sampled by the Gamma point only due to the large size of the supercell. The Re atoms in the bottom three layers of the slab have been fixed to their hcp bulk positions, and the vertical positions of all Re atoms in the topmost layer and the Fe adatom have been optimized by using a force convergence criterion of 0.01 eV/Å acting on the individual atoms. The Fe adatom is found to have a spin magnetic moment of , and it pulls out its three nearest-neighbor (NN) Re atoms slightly from the top Re layer, arriving at a Fe-Re vertical distance of Å with respect to these nearest neighbors. We also find that the top Re layer relaxes toward the substrate which leads to a vertical Re-Re distance of Å between the above-mentioned three NN Re atoms of the Fe adatom and the Re atoms in the subsurface layer, which is smaller than the bulk Re interlayer distance of Å. These Fe-Re and Re-Re vertical distances were used in the subsequent KKR calculations for the Fe adatom and the atomic chains on Re(0001).
II.2 KKR calculations
We used the Green’s function embedding technique based on the KKR multiple scattering theory Lazarovits et al. (2002) to determine the electronic and magnetic properties of the Fe clusters. The Re(0001) surface has been modeled as an interface region between semi-infinite bulk Re and vacuum consisting of eight atomic layers of Re and four atomic layers of empty spheres (vacuum). The energy integrals were performed using 16 points along a semicircle contour in the upper complex semiplane and a sampling of up to 3282 points in the Brillouin zone was used to calculate the Green’s function of the host. The Ceperley–Alder-type of exchange-correlation functionals Ceperley and Alder (1980) as parametrized by Perdew and Zunger Perdew and Zunger (1981) and an angular momentum cutoff of was considered in the KKR calculations, similarly to Ref. Lászlóffy et al. (2017). A single Fe adatom and chains consisting of five, 10, and 15 Fe atoms were calculated by embedding them in the first vacuum layer with the layer relaxations described in the previous section.
Three different methods have been used to investigate the magnetic properties of the systems. First, the relativistic torque method Udvardi et al. (2003); Ebert and Mankovsky (2009) was applied to determine parameters of a classical spin model restricted to two-spin interactions. The energies of magnetic configurations within this description were compared by atomistic spin model simulations. The spin model is discussed in Sec. II.3, while the method for fitting the parameters is given in Appendix A. Second, the energies of selected spin configurations were compared, such as collinear states with different magnetic orientations or spin spirals with different periods. In the spirit of the magnetic force theorem (MFT) Liechtenstein et al. (1987), these energy differences between magnetic configurations are calculated with fixed electronic potentials based on the band energy, which is obtained by using Lloyd’s formula Lloyd (1967). Third, the ground state of the Fe chain was also determined completely within the ab initio formalism, by updating the spin directions based on the torque acting on them and also performing self-consistent calculations in the obtained spin configurations. This method enables finding local energy minima, ideally the ground state, in the whole configuration space of the spin directions Balogh et al. (2012). It should be noted that in the second and third methods there is no restriction on the possible types of magnetic interactions apart from those enforced by the symmetry of the system.
First, we performed self-consistent calculations for an Fe adatom in hcp position on the top of the Re(0001) substrate with the embedded cluster KKR technique. We considered clusters of different sizes and concluded that the spin magnetic moment of Fe, , changes by less than 1 % when increasing the size of the cluster from 13 lattice sites including three Re atoms and nine empty spheres in the first NN shell to 122 lattice sites including the first three neighbor shells around the Fe adatom. Note that the obtained spin moment of Fe is about 6% less than the value of from the VASP calculations. The magnetic moment of the Fe atom induces a small () magnetic moment in the Re atoms directly below it, while the induced moments of farther Re atoms are negligible.
We determined the anisotropy energy of the adatom in the spirit of the MFT by calculating the energy difference between the cases where the Fe spin is pointing in-plane () and normal to the plane (). Due to the small value of the induced Re moments the exchange-correlation field was set to zero at the Re sites while calculating the energy differences, and we obtained that the single Fe adatom has easy-plane anisotropy with . We confirmed that taking into account the exchange-correlation field on the Re sites leads to a change within about 5 % in the magnetic anisotropy energy; therefore, in all calculations of the Fe chains in terms of the MFT we chose the above approach for simplicity.
We considered close-packed monatomic chains of five, 10, and 15 Fe atoms along the nearest-neighbor direction on the top of Re(0001). In the following this direction will be denoted by , the in-plane direction perpendicular to by , and the normal-to-plane direction by . Based on our investigations for the adatom, we considered clusters containing the atomic positions in a NN environment relative to the Fe atoms, including 11, 21, and 31 Re atoms, as well as 25, 45, and 65 empty spheres for the chains of five, 10, and 15 Fe atoms, respectively.
For the five-atom-long Fe chain we first performed self-consistent calculations with ferromagnetic (FM) order and used the torque method to generate a spin model. As will be discussed in Sec. III.1, the NN isotropic couplings are strongly antiferromagnetic (AFM), implying that an alternating AFM order is considerably lower in energy than the FM state. In order to check the preference for the AFM state, we recalculated the potentials with the AFM order of the spins and, by using these potentials, we calculated the energy difference between the AFM and FM states within the MFT. Indeed, the AFM state was by 15.7 meV/Fe atom lower in energy than the FM state. Based on the above results, for all the Fe chains under consideration we used the self-consistent potentials obtained from alternating AFM configurations to generate the spin-model parameters.
II.3 Spin model
The adiabatic decoupling of the electronic and spin degrees of freedom and the rigid spin approximation Antropov et al. (1996) make it possible to characterize the energy of a magnetic system by a set of unit vectors describing the directions of atomic magnetic moments, where is the number of magnetic atoms in the system. Since the metallic substrate acts as a particle reservoir for the clusters considered in the calculations, instead of the energy we will consider the grand potential at zero temperature, with and being the Fermi energy of the reservoir and the number of electrons in the cluster, respectively. Taking into account one-spin terms and two-spin magnetic interactions, the spin model can be written as
[TABLE]
where is a constant, the are traceless and diagonal second-order single-ion anisotropy matrices, and the are tensorial exchange interactions Udvardi et al. (2003). The matrices can be decomposed into three parts,
[TABLE]
where
[TABLE]
is the isotropic exchange interaction,
[TABLE]
is the traceless symmetric part of the matrix, with denoting the transpose. This is known to contribute to the so-called two-ion magnetic anisotropy of the system. The antisymmetric part of the matrix,
[TABLE]
is related to the DM interaction Dzyaloshinsky (1958); Moriya (1960),
[TABLE]
with the DM vector , being the Levi–Civita symbol and denoting Cartesian components.
Following Ref. Lászlóffy et al. (2017), site-resolved easy-axis directions and anisotropy energies have been determined for the monatomic chains from the spin model, taking into account both single-ion and two-ion contributions. Since the nearest-neighbor isotropic interactions are found to be antiferromagnetic (see Sec. III.1), in order to characterize the magnetic anisotropy we consider alternating local moments . The grand potential of the system can then be expressed as
[TABLE]
with
[TABLE]
and the effective anisotropy matrices
[TABLE]
The normalized eigenvectors of the symmetric matrices in Eq. (9), , , and correspond in order to the easy, intermediate, and hard directions, with the respective energy eigenvalues . For illustrating the site-specific easy directions together with the magnetic anisotropy energies, we will use the following vector:
[TABLE]
The ground state of the spin model was determined by zero-temperature Landau–Lifshitz–Gilbert (LLG) spin dynamics simulations where only the damping term was kept. This is described by the time integration step
[TABLE]
where
[TABLE]
is the effective magnetic field, and a small damping parameter, , was chosen. The new spin vectors were normalized after each step to preserve the unit length of the vectors. The simulations were stopped when the spin components changed less than in subsequent LLG steps. For each system ten runs with independently chosen random initial configurations were performed which all led to the same final state, providing a strong indication that this is the actual ground state of instead of a local minimum.
III Results
III.1 Spin-model parameters for the Fe chains
In this section we discuss the parameters of the spin model containing two-spin interactions described in Sec. II.3, calculated in terms of the KKR method and the relativistic torque method detailed in Sec. II.2 and in Appendix A, respectively. The variation of NN and next-nearest-neighbor (NNN) isotropic interactions from Eq. (3) along the chains can be seen in Fig. 1. For all chains, the NN isotropic interactions are the strongest, and their negative sign means AFM coupling. The isotropic interactions become more and more homogeneous at the middle of the chain as the length of the chain is increased. It is also apparent from Fig. 1 that the isotropic NN interactions are considerably smaller in magnitude at the edges of the chain than inside the chain for all chain lengths.
The ferromagnetic NNN interactions are more than one order of magnitude smaller than the NN ones as shown in Fig. 1. This also holds true for the interactions for farther neighbors as can be seen in Fig. 2(a), where calculated values for the middle spin in the 15-atom-long chain are displayed. However, if one summarizes the effect of farther interactions, it turns out that they play an important role in determining the ground state. This is demonstrated by introducing the Fourier transform of the isotropic interactions as
[TABLE]
where is the number of neighbors taken into account. If one assumes that the interactions in the middle of the chain will no longer be significantly modified as the chain length is increased, then may be used as an approximation for the energy contribution of the isotropic interactions to homogeneous spin spiral states in infinitely long chains, where corresponds to the FM and to the AFM state. The most favorable state is given by the maximum of . It can be seen in Fig. 2(b) that up to this corresponds to the collinear AFM state. However, considering more shells () in the sum in Eq. (13) a spin spiral state becomes the most favorable, which can be regarded as a long-wavelength modulation of the AFM state. This is a consequence of the frustration of the isotropic interactions, which in the AFM state is indicated by the fact that the isotropic interaction between atoms at the distance of an odd multiple of the lattice constant becomes FM and at an even it becomes AFM, although the spins at odd and even positions should be antiparallel and parallel in the AFM state, respectively. It is shown in Fig. 2(a) that such kind of frustration occurs for , explaining how the spin spiral state is formed as the number of shells is increased.
The NN and NNN DM vectors in the chains, see Eq. (6), are drawn in Fig. 3. Similarly to the isotropic exchange interactions, the DM vectors at the middle of the chain become stabilized as the chain length is increased. The DM vectors between the three atoms at the edges of the chains significantly differ from those in the middle of the chains, while these DM vectors are similar between the five-, 10-, and 15-atom-long chains. The obtained DM interactions satisfy the symmetry rules of Moriya Moriya (1960) with respect to the only crystal symmetry of the system, namely the mirroring at the plane intersecting the middle of the chain. Since the DM vector transforms as an axial vector, this symmetry implies
[TABLE]
where is the mirror image of site in the chain of length . Note that in Fig. 3 the vectors are displayed for and by definition.
In the case of the 15-atom-long chain, the numerical values for the components of the NN and NNN DM vectors are given in Table 1. It can be seen that the NNN DM vectors are the largest in magnitude and they are almost parallel to the direction. Although the components of the NN DM vectors have the same sign, these are actually competing with the NNN vectors due to the short-range AFM order (see Sec. III.2), similarly how the alternating isotropic interactions in Fig. 2(a) are competing with the NN interaction. The components of the NN and NNN DM vectors are mostly positive; they only change sign for the NN atoms at the edge of chain (). In general, is larger for the NNs than for the NNNs, but at the middle of the chain they become roughly similar in size. The components of the DM vectors are very small and they should disappear in the limit of infinitely long chains due to the mirror symmetry with respect to the plane. For the 15-atom-long chain, the mirror symmetry implies .
The site-resolved anisotropy vectors defined in Eq. (10) are visualized in Fig. 4, where the arrows point along the easy directions and their magnitude is proportional to the energy difference between hard and easy axes at the given site. At all sites the easy axis is almost parallel to the direction, while the hard axis is roughly along the direction. With increasing chain length the magnitude of the vectors gets quite homogeneous with the maxima of 7.79, 9.03, and 8.92 meV at the middle of the five-, 10-, and 15-atom-long chains, respectively. However, since the site-resolved magnetic anisotropy energy drops at the edge of the chains, the average length of the anisotropy vectors is 6.25, 7.60, and 7.91 meV for the three chains in order. These values are close to the average anisotropy energies, calculated from Eq. (7), 5.79, 7.37, and 7.76 meV/Fe, respectively, since as noted the local easy and hard axes are very close to the and axes for all the Fe atoms in the chains.
III.2 Ground state
Here the ground states of the magnetic clusters will be discussed, focusing on the chirality of spin rotation inside the structures. We will use the convention that the rotation of the spins in the plane is locally right handed at site if the projection of on this plane may be obtained from the projection of via a right-handed rotation by an angle smaller than around the positive axis, meaning that the angle between the projections is smaller than when rotating from the positive towards the positive direction. The opposite chirality is called left handed, in agreement with previous definitions of the chirality for spin spirals close to the ferromagnetic state in Ref. Heide et al. (2008). Analogously, we call the rotation in the plane locally right handed if the projections of the spins rotate from the positive towards the positive direction.
First we determined the ground states of the chains from the spin model following the method described in Sec. II.3. The obtained configurations can be seen in Figs. 5-5. The five-atom-long chain prefers AFM ordering due to the strong AFM coupling between the NN spins. The component of the DM vectors is responsible for a slight noncollinearity in the AFM state. The negative value in Fig. 3 causes a slight left-handed rotation between spins 1 and 2, while the positive causes a right-handed rotation between spins 2 and 3. The ground state is tilted away from the plane through a rotation around the axis by about 12.8∘, so the spins with positive component now have positive component, too. The tilting is caused by the competition of the easy axis anisotropy (see Fig. 4) and the large components of the DM vectors (similar to those in Table 1) preferring a rotation of the spins in the plane.
The ground state of the 10-atom-long chain in Fig. 5 is again almost collinear AFM due to the strong NN AFM coupling between the spins. The components of the DM vectors shown in Fig. 3 cause a rotation of the spins around the axis along the whole chain, but no full period of a spin spiral state can be observed. The ground state is now tilted from the plane through a rotation around the axis by .
In both the five- and 10-atom-long chains, the strong anisotropy confines the systems close to the plane. The chirality is right handed over the middle three atoms in the five-atom-long chain and over the middle eight atoms in the 10-atom-long chain. Note that only looking at every second spin in the chain, this visually corresponds to a left-handed rotation, since the angle between the neighboring spins is close to because of the strong AFM NN couplings. This chirality is determined by the components of the DM vectors shown in Fig. 3. The direction of the tilting is defined by the chirality in the plane on the one hand and the components of the DM vectors on the other hand, the latter influencing the rotational sense in the plane. In both systems the NN and NNN DM vectors are characterized by large negative components, both of which would prefer a left-handed rotation if the angle between the NN spins would be small. However, since this angle is larger than in the present case, the apparent left-handed rotation between the NNN spins actually corresponds to a right-handed rotation between the NN spins, meaning that the NN and NNN DM vectors are competing in this AFM spin structure. In the five-atom-long chain the rotational plane is tilted around the axis by a positive angle, leading to a left-handed chirality in the plane enforced by the NN DM vectors. For the longer chain length of atoms with the same chirality in the plane the influence of the NNN DM vectors becomes stronger, leading to a tilting around the axis by a negative angle and a right-handed chirality in the plane.
The ground state of the 15-atom-long chain shown in Fig. 5 can much better be characterized as a spin spiral state. As described in the context of Fig. 2, the frustrated isotropic interactions induce a spin spiral with a wave number of (see the curve), which corresponds to a wavelength of about , slightly larger than the length of the chain. In Fig. 5 the spins from positions 4 to 11 visually form a full period, which can be understood as a wavelength modulation of the AFM state. This shorter modulation period might easily be caused by the components of the NN DM vectors, which are not included in Fig. 2. The large negative components of the NNN DM vectors play an important role in tilting the rotational plane out from the plane by around the axis, for a simple explanation see Appendix C. A similar tilted spin spiral state was already attributed to the interplay of the DM interaction and easy-plane magnetic anisotropy in Ref. Simon et al. (2018). Similarly to the 10-atom-long chain, the DM vectors imply right-handed rotation both in the plane and in the plane over the whole chain. This ground state cannot satisfactorily be reconciled with the experimental observation of a four-atomic period in a chain of 40 Fe atoms reported in Ref. Kim et al. (2018), which would correspond to a spin spiral state where neighboring spins are perpendicular to each other.
The local rotational sense of the spins in a spin spiral discussed above can be quantitatively described by the site-dependent chirality vector defined as
[TABLE]
If only NN DM vectors were included in the model, would be parallel to the direction of . The chirality is right-handed in the plane and in the plane for and , respectively. In Fig. 6 the magnitude of the local chirality vectors , where is the angle between the NN spins, as well as the and components of the chirality vectors are displayed for the 15-atom-long chain. The angle between the spins at the edges of the chain is almost and also the sign of is switched compared to the middle of the chain, which can be explained by the edge effects in the interaction parameters discussed in Sec. III.1. In the middle of the chain the local environment of the sites is similar, leading only to slight variations in . The component of the chirality vectors takes a value between 0.1 and 0.2 for the spins 2 to 13, indicating that the spins tilt away from the plane as argued in Appendix C.
For comparison with the ground state obtained from the spin model, we calculated the energy of the 15-atom-long Fe chain in the homogeneous spin spiral configuration,
[TABLE]
in the spirit of the MFT by keeping the AFM potentials fixed. Here is the modulation angle of the AFM state, is the tilting angle of the spiral from the plane and is a phase factor. The NN spin angle is , indicating right-handed rotation in the and left-handed rotation in the planes for and , respectively. Both rotational senses switch under a sign change of , and the rotation in the plane proceeds in the opposite direction for . We did not consider an additional angle variable which would differentiate between left- and right-handed rotational senses in the plane, since in an infinitely long chain these two chiralities are equivalent due to the mirror symmetry with respect to the plane. The phase factor was determined by minimizing the anisotropy energy assuming a homogeneous magnetic anisotropy with easy axis, i.e., maximizing for every .
The energy of the spin spirals normalized to one Fe atom is shown in Fig. 7 as a function of and , where the zero level corresponds to the state with the lowest energy. The obtained minimum is at and , with the corresponding spin configuration shown in Fig. 5. While the period of this spin spiral, , shows good agreement with that obtained from the simulations based on the spin model shown in Fig. 5, the actual values of and indicate a right-handed rotation in the and a left-handed rotation in the planes (see above), the latter being the opposite of the spin model simulation results. This can clearly be seen in Fig. 6(c), where for the ground state of the spin model and for the spin spiral with lowest energy are opposite in sign.
In the case of the 15-atom-long chain we also performed a fully ab initio spin dynamics energy minimization as described in Sec. II.2. The energetically most favorable configuration found by this method is shown in Fig. 5, which also resembles a flat spin spiral state apart from small deviations from the coplanar spin arrangement. This method predicts a right-handed rotation of the spins in the and a left-handed rotation in the planes, in agreement with the MFT calculations performed for the homogeneous spin spiral states. This approach results in a wavelength of () of the spin spiral modulation which is significantly shorter than those obtained from the previous two methods. On top of the AFM state, this visually leads to a period of the spin structure, see spins 3, 8, and 13 in Fig. 5, which fits the experimental observation the most.
III.3 Four-spin chiral interactions
It was found in Sec. III.2 that the spin model Eq. (1) and the MFT calculation of homogeneous spin spirals yield opposite rotational senses of the spin components in the plane for the 15-atom-long chain. This most likely indicates that multispin interactions play an important role in the present system, since these were not taken into account in the spin model, but are implicitly included in the MFT calculations. In order to estimate the magnitude of these interactions, it is worthwhile to calculate the chiral interactions from energy differences based on the MFT directly and compare them to the values of the spin model shown in Table 1. This method is illustrated for the component in Fig. 8, where four configurations in the plane are shown. In the configuration denoted by , during a global rotation of the spins around the axis by the angle most spins are rotated according to , while the perpendicular spin at site will follow . The configurations and are obtained by switching the signs of spins or as illustrated on the right side of Fig. 8.
Assuming the spin model containing only two-spin interactions, from the grand potentials associated to the configurations - one obtains
[TABLE]
as a function of the rotation angle . Note that due to the choice of configurations and the switching of the spin directions only the interaction between sites and remains in Eq. (17). Averaging Eq. (17) over the angle we define the component of the chiral interaction vector as
[TABLE]
which, by comparing with Eq. (6), corresponds to the component of the DM vector , if only two-spin interactions are considered in the spin model. Here the index r denotes that the chiral interaction vector was obtained from the rotational scheme depicted in Fig. 8. Analogously, the quantities and corresponding to the other two components of the DM vector can be calculated by performing the rotation in the and planes, respectively.
The calculated values for () are collected in Table 2 for NN and NNN spins in the middle of the 15-atom-long chain. In addition, the values obtained from the torque method, defined in Eq. (41) in Appendix A, are listed in the table, which within the spin model Eq. (1) should also coincide with (cf. Table 1). Due to the mirror symmetry with respect to the plane going through atom , the symmetry rules for the DM vectors, Eq. (14), imply and . Remarkably, these symmetry relations do not apply to the corresponding chiral interaction vectors obtained from the rotational method; in particular, does not vanish, but it has a comparable value to the other components. This means that the spin model parametrization of the band energy surface of the present system is not compatible by taking into account two-spin DM interactions only.
In order to explain the differences between and in Table 2, it is necessary to include multispin chiral interactions in the model description. Consider a grand potential of the form
[TABLE]
where the last three terms represent two-, three-, and four-site four-spin chiral interactions combining isotropic (scalar product) and DM (cross product) contributions. In the sums in Eq. (19), the , , , and indices run over all lattice sites, and in each sum the different indices label different atoms. Equation (19) may be rewritten in an alternative way where the summations are performed over all pairs, three-site and four-site clusters; this is presented in Appendix B.
The two-site four-spin chiral interactions were recently investigated in Ref. Brinker et al. (2019). Following from the definition Eq. (19), they are antisymmetric in their indices,
[TABLE]
similarly to the two-spin Dzyaloshinsky–Moriya interaction in Eq. (6). For the three-site interactions we will consistently use the notation where the second and third site indices coincide, in which case there is no intrinsic symmetry relation connecting the coefficients in the sum in Eq. (19). By definition, the four-site interactions satisfy the symmetry relations
[TABLE]
therefore, a prefactor of is introduced in the last term of Eq. (19). Moreover, the mirror symmetry on the plane in the center of the chain implies
[TABLE]
where was defined in the context of Eq. (14). Equation (23) is satisfied for two-, three-, and four-site chiral interactions.
If Eq. (18) is evaluated based on the model Eq. (19), one obtains
[TABLE]
Note that the two-site four-spin interaction does not contribute to the grand potential of the configurations shown in Fig. 8, since it is only finite between pairs of spins which are not parallel and not perpendicular to each other. Considering the chiral interaction vectors in Table 2 it is possible to derive
[TABLE]
which generally does not vanish since the three-site and four-site chiral interactions are not antisymmetric with respect to their first and fourth indices. This indicates the presence of four-spin chiral interactions in the system on the order of , which is not negligible compared to the total value of chiral interactions also containing two-site contributions displayed in Table 2. The relation , which breaks the symmetry rules for two-spin DM interactions, may similarly be explained by the presence of the four-spin chiral interactions.
Changing from the two-spin model in Eq. (1) to the model containing also four-spin interactions in Eq. (19) naturally modifies the interpretation of the chiral interaction energies obtained from the torque method,
[TABLE]
for details of the derivation see Appendix A. It should be noted that contrary to Eq. (24), Eq. (26) is antisymmetric with respect to the site indices and , thus preserving the symmetries of the two-site DM vectors. Most importantly, the different treatment of the two-, three-, and four-site four-spin interactions between the rotational scheme in Eq. (24) and the torque method in Eq. (26) can explain why the components of the two kinds of chiral interaction vectors have different signs in Table 2. While the torque method supports positive or right-handed chirality in the plane, the rotational scheme supports negative or left-handed chirality in the same plane. The chirality derived from the rotational method is in agreement with the lowest-energy spin spiral state obtained from MFT calculations and the ground state found by the ab initio energy minimization discussed in Sec. III.2.
IV Conclusion
Ab initio electronic structure calculations were performed to study the magnetic properties of Fe monatomic chains on the Re(0001) substrate. For all the considered chains strong antiferromagnetic couplings between the nearest-neighbor spins were observed, which in the case of the five-atom-long chain led to a nearly collinear antiferromagnetic ground state with the spins approximately aligned perpendicular to the chain and to the surface normal. As the length of the chain is increased, the frustration of the isotropic exchange interactions at farther neighbors transforms the ground state into a spin spiral state, with a single modulation period observable in the 15-atom-long chain. The experimental investigations in Ref. Kim et al. (2018) also concluded on a spin spiral ground state of 40-atom-long Fe chains, although with a smaller period.
For the 15-atom-long chain the spin components in the plane were found to follow a right-handed rotation in the spin spiral. Regarding the rotation in the plane, calculations based on a spin model only containing two-spin interactions yielded a right-handed rotation, while determining the optimal homogeneous planar spin spiral state using magnetic force theorem calculations and performing an optimization of the configuration directly within the ab initio scheme both indicated a left-handed chirality. This discrepancy was resolved by considering chiral multispin interactions in the spin model, the presence of which in the system is supported by calculations of specific rotated spin configurations sensitive to the chirality.
Since the experiments were carried out in the superconducting phase of the Re substrate, future ab initio calculations including the superconducting state of Re by solving the Bogoliubov–de Gennes equation Csire et al. (2015, 2018) may reveal the reasons behind the observed discrepancies between theory and experiment regarding the spin spiral period. Such first-principles calculations would also enable the investigation of the interplay between exotic magnetic states and topological superconductivity in clusters of magnetic atoms on a superconducting substrate.
Acknowledgements.
The authors would like to thank R. Wiesendanger for useful discussions. Financial support of the Nemzeti Kutatási Fejlesztési és Innovációs Hivatal (Hungary) under Projects No. K115575 and No. FK124100, the BME-Nanotechnology FIKP grant of Emberi Erőforrások Minisztériuma (BME FIKP-NAT), the Alexander von Humboldt Foundation, and the Deutsche Forschungsgemeinschaft via SFB668 are gratefully acknowledged.
Appendix A Torque method
Here we give a short summary of how the parameters of the classical spin model in Eq. (1) were determined by using the torque method; for more details see Ref. Rózsa (2016). As shown in Fig. 9, at each site the spin direction will be denoted by , and two orthogonal vectors , are defined which form a right-handed basis together with , and around which is rotated by the infinitesimal angles and , respectively. The derivatives with respect to these angles may be expressed as
[TABLE]
Supposing the model of Eq. (1), the second derivatives of with respect to the angles and can be calculated as
[TABLE]
[TABLE]
[TABLE]
[TABLE]
[TABLE]
[TABLE]
[TABLE]
where and label summations over Cartesian indices.
Within the torque method the derivatives on the left-hand sides of Eqs. (29)–(35) are obtained directly from first principles, using the expressions derived from Lloyd’s formula given in Ref. Udvardi et al. (2003). The diagonal elements of the anisotropy tensor were determined from Eqs. (33) and (34) via
[TABLE]
which simplifies to if is pointing along the direction. This way it is only possible to determine the differences between the diagonal elements, but this information is sufficient since was defined as a traceless tensor because its trace only shifts the grand potential by a constant factor due to the normalization of the spins. The two independent components were computed by considering spin configurations where all spins are pointing along the and directions.
For calculating the exchange interaction tensor one has to rely on Eqs. (29)–(32). By introducing the tensor products
[TABLE]
for , and the notation for the derivatives
[TABLE]
with the opposite angle index, Eqs. (29)–(32) can be summarized as
[TABLE]
where is rewritten as a vector in the tensor product space. By introducing , the orthogonal projection onto the subspace of , summing over Eqs. (29)–(32) one obtains the system of linear equations
[TABLE]
Since is a rank-1 matrix, the sum over four equations in Eq. (40) only enables the calculation of four components of as described in Ref. Udvardi et al. (2003). In order to determine the full tensor, Eq. (40) has to be summed up over calculations performed for at least three linearly independent directions of the vectors, corresponding to a least-squares fitting procedure for . Due to the symmetry of the Re(0001) surface, during the calculations four ferromagnetic configurations of the vectors were taken into account, including three NN directions parallel to the axis and at angles and with respect to this direction, as well as one along the out-of-plane direction.
The chiral interaction vectors introduced in Sec. III.3 were calculated in ferromagnetic configurations with all spins pointing along one of the , , or directions, i.e. in Fig 9. The components of are defined as
[TABLE]
which within the model Eq. (1) containing only two-spin interactions simplifies to the DM vector [cf. Eqs. (30) and (31)],
[TABLE]
On the other hand, inserting the spin model Eq. (19) instead of Eq. (1) into Eq. (41) and using the relations Eqs. (27) and (28) to calculate the second derivatives yields Eq. (26) in Sec. III.3,
[TABLE]
Appendix B Alternative notation for the four-spin chiral interactions
Equation (19) follows the convention where the summations are performed for each index separately over all lattice sites, treating the cases where some of the indices coincide individually. This is in agreement, e.g., with the notation for the two-site two-spin and four-spin chiral interactions used in Ref. Brinker et al. (2019). Another way of expressing the grand potential or the Hamiltonian is by performing the summations over all different plaquettes, i.e. pairs, triangles, and quadrilaterals in the case of the two-, three-, and four-site interactions, respectively. Such a convention is used, e.g., in Ref. Hoffmann and Blügel (2018) for the isotropic four-spin interactions.
Following this convention, Eq. (19) may be rewritten as
[TABLE]
Once again, the , , , and indices all label different atoms. The sums over pairs contain half as many terms as the two-site summations in Eq. (19); however, this is resolved by taking into account the intrinsic symmetry relations and Eq. (20), which enable canceling the prefactor. As discussed in the main text, no such intrinsic symmetry relations exist for the three-site four-spin chiral interaction, which necessitates a summation over six different terms in Eq. (LABEL:eqn60), corresponding to the possible permutations of the site indices in a triangle. For the four-site interactions, the symmetry relations in Eq. (22) cancel with the prefactor in Eq. (19), simplifying the sum over permutations in a quadrilateral to six different terms in Eq. (LABEL:eqn60).
Note that Eq. (LABEL:eqn60) is the most general form of the grand potential containing four-spin chiral interactions. Crystal symmetries may reduce the number of independent coefficients for specific plaquettes. For example, the mirror symmetry defined in Eq. (23) implies that in the triangle with and there are only three inequivalent three-site four-spin chiral interactions instead of six in the general case.
Appendix C Tilted spin spirals
In this section we summarize why the spin spiral ground state becomes tilted away from the plane due to the component of the DM vectors. We will consider a simplified spin model consisting of sites where the interactions are homogeneous along the chain, with anisotropy accounting for the easy direction and DM interaction between NN spins. We will compare the energies of harmonic spin spiral configurations as defined in Eq. (16), simplifying the description to two parameters and . It is assumed that a spin spiral state with a specific value is formed by the frustration of the isotropic exchange interactions, as was discussed in Sec. III.2, and only the dependence of the grand potential of the spin model on the parameter is considered. This is given by the expression
[TABLE]
where now describes the contributions which do not depend on , such as the isotropic interactions. For , the average of with respect to atomic indices equals , and using a simple addition formula it is possible to arrive at
[TABLE]
One can obtain the value of minimizing the grand potential by differentiation,
[TABLE]
for , and
[TABLE]
otherwise. This indicates that for an arbitrarily small value of , the most preferred state is tilted away from the plane where . We note that including the component of the DM vectors influences the dependence of the grand potential on the parameter, but does not change this qualitative conclusion.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gambardella et al. (2002) P. Gambardella, A. Dallmeyer, K. Maiti, M. C. Malagoli, W. Eberhardt, K. Kern, and C. Carbone, Nature (London) 416 , 301 (2002).
- 2Khajetoorians et al. (2011) A. A. Khajetoorians, J. Wiebe, B. Chilian, and R. Wiesendanger, Science 332 , 1062 (2011) . · doi ↗
- 3Dzyaloshinsky (1958) I. Dzyaloshinsky, J. Phys. Chem. Sol. 4 , 241 (1958) . · doi ↗
- 4Moriya (1960) T. Moriya, Phys. Rev. 120 , 91 (1960) . · doi ↗
- 5Menzel et al. (2012) M. Menzel, Y. Mokrousov, R. Wieser, J. E. Bickel, E. Vedmedenko, S. Blügel, S. Heinze, K. von Bergmann, A. Kubetzka, and R. Wiesendanger, Phys. Rev. Lett. 108 , 197204 (2012) . · doi ↗
- 6Alicea (2012) J. Alicea, Rep. Prog. Phys. 75 , 076501 (2012) .
- 7Nadj-Perge et al. (2014) S. Nadj-Perge, I. K. Drozdov, J. Li, H. Chen, S. Jeon, J. Seo, A. H. Mac Donald, B. A. Bernevig, and A. Yazdani, Science 346 , 602 (2014) . · doi ↗
- 8Kim et al. (2018) H. Kim, A. Palacio-Morales, T. Posske, L. Rózsa, K. Palotás, L. Szunyogh, M. Thorwart, and R. Wiesendanger, Sci. Adv. 4 , eaar 5251 (2018) . · doi ↗