aRrayLasso: a network-based approach to microarray interconversion
Adam S. Brown, Chirag J. Patel

TL;DR
aRrayLasso is a new method that improves the conversion of microarray data between different platforms using a statistical model.
Contribution
aRrayLasso introduces a Lasso-penalized model for accurate microarray interconversion without relying on manufacturer annotations.
Findings
aRrayLasso achieves high fidelity in predicting expression levels comparable to technical replicates.
The method enables integration of datasets from different microarray platforms using open-source R functions.
It outperforms existing methods that rely on direct probe alignment or incomplete annotations.
Abstract
Summary: Robust conversion between microarray platforms is needed to leverage the wide variety of microarray expression studies that have been conducted to date. Currently available conversion methods rely on manufacturer annotations, which are often incomplete, or on direct alignment of probes from different platforms, which often fail to yield acceptable genewise correlation. Here, we describe aRrayLasso, which uses the Lasso-penalized generalized linear model to model the relationships between individual probes in different probe sets. We have implemented aRrayLasso in a set of five open-source R functions that allow the user to acquire data from public sources such as Gene Expression Omnibus, train a set of Lasso models on that data and directly map one microarray platform to another. aRrayLasso significantly predicts expression levels with similar fidelity to technical replicates…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
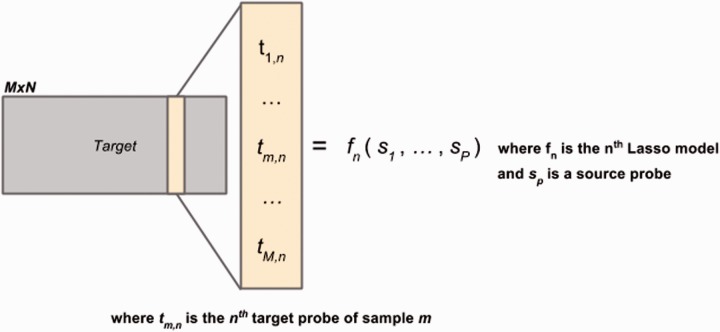 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGene expression and cancer classification · Molecular Biology Techniques and Applications · Bioinformatics and Genomic Networks
1 Introduction
A pressing issue in translational biology is the ability to reference and utilize historical microarray datasets for large-scale discovery programs (Tsiliki et al., 2014). The appeal of using historical datasets includes capturing previous investment to construct larger cohorts. Despite interest in both industry and academia (Tsiliki et al., 2014; Yengi, 2005), few groups have attempted to tackle the problem of platform integration. Current approaches primarily rely upon passing different microarray platforms through a common identifier system, such as EntrezGene IDs, using specially designed packages (Alibes et al., 2007; Mohammad et al., 2012) or online tools (Huang et al., 2009). While these systems work well in cases where manufacturers have maintained annotations of their microarray databases, ID-based conversion methods fail for deprecated and undermaintained microarray platforms. Another approach to convert between platforms is sequenced-based, wherein each sequence tag is aligned to the genome or transcriptome and annotated (Fumagalli et al., 2014; Liu et al., 2007). Unfortunately, it is often the case that de novo annotations do not capture the complexity of the transcriptome (e.g. for genes with alternative splice variants Gambino et al., 2015).
To address the shortcomings of both annotation- and sequence-based conversion methods, we have developed aRrayLasso, a Lasso-regression based network model. Our method directly predicts the probe expression levels of the target platform. To demonstrate the accuracy of our method, we show that predictions made using aRrayLasso are of similar accuracy to technical replicates from the 6 same mRNA pool. Our methodology allows users to utilize currently available methodologies for integrating cross-experiment microarray datasets (Tsiliki et al., 2014) and allow for the construction of large-cohort retrospective studies.
2 Methods
To convert from a source to a target microarray platform, we chose to model each individual sequence tag in the target platform as a linear combination of all sequence tags from the source platform (see Fig. 1 and Supplementary Methods). Because microarrays have greater than 10 000 individual probes, we chose to use the Lasso algorithm for generalized linear regression (Friedman et al., 2010). The Lasso algorithm allows the resulting linear model to be ‘sparse’ in that only the most relevant and robust (by cross-validation) predictors are assigned non-zero values. This optimization allows the model to outperform similar models that require all predictors to be assigned non-zero coefficients (Tibshirani et al., 2010). Lasso is implemented in the R package ‘glmnet,’ allowing for ease of use (Friedman et al., 2010). Fig. 1.Schematic of the aRrayLasso algorithm. aRrayLasso takes in an MxN target matrix containing M samples and N probes. A Lasso model, f_n_, is then constructed for each target probe using all probes in the MxP source matrix (M samples, P probes)
We first generate a list of lasso models for each sequence tag in the target microarray platform. Our implementation can take as input a variety of data formats, including expression matrices, R expressionSet objects and Gene Expression Omnibus accession numbers (Edgar et al., 2002). Once the full list of models has been computed, we provide functions that allow either the straightforward prediction of sequence tag values or the validation of the model list by calculation of Pearson product-moment correlation coefficients.
To demonstrate the utility of our methodology, we utilized three datasets: (i) GSE6313, containing C57/B6 adult mouse retina cDNA profiles (Liu et al., 2007), (ii) GSE7785, containing PANC-1 derived cDNA profiles (Tan et al., 2003) and (iii) GSE4854, containing mouse cortex expression profiles (Kuo et al., 2006). Each dataset is composed of multiple technical replicates for several distinct microarray platforms (see Supplementary Table S1). For both datasets, we used aRrayLasso to first train models to intraconvert between each individual platform and then predicted intraconversions between each pair of platforms for all technical replicates. To assess the accuracy of our conversions, we calculated the average Pearson’s r between the predicted values and actual experimental values for each platform and replicate. We also calculated the average inter-replicate Pearson’s r for each platform (see Supplementary Table S2).
3 Results
To explore the performance of aRrayLasso, we began by comparing our method’s ability to predict expression to the biological variation between replicates on the same platform. We assessed the degree to which aRrayLasso could accurately predict platform interconversions in three datasets, representative of different experimental systems, organisms and platforms. For the five platforms tested, aRrayLasso predictions are within the technical variation of each microarray platform when compared with technical replicates from the same cDNA pool, even when subjected to multiple sequential conversions (Supplementary Table S2). In addition, once built, aRrayLasso models can be used between experimental conditions: using the models built on GSE6313, we predicted expression levels in GSE4854 with no significant loss of signal (Pearsons product-moment correlation, P < 0.38). While the results presented here do not guarantee similar results for all training and testing datasets, these analyses serve as a promising proof of concept. Furthermore, our success with a relatively small dataset suggests that aRrayLasso may reach even higher levels of performance as the size of the datasets involved increases.
4 Discussion
Implementation: In this investigation, we propose a data-driven method for integrating across high-throughput genomic measurement modalities that avoids the use of annotation- or sequence alignment-based tools. We have implemented a Lasso regression-based modeling approach to model the expression level of each sequence tag in a target microarray as a linear combination of all sequence tags in a source microarray. Our implementation represents a straightforward, easy-to-use and open-source methodology for conversion between microarray platforms.
Limitations: One drawback of our method is the need for extant or newly generated matched samples in the source and target platforms. In our experience, however, there are a large number of datasets available that have matched samples with replicates for a number of popular microarray platforms. A second limitation to our method is in conversion which lack overlap in gene coverage. In these cases, as with currently available methodologies, our method will fail to provide meaningful conversions. Lastly, while we have shown in one case that interexperiment conversions are feasible, we caution that systematic technical error in a single experiment may lead to the creation of a biased model. In general, however, when coupled with one of several cross-experiment dataset integration tools, aRrayLasso will enable mining of the remarkable and untapped historical pool of microarray datasets for large-scale metastudies for well-powered discovery.
Supplementary Material
Supplementary Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alibes A. (2007) I Dconverter and ID Clight: conversion and annotation of gene and protein I Ds. BMC Bioinformatics, 10, 8–9. 10.1186/1471-2105-8-9PMC 177980017214880 · doi ↗ · pubmed ↗
- 2Edgar R. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res., 30, 207–210.1175229510.1093/nar/30.1.207PMC 99122 · doi ↗ · pubmed ↗
- 3Friedman J. (2010) Regularization paths for generalized linear models via coordinate descent. J. Stat. Software, 33, 1–22.PMC 292988020808728 · pubmed ↗
- 4Fumagalli D. (2014) Transfer of clinically relevant gene expression signatures in breast cancer: from Affymetrix microarray to Illumina RNA-Sequencing technology. BMC Genomics, 15, 1008–1020.2541271010.1186/1471-2164-15-1008 PMC 4289354 · doi ↗ · pubmed ↗
- 5Gambino G. (2015) Characterization of three alternative transcripts of the BRCA 1 gene in patients with breast cancer and a family history of breast and/or ovarian cancer who tested negative for pathogenic mutations. Int J Mol Med., 35, 950–956.2568333410.3892/ijmm.2015.2103 PMC 4356434 · doi ↗ · pubmed ↗
- 6Huang D.W. (2009) Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc., 4, 44–57.1913195610.1038/nprot.2008.211 · doi ↗ · pubmed ↗
- 7Kuo W.P. (2006) A sequence-oriented comparison of gene expression measurements across different hybridization-based technologies. Nat. Biotechnol. 24, 832–840. 1682337610.1038/nbt 1217 · doi ↗ · pubmed ↗
- 8Liu F. (2007) Comparison of hybridization-based and sequencing-based gene expression technologies on biological replicates. BMC Genomics, 8, 153–167.1755558910.1186/1471-2164-8-153PMC 1899500 · doi ↗ · pubmed ↗