RM1 Semiempirical Quantum Chemistry: Parameters for Trivalent Lanthanum, Cerium and Praseodymium
José Diogo L. Dutra, Manoel A. M. Filho, Gerd B. Rocha, Alfredo M. Simas, Ricardo O. Freire

TL;DR
This paper introduces a new quantum chemistry model for trivalent lanthanides La(III), Ce(III), and Pr(III), offering higher accuracy than previous methods.
Contribution
The paper presents a novel semiempirical quantum chemistry model parameterized for trivalent lanthanides with improved accuracy.
Findings
The new RM1 model is more accurate than previous sparkle models for lanthanide complexes.
The model achieves an accuracy of about 0.07Å for La(III) and Pr(III), and 0.08Å for Ce(III).
The model is essential for coordination types beyond oxygen and nitrogen ligands.
Abstract
The RM1 model for the lanthanides is parameterized for complexes of the trications of lanthanum, cerium, and praseodymium. The semiempirical quantum chemical model core stands for the [Xe]4fn electronic configuration, with n =0,1,2 for La(III), Ce(III), and Pr(III), respectively. In addition, the valence shell is described by three electrons in a set of 5d, 6s, and 6p orbitals. Results indicate that the present model is more accurate than the previous sparkle models, although these are still very good methods provided the ligands only possess oxygen or nitrogen atoms directly coordinated to the lanthanide ion. For all other different types of coordination, the present RM1 model for the lanthanides is much superior and must definitely be used. Overall, the accuracy of the model is of the order of 0.07Å for La(III) and Pr(III), and 0.08Å for Ce(III) for lanthanide-ligand atom distances…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
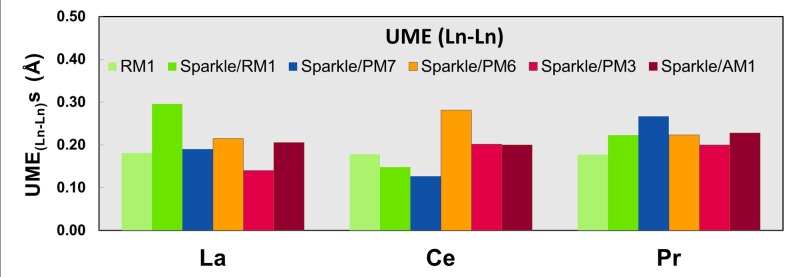 Fig 1
Fig 1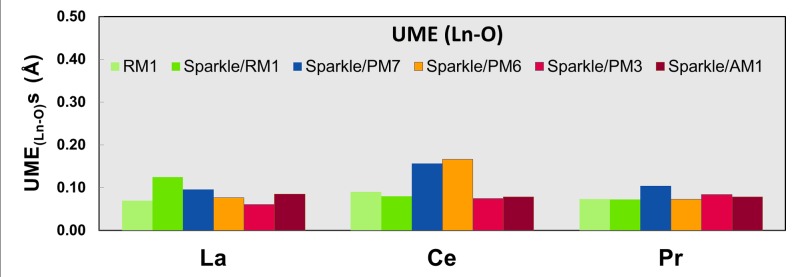 Fig 2
Fig 2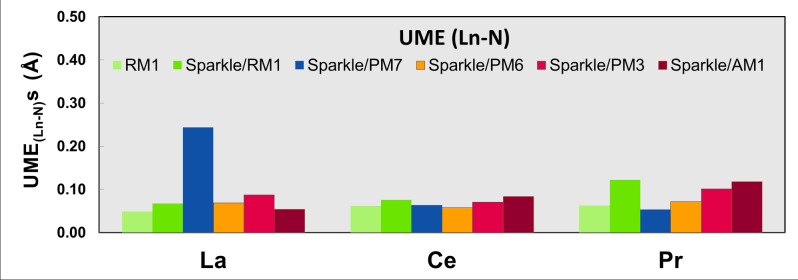 Fig 3
Fig 3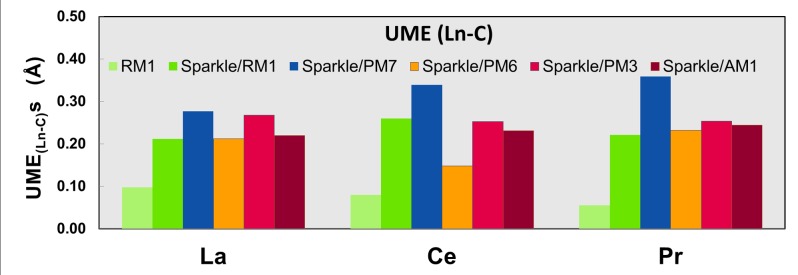 Fig 4
Fig 4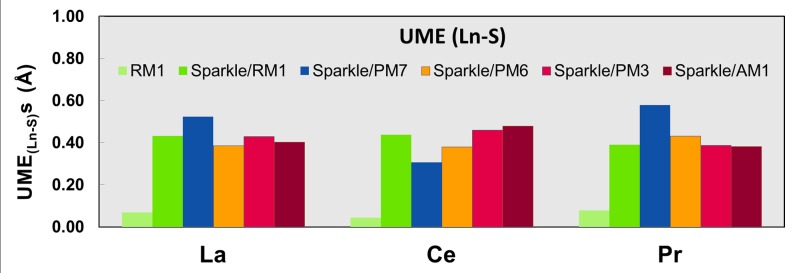 Fig 5
Fig 5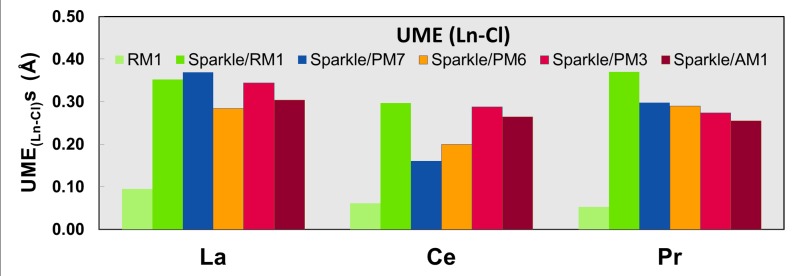 Fig 6
Fig 6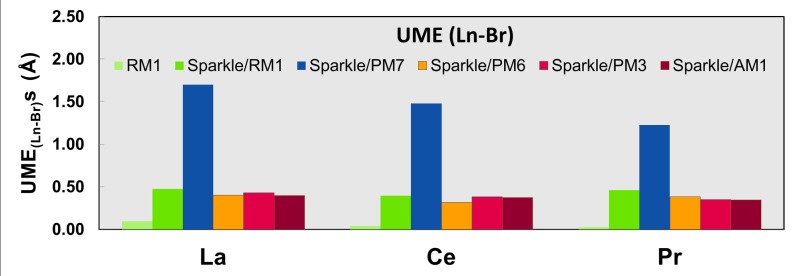 Fig 7
Fig 7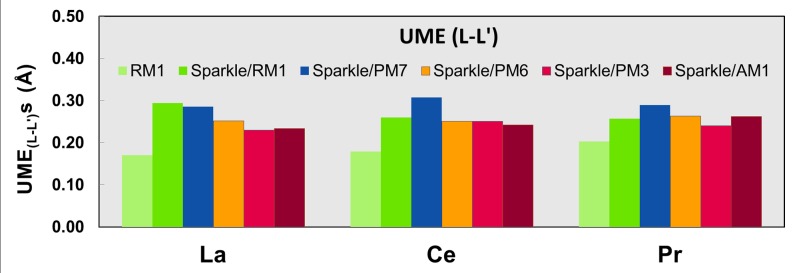 Fig 8
Fig 8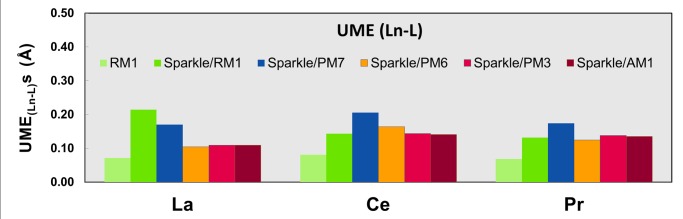 Fig 9
Fig 9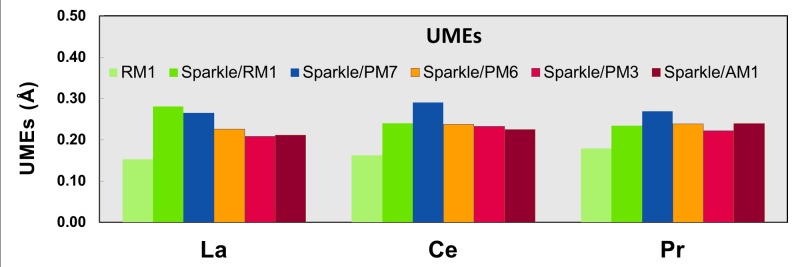 Fig 10
Fig 10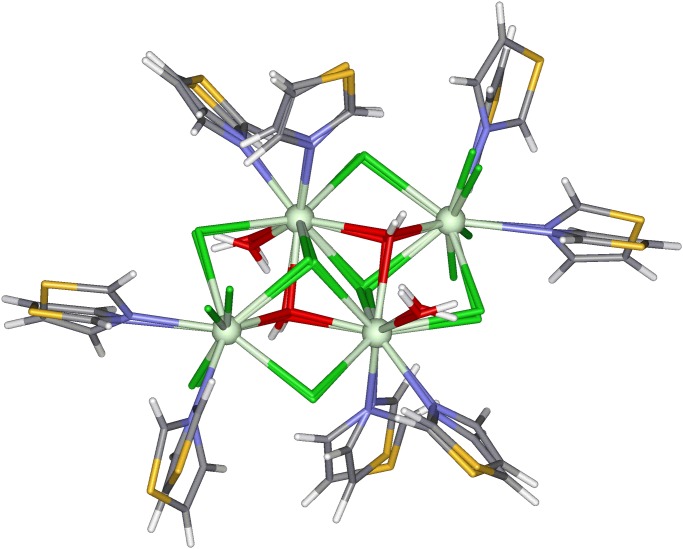 Fig 11
Fig 11Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLanthanide and Transition Metal Complexes · Lanthanide and Transition Metal Complexes · Organometallic Complex Synthesis and Catalysis
Introduction
Lanthanum complexes find their usage as catalysts, for example, in the transesterification of triglycerides to monoesters [1], important in the making of biodiesel fuel, in the synthesis of novel antioxidants with high superoxide scavenging activity [2], in asymmetric epoxidation reactions [3], in P_4_ activation by lanthanum naphthalene complex [4], etc. Furthermore, lanthanum complexes may serve as extreme pressure lubrication additives in paraffin oil [5], they may display pH sensitivity [6], and are of interest to studies on chelator design [7] and polymer build up [8].
Cerium(III) complexes display low toxicity when compared to other lanthanide ions and are, for example, of interest in the design of new drugs targeting DNA [9]. They, of course, may also be used as catalysts, for example in the catalytic cleavage of phosphate esters, an important reaction which mimetizes the hydrolytic cleavage of DNA [10]. Also, due to the relative ease by which they can convert to Ce(IV), Ce(III) complexes may act as antioxidation agents, for example, as a hydroxyl radical quencher in fuel cell electrolyte membranes [11]. The structure of novel Ce(III) complexes are also thoroughly studied due to their several potential applications [12,13].
The 4f^2^ electronic configuration of Pr(III) gives rise to a series of electronic states, and therefore luminescence of Pr(III) covers a wide range of wavelengths, ranging from the ultraviolet to the near infrared. However, Pr(III) luminescence is rarely observed in the visible region[14]–the most important use of Pr(III) complexes in luminescent and electroluminescent devices being as near infrared emitters [15]. Biological activities of praseodymium complexes have also been observed due to their substantial affinities for many biomolecules, as are the cases of DNA-binding [16] and their presence in Schiff-base complexes [17]. Further, the large variety of architectures of Pr(III) complexes have been receiving continued interest, as novel complexes are being reported [18], including a complex of Pr(III) with organofluorotitanate ligands with coordination number 12 [19]. Furthermore, larger clusters are being prepared, as is the case of the tetramer [Pr_4_Cl_10_(OH)2(thiazole)8(H_2_O)2], the first thiazole complex of a lanthanide ion reported [20], and of the three-dimensional 5-aminoisophtalate Pr(III) polymeric complex which presents good gas storage capabilities [21].
Therefore, the theoretical modeling of complexes of La(III), Ce(III), and Pr(III) is an open area of research, important for the selection of ligands, of counter ions, of specific coordination geometries, of metal to ligand ratios, of polymer framework topologies, of thermal and photostability, for the fine tuning of optical and magnetic properties, and so on.
Previously, our research group introduced the Sparkle Model [22,23], originally to allow the calculation of Eu(III) complexes within the semiempirical AM1 model [24], together with a prescription to compute the UV-Visible electronic spectra of the complexes using another semiempirical model, INDO/S [25–28]. The model was then successfully applied to the design of luminescent Eu(III) complexes [29–32] and proven useful for the prediction of ligand field parameters [33]. In 2004, we introduced Gaussian functions in the core-core repulsion of our sparkle model in order to make it consistent with AM1, something that greatly improved the accuracy of the model [34]. Thus, in 2005 we introduced the new model, we called Sparkle/AM1 for all trivalent lanthanide ions [35]. Since then, we have further introduced the Sparkle Model for La (III), Ce (III), and Pr (III) for PM3 [36–38], for PM6 [39], for PM7 [40], and RM1 [41].
The sparkle model proved very accurate for more ionic bonds of the hard-hard type, such as the cases of directly coordinated oxygen and nitrogen atoms. However, the sparkle model fails when other types of atoms are directly coordinated to the lanthanide ion, as is the cases of carbon, sulfur, and the heavier halogens.
In order to be able to address all types of bonds between the central trivalent lanthanide ion and its ligands, we introduced in 2013 a new and more perfected model, we called the RM1 model for the lanthanides, and presented parameters for Eu(III), Gd(III), Tb(III) [42], Dy(III), Ho(III), and Er(III) [43]. In this model, we considered the lanthanide ion to be a regular neutral atom within RM1 [44] where the semiempirical core represents the electronic configuration [Xe]4f^n^, with n varying from 0 for La (III) to 14 for Lu (III), and we add three electrons to a valence shell comprised of the semiempirical atomic orbitals 5d, 6s, and 6p. This new RM1 model for the lanthanides proved to be very general and capable of much more accurately describing the multitude of different bonds that show up in lanthanide chemistry.
In the present article, we further extend the RM1 model for the lanthanides to complexes of La(III), Ce(III), and Pr(III).
Method
As indicated before, the RM1 model for the lanthanides assumes that the electronic configuration [Xe]4f^n^ with n = 0,1,2, for La(III), Ce(III) and Pr(III), respectively, can be correctly described by the semiempirical core of charge +3. In addition, the model attaches a set of semiempirical 5d, 6s, and 6p orbitals to describe the valence shell, which always contains 3 electrons for all lanthanide trications. As a result, 22 parameters need to be optimized for each of the lanthanides.
A usual and recurrent criticism of semiempirical models is that they tend to perform well for systems for which they were parameterized, and tend to perform poorly or even badly for other systems. In order to minimize this, we created, in our research group, a method of parameterization which seeks to obtain much more robust models [34,35,45]. We start by collecting all existing complexes of the lanthanide ion of interest that can be found in the Cambridge Structural Database (CSD) [46–48]. In order to guarantee quality in our parameters, we restrict ourselves to collect only complexes of high crystallographic quality (R < 0.05). Of course, we understand that, due to their unique characteristics, each lanthanide metal has a particular palette of applications, each requiring their own specific type of ligands. Therefore, we assume that the more useful complexes will be naturally more numerous in the universe of high quality structures of the CSD database for each particular metal. Having collected that, we note that there is no point in parameterizing the model for all existing high quality CSD complexes simultaneously because, there could be many repeating ligands, which would be overrepresented in the parameterization set and which could cause an imbalance in the parameters. Therefore, at this point, we need to select sub-sets of complexes to serve as parameterization sets. In addition, this selection must take into account the relative difficulty of predicting, from quantum chemical calculations, the geometries of the complexes. For the purpose of this selection, we assume that a good measure of this difficulty is the difference between the crystallographic geometries and geometries obtained by our previous model Sparkle/AM1 [37,38,49]. Thus, for each complex i, we define the following measure R_i_:
where d refers to distances, and θ to angles; CSD refers to data obtained from the CSD, and Calc refers to data obtained from our previous model calculation (Sparkle/RM1); j runs over all types of bonds, e.g. Ln-O, Ln-N, Ln-C, etc, and k runs over all bonds of the j type; is the standard deviation of all differences between CSD and Sparkle/AM1 (calc) for all bonds of the j type; l runs over all angles; and is the standard deviation of all angle differences between CSD and Sparkle/AM1 (calc). The set of measures R_i_ was then used as input for a divisive hierarchical clustering analysis, DIANA [50], from which we selected two parameterization sets from the universes of complexes for each lanthanide metal: one we call the small set, with only 15 complexes for La(III), 8 complexes for Ce(III), and 7 complexes for Pr(III); and another one we call the large parameterization set, with 38 complexes for La(III), 18 for Ce(III), and 16 for Pr(III). The next step, is the optimization of the model where, by means of a combination of a few non-linear optimization techniques, we seek to minimize a response function, which is the sum of all R_i_s of Eq (1), with the difference that calc will now refer to the particular distance or angle calculated by means of the intermediary set of parameters of the optimization procedure. When the nonlinear optimization process converges for the small set of complexes, we start it all over again with the large set. Finally, we declare the process of nonlinear optimization to be finished when it converges for the large set.
Assessments of the accuracy of the model can be made via the unsigned mean error, UME_i_, defined for each complex i as
where CSD and Calc are as in Eq (1), and the summation runs over all the n bonds being considered. As before, we use two different measures: UME_(Ln-L)i_ and UME_i_. The first contains all j distances between the lanthanide ion and its directly coordinated atoms. The second, includes, in addition, all distances between all directly coordinated atoms and indirectly also reflects a measure of the accuracy of the predicted angles within the coordination polyhedron.
The next step in verifying the robustness of the parameterization is to determine if the distribution of unsigned mean deviations between the predicted and crystallographic geometries can be adequately described by a gamma distribution function. That can be ascertained, by means of the one-sample nonparametric Kolmogorov-Smirnoff test whose p-value must be larger than 0.05, indicating that usage of the mean and variance of the gamma distribution fit as accuracy measures of the models are statistically justified within a 95% level of confidence.
Table 1 presents the three sets of 22 RM1 parameters found for Ln(III), Ce(III), and Pr(III). And Tables 2 and 3 present the mean and variance of the gamma distribution fits for the both types of unsigned mean errors for the universe of complexes, together with the p-value which is larger than 0.05 for all cases. All that indicates that the RM1 models here advanced for La(III), Ce(III), and Pr(III) are capable of predicting the geometries of the corresponding complexes in a reliable manner, and that the eventual deviations from the experiment behave as random around the correct values.
Table 1: Parameters for the RM1 model for the trications of La, Ce and Pr.*
Table 2: Means and variances of the γ distribution fits for the UME(Ln-L)s computed for the N complexes for each lanthanide trication.
Table 3: Means and Variances of the γ distribution fits for the UMEs computed for the N complexes for each lanthanide trication.
Results and Discussion
Table 4 presents unsigned mean errors for each of the specific types of distances between the lanthanide ion and its directly coordinated atoms found in the universe of complexes for La(III), both for the present RM1 model for the lanthanides and for each of the previous sparkle models. In order to facilitate interpretation of the table, the smallest error in each line is being bolded. Clearly, for dinuclear complexes, the La-La bond is more accurately predicted by Sparkle/PM3. However, its error is relatively close to the RM1 error. The same happens for La-O bonds, where Sparkle/PM3 is again the best model. However, its unsigned mean error of 0.0610Å is too close to the RM1 error of 0.0698Å. However, for all other distances, RM1 presents the smallest errors while the previous Sparkle models sometimes display huge errors as is the case of La-S bonds when the average errors of the Sparkle models is 0.4345Å, a value more than 6 times larger than the RM1 error of 0.0680Å. In Table 4, La-L refers to the unsigned mean error of all distances of all types between the central lanthanum ion and its directly coordinated other atoms, whereas L-L includes all interatomic distances between all directly coordinated atoms, and is, indirectly, a measure of the angles within the coordinated polyhedron. Clearly, RM1, with its unsigned mean error of 0.1704Å is 52% smaller than the average of the previous sparkle models, a situation similar to what happens to the next unsigned mean error, which includes all 5315 types of distances for all lanthanum complexes considered: La-L, La-La, and L,L’, when RM1 displays an error which is 56% smaller than the average error of all previous sparkle models.
Table 4: Sparkle/AM1, Sparkle/PM3, Sparkle/PM6, Sparkle/PM7, Sparkle/RM1 and RM1 unsigned mean errors for lanthanum(III) complexes.
Tables 5 and 6 show equivalent results for Ce(III) and for Pr(III) complexes. For Ce(III) complexes, RM1 is more accurate when compared to the other sparkle models with respect to all measures except for Ce-O distances. Likewise, for Pr(III) complexes, RM1 is more accurate than the other sparkle models except for Pr-O and Pr-N distances. Even in these cases, the accuracy of RM1 is close to the best accuracy available from the other sparkle models. A noteworthy case, are the distances of the three lanthanide ions and bromine, where Sparkle/PM7 displays enormous unsigned mean errors, larger than 1Å, suggesting there might be perhaps some difficulties with the parameterization of bromine in PM7.
Table 5: Sparkle/AM1, Sparkle/PM3, Sparkle/PM6, Sparkle/PM7, Sparkle/RM1 and RM1 unsigned mean errors for cerium(III) complexes.
Table 6: Sparkle/AM1, Sparkle/PM3, Sparkle/PM6, Sparkle/PM7, Sparkle/RM1 and RM1 unsigned mean errors for praseodymium(III) complexes.
Figs 1–10 present the data in Tables 3–5 in pictorial form in order to provide the user with an instant comprehension of the relative accuracies of the present RM1 model for the lanthanides (light green bar) and the previous sparkle models. Note that the Sparkle/RM1 green bar lies next, followed by the blue bar of Sparkle/PM7. Thus, the situation highlighted above on the inadequacy of Sparkle/PM7 for any of the parameterized lanthanide ions when directly bonded to bromine can be immediately detected in Fig 7. On the bright side, the good accuracy of all previous sparkle models for directly coordinated oxygen and nitrogen bonds is very clearly manifested (Figs 2 and 3).
UME(Ln-Ln)s obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated Ln-Ln interatomic distances, summed up for all complexes, for each of the lanthanides.
UME(Ln-O)s obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated Ln-O interatomic distances, summed up for all complexes, for each of the lanthanides.
UME(Ln-N)s obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated Ln-N interatomic distances, summed up for all complexes, for each of the lanthanides.
UME(Ln-C) obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated Ln-C interatomic distances, summed up for all complexes, for each of the lanthanides.
UME(Ln-S) obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated Ln-S interatomic distances, summed up for all complexes, for each of the lanthanides. There are no Ho-S distances in the universe of Ho(III) complexes considered.
UME(Ln-Cl)s obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated Ln-Cl interatomic distances, summed up for all complexes, for each of the lanthanides.
UME(Ln-Br)s obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated Ln-Br interatomic distances, summed up for all complexes, for each of the lanthanides.
UME(L-L’)s obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).The UMEs are calculated as the absolute value of the difference between the experimental and calculated interatomic distances between the coordinated atoms, L-L’, summed up for all complexes, for each of the lanthanides.
UME(Ln-L’)s obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).
UMEs obtained using the RM1 model for the lanthanides and all five versions of the Sparkle Model: Sparkle/RM1, Sparkle/PM7, Sparkle/PM6, Sparkle/PM3 and Sparkle/AM1 for all complexes of the universe set for each of the lanthanide trications: La(III), Ce(III) and Pr(III).
Finally, the raw data used to arrive at the values presented in Tables 4–6, can be found in Tables 7, 8, and 9, which show individual unsigned mean errors for each of the complexes considered, and identifies by a underlined and bolded codes, the complexes used in the small and large parameterization sets.
Table 7: Unsigned mean errors, UME(Eu-L)s and UMEs, for RM1 model for lanthanides, as compared to the respective experimental crystallographic values, obtained from the Cambridge Structural Database, for each of the 84 lanthanum(III) complexes.
Table 8: Unsigned mean errors, UME(Gd-L)s and UMEs, for RM1 model for lanthanides, as compared to the respective experimental crystallographic values, obtained from the Cambridge Structural Database, for each of the 57 cerium(III) complexes.
Table 9: Unsigned mean errors, UME(Tb-L)s and UMEs, for RM1 model for lanthanides, as compared to the respective experimental crystallographic values, obtained from the Cambridge Structural Database, [46–48] for each of the 65 praseodymium(III) complexes.
Case Study
The new RM1 model was applied to predict the structure of tetramer of praseodymium, [Pr4Cl10(OH)2(thiazole)8(H2O)2][20]. The RM1 structure was calculated using MOPAC 2009 software and keywords used were the following: RM1 (the Hamiltonian used), PRECISE, GNORM = 0.25, SCFCRT = 1.D-10 (in order to increase the SCF convergence criterion) and XYZ (the geometry optimizations were performed in cartesian coordinates).
Fig 11 shows the overlapping of the RM1 and crystallographic structures. The good match observed visually can be confirmed by the low value obtained for the root mean square deviation (RMSD) of 0.034Å, obtained via a RMSD fit and alignment.
Root mean square deviation fit and alignment of crystallographic and RM1 fully optimized structures of the tetramer of praseodymium, [Pr4Cl10(OH)2(thiazole)8(H2O)2][20].
A detailed analysis reveals that for the RM1 structure, the average bond length between the Pr3+ ions is 4.54Å whereas the average obtained from crystallographic structure is 4.58Å. The UME considering all Pr3+—L distances (where L = Pr3+, O, N and Cl) is 0.12Å. It is important to highlight that the CPU time for the full geometry optimization using the RM1 model was very fast, less than 3 minutes using a laptop core i7 with 8GB of RAM memory.
Conclusion
The overall advantage of the RM1 model for the lanthanides presented in this article is that it can perform a full geometry optimization on a complex such as the tetramer of praseodymium, [Pr_4_Cl_10_(OH)2(thiazole)8(H_2_O)2], with relative ease; something that would be exceedingly difficult for an ab initio type calculation. The same can be said of calculations on the three-dimensional 5-aminoisophtalate Pr(III) polymeric complex, which presents good gas storage capabilities [21]. Even if ab initio calculations would be later needed for specific properties that could not be obtained at useful accuracy levels by any other means, they could be carried out on RM1 optimized geometries—something that could save an enormous amount of computing time and resources.
In conclusion, the previous sparkle models seem to be very good models provided the complex has only nitrogen or oxygen directly coordinated to the lanthanide ion. However, if the complex of interest has other types of atoms directly coordinated to the lanthanide ion, then the RM1 model for the lanthanides, introduced in this article, must be the method of choice.
Supporting Information
S1 Supporting InformationInstructions on how to run the RM1 model for the lanthanides in MOPAC2012, together with sample calculations on complexes of each of the parameterized lanthanide trications: La(III), Ce(III), and Pr(III).(DOCX)Click here for additional data file.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yagyu T, Mizuno Y, Jitsukawa K. Transesterification catalyzed by lanthanum complex grafted upon hydrotalcite as a macroligand. Inorg Chim Acta. 2014; 412: 114–120.
- 2Kobayashi S, Kanai S. Superoxide Scavenging Effects of Some Novel Bis-Ligands and Their Solvated Metal Complexes Prepared by the Reaction of Ligands with Aluminum, Copper and Lanthanum Ions. Molecules. 2013; 18: 6128–6141. 10.3390/molecules 18066128 23702919 PMC 6270574 · doi ↗ · pubmed ↗
- 3Li XY, Shen QC, Zhang GB, Zhang DH, Zheng AQ, Guan F, et al Layered zinc and lanthanum hydroxide nitrates hosting chiral sulphonato-(salen)manganese(III) complex catalyzed asymmetric epoxidation reactions. Catal Commun. 2013; 41: 126–131.
- 4Huang WL, Diaconescu PL. P-4 Activation by Lanthanum and Lutetium Naphthalene Complexes Supported by a Ferrocene Diamide Ligand. Eur J Inorg Chem. 2013; 2013: 4090–4096.
- 5Rastogi RB, Maurya JL, Jaiswal V, Tiwary D. Studies on Lanthanum Complexes of 1-Aryl-2,5-Dithiohydrazodicarbonamides in Paraffin Oil as Extreme Pressure Lubrication Additives. J Tribol-T Asme. 2013; 135.
- 6Jimenez-Reyes M, Solache-Rios M. The influence of p H on the stability constants of lanthanum and europium complexes with humic acids. J Radioanal Nucl Ch. 2012; 293: 273–278.
- 7Akbar R, Baral M, Kanungo BK. Experimental and theoretical approach of photophysical properties of lanthanum(III) and erbium(III) complexes of tris(methoxymethyl)-5-oxine podant. Spectrochim Acta A. 2014; 129: 365–376.10.1016/j.saa.2014.03.04524747862 · doi ↗ · pubmed ↗
- 8Sharif S, Khan IU, Sahin O, Ahmad S, Buyukgungor O, Ali S. Synthesis and Crystal Structures of a Lanthanum(III) 1D Polymer and a Mixed-Ligand Cerium(III) Binuclear Complex Derived from Pyridine-2,6-dicaboxylic Acid. J Inorg Organomet P. 2012; 22: 1165–1173.