The Homeobox Genes: Classification, Regulation, Biological Functions, and Diseases
Maedeh Dadzadi, Shahin Ramazi, Mona Darvazi, Sepideh Yoosefi, Melika Abbasi, Shirin Farsad

TL;DR
This review summarizes the biology of homeobox genes and their roles in development, disease, and cancer, focusing on dysregulation and epigenetic mechanisms.
Contribution
An integrative overview of homeobox gene biology and their roles in both noncancerous and cancerous diseases.
Findings
Homeobox genes are master regulators in development and cell differentiation.
Dysregulation of homeobox genes is linked to various diseases, including cancer and neurodegenerative disorders.
Epigenetic mechanisms are central to homeobox gene dysregulation in lung cancer progression.
Abstract
Homeobox genes constitute a large family of transcription factors that act as master regulators involved in multiple fundamental processes such as development and cell differentiation. Consequently, these transcription factors perform diverse functions throughout human life. However, dysregulation of homeobox gene expression, through pathogenic variants or epigenetic alterations, has been increasingly associated with a wide range of human disorders. In particular, correlations between homeobox genes and various types of cancer have been documented in hundreds of studies. This review provides an integrative overview of homeobox gene biology, summarizing their classification as well as their physiological and pathological roles across noncancerous and cancerous diseases. Particular attention is given to how dysregulation of gene expression contributes to various noncancerous diseases…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
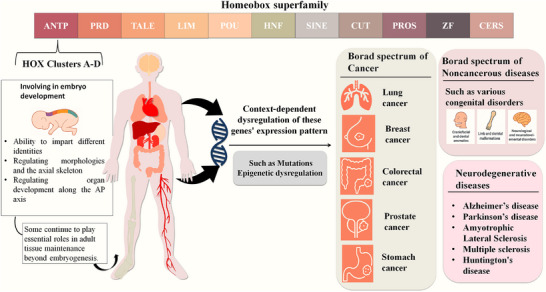 FIGURE 1
FIGURE 1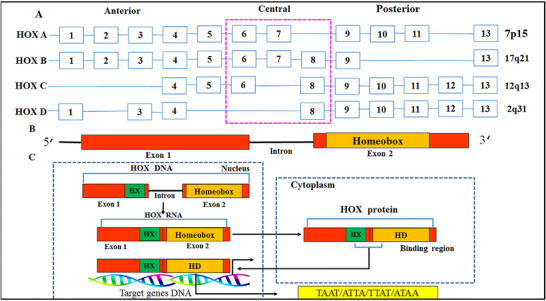 FIGURE 2
FIGURE 2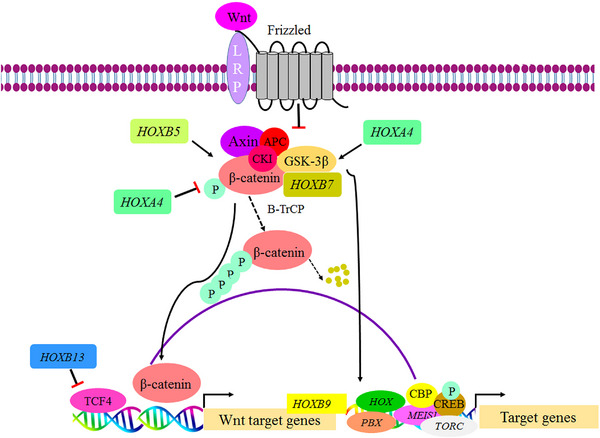 FIGURE 3
FIGURE 3| Class | Subclass | Family/representative genes | Core developmental processes (examples) | References |
|---|---|---|---|---|
| ANTP (Antennapedia) | HOXL | HOX (A–D) | Anterior–posterior patterning, and involve in the development of axial skeleton, hindbrain, and hematopoietic | [ |
| EVX ( |
| [ | ||
| MNX ( |
| [ | ||
| MEOX ( |
| [ | ||
| GBX ( |
| [ | ||
| PDX ( |
| [ | ||
| CDX ( |
| [ | ||
| NKL | BARHL ( |
| [ | |
| BARX ( |
| [ | ||
| DLX ( |
| [ | ||
| MSX ( |
| [ | ||
| NKX ( |
| [ | ||
| HMX ( |
| [ | ||
| LBX ( |
| [ | ||
| VAX ( |
| [ | ||
| VENTX |
| [ | ||
| HHEX |
| [ | ||
| NOTO |
| [ | ||
| PRD (Paired) | PAX |
|
| [ |
| PAXL | OTX ( |
| [ | |
| PITX ( |
| [ | ||
| SHOX ( |
| [ | ||
| ALX ( |
| [ | ||
| PHOX ( |
| [ | ||
| HESX ( |
| [ | ||
|
| The expression of | [ | ||
| PAXL (embryo‐restricted) |
| These genes are predominantly expressed during human preimplantation embryonic stages. | [ | |
| TALE |
| IRX ( |
| [ |
| MEIS ( |
| [ | ||
| Mohawk ( |
| [ | ||
| PBX ( |
| [ | ||
|
|
| [ | ||
| TGIF ( |
| [ | ||
| LIM |
|
|
| [ |
| POU |
|
|
| [ |
| HNF |
|
|
| [ |
| SINE |
|
|
| |
| CUT |
| CUX ( |
| [ |
| ONECUT ( |
| [ | ||
| SATB ( |
| [ | ||
| PROS |
| PROX ( |
| [ |
| ZF (Zinc‐finger homeobox) |
| ZHX ( |
| [ |
| CERS (Cerberus/other) |
|
|
| [ |
| Cluster | Genes | Chromosomal location | Embedded noncoding elements lncRNA | Explanation | References |
|---|---|---|---|---|---|
| HOXA |
| 7p15‐7p14.2 |
|
| [ |
| HOXB |
| 17q21.32 |
|
| [ |
| HOXC |
| 12q13.13 |
|
| [ |
| HOXD |
| 2q31.1 |
|
| [ |
| Biological/cellular function | Examples of homeobox gene | Homeobox‐target gene associations | Function summary | References |
|---|---|---|---|---|
| Embryogenesis and organogenesis |
|
CDX2→
|
CDX2 directs early embryonic lineage commitment by activating TE genes and maintaining
During embryogenesis, | [ |
| Migration |
|
|
| [ |
| Stem cell regulation/maintenance |
|
|
MSX2 contributes to the destabilization of pluripotency in hPSCs by repressing
| [ |
| Differentiation and lineage specification |
|
PAX5→ PBX1→ |
DLX3 promotes osteoblast differentiation by activating PDX1 is essential for pancreatic development and β‐cell identity. During postnatal β‐cell maturation, PDX1 acts together with PAX5 maintains B‐cell identity by activating lineage‐specific transcriptional networks, promoting B‐lineage genes (e.g., PBX1 contributes to lineage specification in B‐cell and megakaryocyte lineages by regulating genes (e.g., | [ |
| Hematopoiesis |
HOXA9 HOXB4 |
HOXA9→
|
| [ |
| Neurogenesis |
|
|
DLX TFs promote the differentiation of GABAergic interneurons in multiple brain regions (e.g., the olfactory bulb and cortex) through activation of LHX2 contributes to human neural differentiation by directly activating CUX2 plays a key role in spinal cord neurogenesis by regulating neural progenitor cell‐cycle dynamics and neuronal fate determination. It directly activates | [ |
| Vasculogenesis and angiogenesis—lymphangiogenesis |
|
|
PROX1 is implicated in the lymphatic development by driving BEC‐to‐LEC differentiation and activating maturation programs through regulation of key genes’ expression within this process. Therefore, PROX1 induces
| [ |
| Tissue integrity and homeostasis |
|
|
PAX5 secures B‐cell lineage commitment by activating B‐cell‐specific programs (e.g., PAX6 participates in regulating CECs differentiation and sustains corneal homeostasis through its downstream target | [ |
| Tissue regeneration and repair |
|
LHX2→
|
LHX2 regulates ectodermal morphogenesis and stem‐cell activity. It is expressed in hair‐follicle buds and, postnatally, in epithelial compartments with abundant stem cells within the follicle (e.g., the secondary hair germ), where LHX2+ cells also express specific stem‐cell markers. After skin injury, IRX1 is expressed in the oral epithelium, localizing to the gingival basal layer and stromal cells. Functionally, IRX1 enhances proliferation by upregulating | [ |
| Immune regulation and inflammatory signaling |
|
|
CDX2 is implicated in intestinal inflammation by regulating PAX5 is essential throughout B‐cell development. During early B‐cell commitment, it regulates TFs, receptors, and signaling genes by binding to promoters and enhancers, including activation of the | [ |
| Pathway | Core physiological function | Representative homeobox–pathway interactions and associated mechanisms | References |
|---|---|---|---|
| Wnt signaling | Cell fate determination, cell proliferation, tissue homeostasis, migration, survival |
| [ |
|
| Embryonic development, hematopoiesis, innate immunity, inflammatory responses |
| [ |
| Notch signaling | Cell fate determination, expansion, survival, self‐renewal, and differentiation during development, along with hematopoiesis, and T cell differentiation |
| [ |
| EGFR signaling | Cell growth, proliferation, differentiation |
| [ |
| BMP signaling | Limb patterning, morphogenesis, axial growth, bone development, skeletal maintenance |
| [ |
| RA signaling | Embryonic patterning, organogenesis, limb development, cellular differentiation |
| [ |
|
| Embryonic development, differentiation, proliferation |
| [ |
| Disease category | Disease/syndrome | Implicated homeobox gene(s) | Mutation type/variant | Molecular effect/mechanism | References |
|---|---|---|---|---|---|
| Congenital malformations → neurological and neurodevelopmental disorders | Hypomyelinating leukodystrophy (severe form) |
| Frameshift (c.606delinsTA, p.Lys202Asnfs*?), nonsense (c.565G>T, p.Glu189*), missense (c.599G>A, p.Arg200Gln) | Biallelic inactivating variants in | [ |
| HCFP3 |
| Missense mutation (c.763C>G, p.Arg255Gly; c.781C>T, p.Arg261Cys) | The identified variants are predicted to reduce the functional capacity of HOXB1. Importantly, this report describes a biallelic combination of pathogenic variants in HCFP3 for the first time. | [ | |
| CCHS |
| Nonsense mutation (c.83C>G, p.Ser28*) | The nonsense mutation occurs in exon 1 and has been identified in patients with CCHS with phenotypic variability. | [ | |
| Limb and skeletal malformations | MDUGA |
| De novo heterozygous variant (c.881T>G, p.Met294Arg) | This variant occurs within the HD, disrupting both DNA binding and protein–protein interactions, thereby impairing HOXA11 activity. These molecular defects are consistent with the forelimb and hindlimb malformations and urogenital abnormalities observed in both in vivo models and patient studies of MDUGA. | [ |
| HFGS |
| Missense mutation (c.1123G>T, p.V375F) | The mutation of | [ | |
| SPD |
| Missense mutation (c.G917T, p.R306L) | The mutation in | [ | |
| LWD/LMD |
| Missense mutations (c.508G>C, p.A170P)/(c.509C>A, p.A170D) | Missense mutations (p.A170P, p.A170D) disrupt nuclear translocation and impair the function of the SHOX protein. Particularly, the A170P mutation induces aberrant subcellular distribution of the protein. Despite being expressed throughout the growth plate, the mutant protein is associated with irregular alignment of chondrocytes. | [ | |
| Tibial hemimelia and mirror‐image polydactyly (lower‐limb malformations) |
| 35 bp deletion in exon 3 (c.765_799del) causing a frameshift mutation (p.Ala256ArgfsX303) | The 35 bp deletion introduces a frameshift and a PTC, leading to haploinsufficiency and loss of the C‐terminal OAR domain, which is essential for DNA binding. This molecular defect impairs PITX1 function, thereby contributing to a spectrum of lower‐limb malformations. | [ | |
| SHFM1 |
| Missense mutation (c.558G>T, p.Gln186His) | The mutation in | [ | |
| Congenital malformations → craniofacial and dental anomalies | Craniosynostosis (syndromic/multsuture) |
| Multiple heterozygous variants such as a missense mutation (c.161A>C, p.Asp54Ala), a nonsense mutation (c.283C>T; p.Arg95∗), and a single‐nucleotide deletion (c.52del) expected to cause a frameshift (p.Arg18Alafs∗23) | Multiple | [ |
| Congenital tooth agenesis |
| Heterozygous deletion (c.433_449del) causing frameshift (p.Trp145Leufs*24) | The frameshift mutation results in a PTC and the production of a truncated protein, ultimately causing impairment of MSX1 function and leading to tooth agenesis. | [ | |
| Craniosynostosis (Boston‐type) |
| Missense mutation (c.443C>T, p.Pro148Leu) | Missense mutation probably changes the DNA‐binding activity of MSX2. | [ | |
| TA |
| >150 variants; >50 mutation types → missense, deletion, nonsense, insertion, frameshift mutations |
| [ | |
| TDO syndrome |
| Frameshift mutation (c.604_605del، p.S202*) | The mutation impairs DLX3 transcriptional activity and downregulates | [ | |
| Frontonasal dysplasia spectrum |
| Nonsense mutation (c.793C>T, p.R265X) | Mutations in the | [ | |
| Ocular malformations | Aniridia |
| Nonsense (c.718C>T, p.Arg240*; c.299G>A, p.Trp100*), frameshift (c.112del, p.Arg38Glyfs*16) mutations | Mutations lead to the production of truncated PAX6 proteins with impaired DNA‐binding, mostly due to PTC variants that result in haploinsufficiency. These mutations are also associated with anterior lens capsule rupture in aniridia. | [ |
| Severe myopia |
| Missense mutation (c.235G>A, p.Glu79Lys) | The mutation is located within the protein's DNA‐binding domain, causing protein destabilization and impairment of its activity. These molecular defects disrupt ocular development and are associated with high myopia and retinal dystrophy. | [ | |
| Autosomal recessive microphthalmia |
| Missense (c.668G>C, p.Gly223 Ala), deletion (c.249delG, p.Leu84SerfsX57) mutations | The missense variant disrupts DNA binding by affecting the conserved CVC motif and is associated with bilateral microphthalmia. Moreover, the deletion results in a truncated VSX2 protein that lacks critical regions (the HD, CVC motif, and C‐terminal). | [ | |
| Corneal staphyloma and corneal fistula |
| Frameshift mutation (c.640_656dup, p.Gly220Profs*95) | The mutation produces a truncated PITX3 protein and has been associated with unilateral buphthalmos, corneal fistula, and corneal staphyloma. | [ | |
| Ear and auditory developmental anomalies | Bilateral nonsyndromic microtia |
| Nonsense mutations (c.637A>T (p.Lys213*; c.703C>T, p.Gln235*) | Nonsense mutations cause loss of HOXA2’s transcriptional activation, leading to reduced expression of its target gene, | [ |
| Sensorineural hearing impairment (AR and dominant) |
| Missense mutation (c.1106T>C, p.Ile369Thr) | The missense variant in the C‐terminal region disrupts hydrophobic interactions between p.Ile369 and residues in the homeodomain, thereby impairing DNA binding and LMX1A’s transcriptional activation. | [ | |
| Endocrine and metabolic anomalies | CH |
| Eight variants → missense (c.177C>A, p.Ser59Arg; c.208A>G, p.Ser70Gly; c.397C>T, p.Arg133Trp; c.397C>T p.Arg133Trp; c.1334C>T, p.Thr445Met), in‐frame indel (c.396_397delCCinsTT, p.Arg133Trp), in‐frame deletion (c.196_198delTAC, p.Tyr66del), and splicing (c.1276+1G>A) mutations |
| [ |
| CPHD/IGHD |
| Missense variants (c.559C>T, p.Pro187Ser; c.658C>A, p.Leu220Met) | The p.Pro187Ser variant destabilizes the HD, disrupting the DNA‐interaction domain, decreasing the protein stability and transcriptional activity, correlating with CPHD, whereas p.Leu220Met is likely benign or associated with IGHD. | [ | |
| CPHD |
| Missense mutations (c.300G>T, p.Gln100His; c.611G>T, p.Trp204Leu; c.251G>A, p.Arg84His) | The mutations alter the structural conformation of LHX4. In particular, the p.Trp204Leu variant disrupts the hydrophobic core within the homeodomain helix, leading to | [ | |
| CPHD |
| Synonymous missense mutation (c.219C>T, p.Ser73Ser) | A synonymous variant in | [ | |
| MODY |
| Heterozygous missense mutation (c.97C>A, p.Pro33Thr) | The mutation lies within the highly conserved transactivation region of | [ | |
| MODY9 |
| Missense mutation (c.487C>T, p.Arg163Trp) | The mutation disrupts the DNA‐binding capacity of the PAX4 protein, causing loss of regulatory control over insulin and glucagon promoters and contributing to dysregulated glucose metabolism and hyperglycemia. | [ | |
| Cardiac developmental defects and anomalies | Nonsyndromic CHD |
| Nonsense mutation (c.342C>A, p.Cys114*) | The nonsense mutation causes PTT, producing a truncated NKX2‐5 protein that impairs its normal function. | [ |
| AF |
| Missense mutations (c.309G>C, p.Gln103His; c.370G>A, p.Glu124Lys) | Mutations located within the PITX2c isoform HD impair transcriptional activity, reducing Nppa promoter activation and disrupting the repression of the | [ | |
| SND and AF |
| Missense mutation (c.98C>G, p.Pro33Arg; c.230G>A, p.Gly77Asp) | Heterozygous missense variants in | [ | |
| Kidney, urinary tract, and renal anomalies and diseases | Bilateral renal agenesis |
| Synonymous mutation (c.792G>A, p.Gln264Gln) | The synonymous variant affects mRNA splicing, causing exon 6 skipping and introducing a PTC in exon 7. This leads to structural disruption of the HD and generates a truncated protein lacking the transactivation domain. The resulting defect compromises DNA‐binding ability, leading to loss of function. | [ |
| CAKUT and bilateral kidney hypoplasia |
| Nonsense mutation (c.992C>A, p.Ser331*) | The mutation occurs at the end of exon 6, a region associated with the HD, resulting in a truncated PBX1 protein. | [ | |
| Uterine, Müllerian duct, and ovarian anomalies | Septate uterus |
| Missense mutation (c.763C>A, p.Glu255Lys) | The missense mutation occurs within the HD, impairing the DNA‐binding affinity and transactivation function of HOXA11, which impairs Müllerian duct development and leads to a septate uterus due to incomplete medial septum regression. | [ |
| CAUV |
| Missense mutation (c.G1108A, p.Ala370Thr) | The mutation lies within the C‐terminal transcriptional activation domain and alters the transcriptional activity of LHX1, disrupting regulation of its downstream target gene, particularly the GSC promoter, thereby affecting urogenital system development. | [ | |
| POI |
| Missense mutation (c.131G > T, p.Arg44Leu; c.271G > T, p.Gly91Trp; c.454G > A, p.Gly152Arg; c.1354G > A, p.Asp452Asn; c.349C > T, p.Arg117Trp) | Various | [ |
| Neurodegenerative disease | Implicated homeobox gene(s) | Expression change in disease (↑/↓) | Functional and mechanistic implications | References |
|---|---|---|---|---|
| Alzheimer's disease (AD) |
| ↑ Upregulated | Aβ activates E2F1 and its downstream effector c‐Myb, both of which synergistically transactivate PAX6. PAX6 subsequently upregulates GSK‐3β transcription, leading to increased tau phosphorylation, thereby promoting neuronal death. | [ |
|
| ↓ Downregulated | Aberrant hypermethylation spanning the HOXA gene cluster, most notably within | [ | |
|
| ↓ Downregulated | Reduced | [ | |
| Parkinson's disease (PD) |
| ↓ Downregulated |
| [ |
|
| ↓ Downregulated |
| [ | |
|
| ↓ Downregulated |
| [ | |
|
| ↑ Upregulated | Experimental studies have demonstrated that | [ | |
| Amyotrophic lateral sclerosis (ALS) |
| ↓ Downregulated | In vitro studies in human iPSC‐derived motor neurons have demonstrated that TARDBP mutations reduce both axonal | [ |
|
| ↑ Upregulated |
| [ | |
| Multiple sclerosis (MS) |
| ↑ Upregulated | MS patient‐derived myelin‐reactive Th17 cells exhibit an upregulation of | [ |
| Huntington's disease (HD) |
| ↑ Upregulated | Upregulation of | [ |
|
| ↓ Downregulated |
| [ |
| Cancer type | Homeobox gene(s) | Expression change in cancer (↑/↓) | Oncogene versus tumor suppressor | Signaling pathway/axis | Functional evidence/key findings | References |
|---|---|---|---|---|---|---|
| Breast cancer |
|
| Oncogene | Cell‐cycle progression; activation of NF‐κB signaling |
| [ |
|
| ↓ Downregulated | Tumor suppressor | Adipocytokine/PPAR signaling pathway |
| [ | |
|
| ↓ Downregulated (promoter hypermethylation; in primary breast tumors); ↑ Upregulated (in ER+ cells and estradiol‐regulated; context‐dependent) | Tumor suppressor | p53 pathway |
| [ | |
|
|
| Tumor suppressor | EMT/Wnt/β‐cadherin pathway |
| [ | |
|
|
| Oncogene | Activation of Ras–RAF–MAPK pathway, TGFB/SMAD3 signaling |
| [ | |
|
|
| Oncogene | Activation of TGF‐β pathway; EMT; enhancement the expression of angiogenic factors |
| [ | |
|
|
| Tumor suppressor | RA signaling |
| [ | |
|
|
| Tumor suppressor | miR‐10b–HOXD10–RhoC metastasis axis |
| [ | |
| Colorectal cancer |
|
| Oncogene | RA signaling | Dysregulated | [ |
|
|
| Oncogene | IGF1R/PI3K/AKT/HIF1α signaling pathway | IGF1 induces | [ | |
|
|
| Oncogene | CXCR4–ERK1/2–ETS1 signaling pathway | CXCL12 upregulates | [ | |
|
|
| Oncogene | PI3K/AKT and MAPK/ERK signaling |
| [ | |
|
|
| Oncogene | STAT3 pathway |
| [ | |
| Prostate cancer |
|
| Oncogene | ERK1/2 and AKT signaling |
| [ |
|
|
| Oncogene | TGFβ/SMAD signaling |
| [ | |
|
|
| Oncogene | HOXA13–SLC7A11/SLC3A2 axis |
| [ | |
|
|
| Oncogene | p21–RB–E2F signaling pathway |
| [ | |
|
|
| Oncogene | Notch and Wnt pathways |
| [ | |
|
|
| Tumor suppressor | NF‐κB signaling, miR‐196b‐5p–HOXC8–NF‐κB axis |
| [ | |
|
|
| Oncogene | β‐catenin/TCF4 signaling |
| [ | |
| Stomach (gastric) cancer |
|
| Oncogene | JAK1/STAT3 signaling |
| [ |
|
|
| Oncogene | FN1‐mediated FAK/Src axis, HOXA13–FN1–FAK/Src axis; Akt/Erk1/2 activation (PI3K–Akt/MAPK, mTOR signaling) |
| [ | |
|
|
| Oncogene | EGFR‐dependent pathway |
| [ | |
|
|
| Oncogene | OPN‐dependent AKT/ERK signaling pathway | High | [ | |
|
|
| Oncogene | Wnt/β‐catenin signaling pathway | High | [ | |
|
|
| Oncogene | HOXD9–RUFY3 axis | High expression of | [ | |
|
|
| Oncogene | Reg IV/SOX9 signaling |
| [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpigenetics and DNA Methylation · Microtubule and mitosis dynamics · Genetics and Neurodevelopmental Disorders
Introduction
1
It has been four decades since researchers first identified a short but pivotal DNA sequence, later termed the homeobox. Across the animal kingdom, homeobox genes give rise to a broad class of transcription factors (TFs) that play central roles in developmental pathways across tissues and stages of life. The initial discovery of these genes was made in Drosophila melanogaster, where they were found to regulate homeotic transformations, defining the identity of body segments [1, 2, 3, 4]. Since then, hundreds of homeobox‐containing genes have been identified in a wide range of species. Homeobox genes constitute one of the largest TFs superfamilies in the human genome, with over 200 members characterized to date. Among vertebrates, the HOX gene family is one of the best‐studied subsets of the homeobox gene superfamily [5, 6]. This homeobox genes have conserved region, approximately 180 base pairs long, that encodes a ∼60‐amino‐acid DNA‐binding domain known as the homeodomain (HD). Therefore, a defining feature of most homeobox genes is their conserved HD, which enables sequence‐specific DNA binding and precise regulation of gene expression essential for embryogenesis and cell differentiation. Accordingly, numerous studies have established that TFs encoded by homeobox genes serve as master regulators of developmental processes, functioning at the top of gene regulatory hierarchies. Through this hierarchical control, they initiate broad genetic cascades that govern the expression of numerous downstream genes (e.g., effector genes), eventually contributing to the formation of tissue and organ [3, 7, 8, 9]. In particular, HOX genes encode evolutionarily conserved TFs that are indispensable for the proper development of bilaterian body plans. Notably, more posterior body regions typically express a larger complement of HOX genes than do anterior regions. This spatio‐temporal collinearity of HOX genes expression highlights how genomic regulation underpins their role as master regulators in developmental patterning [7]. Importantly, some homeobox genes, chiefly those within the HOX clusters, remain transcriptionally active well beyond embryogenesis. Their region‐specific expression is stably preserved in adult cell types, such as mesenchymal stem cells (SCs) and fibroblasts, where it forms an epigenetically maintained “positional memory” that preserves in embryonic axial information and continues to shape tissue physiology across the lifespan [10, 11, 12, 13]. High‐resolution transcriptomic profiling of fibroblasts isolated from anatomically precise sites confirms that these cells retain a distinctive “HOX code” (the position specific‐pattern of HOX genes’ expression), mirroring the embryonic pattern of these genes’ expression well into adulthood [11, 13, 14].
Despite their well‐established developmental roles, growing evidence indicates that homeobox gene dysregulation underlies a wide spectrum of human pathologies, encompassing both noncancerous diseases and multiple cancer types. Alterations in homeobox gene dosage, structure, or transcriptional regulation have also been implicated in diverse noncancerous disorders, ranging from congenital malformations and metabolic syndromes to neurodegenerative conditions, as well as in cancer, where their dysregulation contributes directly to tumorigenesis [15, 16]. Decades of research have demonstrated that aberrant expression of specific homeobox genes promotes tumor invasion, metastasis, and poor prognosis, frequently correlating with adverse clinicopathological features and poorer survival across tumor types [3, 17, 18]. Notably, accumulating studies reveal that mechanisms such as aberrant DNA methylation, overexpression, and regulation of expression by long noncoding RNAs (lncRNAs)/microRNAs (miRNAs) play critical roles in modulating homeobox gene activity and, consequently, influence oncogenic signaling pathways. Evidence increasingly supports that homeobox function is highly context dependent, with the same gene exerting opposing effects depending on the cell type, tissue microenvironment, and interacting molecular networks [19].
Building on these insights, the present review aims to provide a comprehensive and integrative synthesis of homeobox gene biology across physiological and pathological contexts. Specifically, this review (i) systematically reviews homeobox gene expression and function in normal physiology, emphasizing their hierarchical control of differentiation and tissue homeostasis; (ii) delineates the genetic and epigenetic mechanisms through which homeobox dysregulation contributes to disease pathogenesis across congenital, systemic, and neurodegenerative disorders; (iii) reviews the role of homeobox genes in malignancies, with particular focus on the five highest incidence of human cancers lung, breast, colorectum, prostate, and stomach with an in‐depth discussion of lung cancer, where homeobox deregulation is especially prominent; and (iv) explores unifying mechanistic themes linking homeobox gene regulation, tissue‐specific function, and disease etiology.
Homeobox Genes
2
Classification of Homeobox Genes
2.1
Homeobox genes have been classified into distinct categories by generally examining both the evolutionary relationships of their homeodomain amino acid sequences and the association of the homeodomain with additional protein domains [20]. Another important criterion for classification of homeobox genes into specific families involves assessing their sequence similarity, as genes with higher degrees of homology are typically placed within the same family [21, 22]. In the human genome, homeobox genes are broadly categorized into 11 core classes, each further subdivided into subclasses comprising multiple gene families (Figure 1 and Table 1). Within these families, individual genes share conserved sequences and structural domains that reflect their functional and evolutionary relationships. It should be noted that members of the Antennapedia (ANTP) class are restricted exclusively to multicellular animals (Metazoa) and have not been identified in unicellular eukaryotes, even though homeobox genes do exist in the latter. In humans, the ANTP class constitutes the most expansive and functionally versatile category within the homeobox gene superfamily. It is subdivided into two primary subclasses: the HOX‐like (HOXL) genes, most notably the HOX genes, and the NK‐like (NKL) genes, which are classified as non‐HOX. Beyond the well‐known HOX clusters, the HOXL subclass also includes other key members, such as the ParaHox genes (e.g., caudal‐type homeobox (CDX), and genomic screened homeobox (GSX)) and a group of extended HOX genes [7, 20, 23, 24]. Among these, the ParaHox genes stand out not only for their developmental significance but also for their evolutionary origin. The term “ParaHox cluster” was introduced to reflect its sequence similarity and evolutionary correspondence to specific paralogous groups within the HOX gene clusters. Beyond their structural complexity, the evolutionary trajectories of HOX and ParaHox genes offer key insights into the ancient origins of the ANTP class. Comparative genomic analyses suggest that the remarkable diversification of this class arose through early tandem gene duplication events. These duplications are thought to have produced distinct lineages, including the NK subclass and a putative ProtoHox cluster. The latter is proposed to have undergone further duplication prior to the evolutionary split between deuterostomes and protostomes, ultimately giving rise to both the HOX and ParaHox clusters. This functional compartmentalization reflects the evolutionary refinement of ANTP‐derived clusters toward distinct developmental trajectories [23, 25].
Overview of the homeobox gene superfamily and its pathophysiological roles. The homeobox superfamily comprises several major classes, including ANTP, PRD, TALE, LIM, POU, and others. Among these, the HOX clusters (A–D) play critical roles in embryonic development specifically in conferring cellular identity, regulating morphogenesis, and guiding axial patterning. Beyond development, HOX genes remain active in adult tissue homeostasis. Dysregulation of their expression, whether through mutation or epigenetic alteration, is implicated in a broad spectrum of diseases, including multiple cancers and neurodegenerative disorders.
Among the major homeobox gene classes, the Paired (PRD) class represents the second most extensive group after ANTP in the human genome. Genes in this class are critically involved in regulating developmental processes, particularly in early embryogenesis. This class is generally subdivided into two distinct subclasses: the Paired‐box (PAX) (paired‐type homeobox) subclass, which includes genes directly related to the PAX family, and the PAX‐like (PAXL) subclass, encompassing genes that are structurally or functionally divergent from PAX but still share homology within the paired‐like domain. The PAXL subclass comprises a diverse set of approximately 28 gene families and also consists of pseudogenes. This subclass is also commonly referred to in the literature as PRD‐like homeobox genes. Some notable examples of key gene families within this subclass include Aristaless‐related homeobox (ALX), Arginine‐fifty homeobox (ARGFX), Paired‐like homeobox (PHOX) genes, Cytoplasmic polyadenylated homeobox (CPHX), Orthodenticle homeobox (OTX), Divergent paired‐related homeobox (DPRX), short stature homeobox (SHOX), Paired‐like homeodomain TFs (PITX), Double homeobox (DUX), and Tetra‐peptide repeat homeobox (TPRX) [23, 53]. In humans, several members of the PRD‐like subclass are preferentially expressed in the germline and during the earliest stages of development, including in oocytes and zygotes, highlighting their potential role in preimplantation development. For instance, transcripts of genes such as DPRX, NOBOX oogenesis homeobox (NOBOX), and ARGFX have been detected [20, 53].
Within the PRD class, genes in the PAX subclass are notable for encoding a distinct group of TFs marked by the inclusion of a highly conserved DNA‐binding region known as the paired domain (PD). This domain, composed of approximately 128 amino acids, is structurally unique among homeobox proteins due to the inclusion of a paired‐box motif a signature feature that has remained remarkably conserved across evolutionary lineages and sets PAX proteins apart from other homeobox families. The strong evolutionary conservation in the PD domain underscores its critical function in mediating the DNA‐binding activity of PAX proteins during gene regulation. Owing to this conservation, PAX proteins frequently exhibit similar DNA‐binding motifs; nonetheless, each member of the family achieves functional specificity by regulating a distinct repertoire of target genes. Notably, the expression of PAX genes has been documented across a wide range of organisms, including both vertebrates and invertebrates. Building on their structural features and target specificity, these proteins play essential roles in developmental processes. By directing lineage commitment and promoting cell differentiation, they ensure the coordinated formation of tissues and organs, while also maintaining cell identity to safeguard the long‐term integrity of tissue function. The mammalian PAX family is composed of nine genes (PAX1 to PAX9), which distributed across eight distinct chromosomes, underscoring the evolutionary antiquity of this subclass. These genes are categorized into four structural subgroups, differentiated by the presence of additional conserved domains beyond the PD. Specifically, classification hinges on whether a PAX protein includes a HD, complete or partial, and/or an octapeptide linker. The octapeptide linker contributes to transcriptional repression by interacting with cofactors and other regulatory proteins, thereby attenuating the expression of downstream target genes. Furthermore, each of the nine PAX proteins harbors a C‐terminal transactivation domain. Collectively, this modular organization of these domains has been evolutionarily conserved across higher vertebrates. Group I members, including PAX1 and PAX9, are devoid of the HD and rely solely on the PD for DNA binding, although they also carry an octapeptide linker. Importantly, although the HD domain is dispensable for the core activity of PAX proteins, whether complete or partial it still contributes to the regulation of specific target genes. By cooperating with the PD, the HD further enhances the DNA‐binding capacity of these proteins. Group II proteins, including PAX2, PAX5, and PAX8, possess a partial HD together with the octapeptide. In this configuration, the domain exhibits diminished DNA‐binding capacity. Group III members, including PAX3 and PAX7, contain a complete HD together with the octapeptide, whereas Group IV members, including PAX4 and PAX6, harbor a full HD but lack the octapeptide. This intact domain incorporates a helix–turn–helix configuration that promotes dimer formation, a property that enhances both the strength and stability of DNA binding [68].
While the 11‐class classification provides a comprehensive evolutionary framework, an alternative functional scheme classifies mammalian homeobox genes into two principal categories based on chromosomal organization and structural features. This approach offers additional insights into the regulatory architecture and developmental roles of these genes. The first category, known as Class I, includes the HOX genes, which belong to the HOXL subclass of the ANTP class. These genes are organized into four tightly linked clusters HOXA through HOXD located on separate chromosomes (Figure 2 and Table 2) [20, 69, 70]. In total, 39 HOX genes contribute to anterior–posterior (AP) axis patterning and are grouped into 13 paralogous, numbered HOX1 to HOX13, based on their sequence homology and positional order within clusters, reflecting evolutionary conservation across the clusters [20, 71]. During vertebrate embryogenesis, HOX gene clusters exhibit the principle of collinearity, whereby their chromosomal arrangement parallels both the spatial domains and the temporal sequence of gene activation. In vertebrates, this synchronization extends across the entire clusters a phenomenon often referred to as whole‐cluster spatio‐temporal collinearity (WSTC) [72, 73]. Specifically, spatial collinearity aligns gene position from 3′ to 5′ with expression domains along the AP axis; temporal collinearity ensures that genes are activated sequentially in developmental time following the same order. A third dimension, quantitative collinearity, has been documented in limb formation, wherein genes located more posteriorly within a cluster tend to be expressed at higher levels. These tightly coordinated mechanisms underscore the evolutionary significance of HOX clustering, serving as a robust framework for establishing precise body plan architecture in vertebrates [20]. The second category, referred to as Class II or non‐HOX/ParaHox genes, is composed of a wide range of gene families (e.g., PAX within the PRD class). Unlike the clustered HOX genes of Class I, members of this group are dispersed across different chromosomal locations, reflecting a higher degree of structural variability [20, 69].
(A) Schematic structure of HOX genes. Structurally, HOX genes consist of two exons and a single intron. The second exon encodes a homo domine DNA‐binding domain consisting of 60 amino acids (known as HD). This HD is crucial for the transcriptional regulation of target genes. (B) Transcriptional role of HOX proteins. HOX proteins mediate gene expression by binding to the promoter regions of their target genes, thereby either activating or repressing their transcription. The HD and hexapeptide (HX) motifs present within the HOX proteins are integral for their regulatory activity [74].
The Physiological Function of Homeobox Genes
2.2
As outlined above, homeobox genes constitute a large family of TFs defined by a conserved homeodomain DNA‐binding motif, and they are widely recognized as master regulators of developmental and cellular processes [74]. Their roles are conserved across a broad range of organisms, from invertebrates to vertebrates, including mammals and humans. The expression of these genes is under precisely regulated in space and time, ensuring that embryonic programs unfold in an orderly manner. This remarkable evolutionary conservation underscores their essential contribution to body plan establishment and organogenesis. Importantly, their role is not confined to embryogenesis. While they orchestrate body‐axis formation and tissue patterning in early development, accumulating evidence shows that certain homeobox genes remain active in postnatal and adult tissues, where they continue to participate in diverse cellular processes such as regulating stem cell (SC) function, maintaining tissue integrity, and preserving long‐term homeostasis. Collectively, these TFs also modulate a broad spectrum of fundamental biological activities, including proliferation, lineage specification, differentiation, hematopoiesis, programmed cell death, migration, angiogenesis, tissue repair, and cell‐cycle regulation, although the relative contribution of individual families to each process can vary considerably [87, 88].
Moreover, individual homeodomain families execute specialized biological roles that reflect their structural and regulatory diversity. This diversity stems not only from variations in DNA‐binding specificity, but also from differences in the presence of associated domains, interactions with cofactors, and the broader transcriptional context in which these proteins function. Such combinatorial mechanisms result in distinct gene regulatory programs across different families [89]. For instance, LHX genes represent a crucial subfamily within the homeobox gene family. These genes encode LIM‐homeodomain (LIM‐HD) proteins, which feature two LIM domains in their N‐termini and a centrally located HD. The HD is responsible for binding specific DNA elements in target genes. Extensive research has demonstrated that these genes encode TFs that regulate gene expression during pivotal developmental processes. The LIM domains enable protein–protein interactions, while the HD directly binds to DNA, thereby influencing the transcription of target genes [90, 91]. Another well‐defined example is the CDX family. The CDX TFs are characterized by the presence of a highly conserved homeobox DNA‐binding domain, which allows them to bind to specific regulatory regions and consequently activate or repress the transcription of their target HOX genes. CDX genes’ family encodes a group of TFs that play a critical role in the regulation of HOX genes’ expression during embryonic development. The CDX proteins act as crucial upstream regulators, integrating signals from key signaling pathways such as retinoic acid and Wnt to modulate the activity of HOX genes promoter. This regulatory function of the CDX factors is essential for the proper patterning of the AP body axis. Through this mechanism, the CDX family members, which in humans include CDX1, CDX2, and CDX3, orchestrate the spatiotemporal expression of the HOX genes, ensuring the coordinated development of the body plan along the AP axis [92, 93].
During embryogenesis, certain homeobox genes display prominent, stage‐specific expression patterns, where they act as central regulators of key developmental events. For instance, PRD‐like homeobox genes are selectively expressed during the earliest phases of human embryonic development. Within this group, ARGFX and DPRX have been reported to function as a transcriptional activator and repressor, respectively, and both are directly implicated in the regulation of embryonic genome activation (EGA) during preimplantation development [53, 94]. LEUTX, another example of an early‐expressed gene, encodes a DNA‐binding TF. The complete homeodomain isoform of LEUTX has also been shown to be fully competent in initiating the expression of numerous genes associated with EGA [53, 95]. Additional examples of functional specialization can be found in other homeobox families. Members of the MSX family, for instance, act as key regulators of epithelial–mesenchymal transition (EMT). EMT is a well‐known biological process associated with embryonic development and morphogenesis, playing a vital role in tissue remodeling during organogenesis and tissue regeneration. Conversely, families such as PAX and DLX are more prominently involved in driving tissue‐specific lineage commitment and differentiation. These genes help define developmental trajectories by modulating transcriptional networks in a context‐dependent manner [96, 97, 98]. DLX TFs play essential functions throughout vertebrate embryonic development and act as central regulators of early skeletal morphogenesis and bone homeostasis, controlling key processes such as chondrogenesis and osteogenesis. During early development, they are particularly important for craniofacial formation, whereas HOX genes primarily govern axial and appendicular skeletal patterning. In addition to their developmental roles, DLX genes also participate in adult bone remodeling. In postnatal bone, DLX activity remains essential for skeletal integrity through interactions with osteogenic regulators such as RUNX2 and OSX/SP7. As development progresses, members such as DLX3 and DLX6 expand their functions beyond embryogenesis. For example, sustained expression of DLX3 in chondrocytes has been documented, underscoring its critical role in cartilage development [96, 99].
Beyond embryogenesis, certain homeobox genes, particularly those within the HOX clusters, retain transcriptional activity in certain adult cell types, particularly in adult SCs like mesenchymal stromal cells (MSCs) [11]. For instance, within the homeobox superfamily, the HOX clusters have been identified as a pivotal role in hematopoiesis and hematopoietic SC (HSC) differentiation. HOXA9 is one of the most abundantly expressed TFs in HSCs, where it acts as a key regulator of stemness and differentiation through the regulation of a broad set of target genes (e.g., CDK6, Erg, Foxp1, Gfi1, SOX4, and Lmo2). Notably, many of these genes are also regulated by other cofactors such as meis homeobox 1 (MEIS1) and HOXB4, indicating the existence of a cooperative regulatory network that underpins HSC maintenance and differentiation. Together, HOXA9 and HOXB4 play a particularly prominent role in sustaining stemness within HSC populations. During normal hematopoiesis, the gradual decline in these genes’ expression serves as a molecular signal that drives SCs out of quiescence and promotes their progression toward differentiation to lymphoid and myeloid. Consistent with this, genes in the HOXA cluster including HOXA5, HOXA7, and HOXA9 are progressively downregulated during the differentiation of human pluripotent SCs (hPSCs). Experimental evidence indicates that elevated HOXA9 levels accelerate the hematopoietic differentiation of human embryonic SCs (hESCs), driving hemogenic endothelial precursors toward primitive and CD45^+^ blood cell lineages. In contrast, HOXA9 expression undergoes a marked decline as HSCs progress to fully differentiated blood cells. Therefore, while downregulation of HOXA9 accompanies normal differentiation, its sustained overexpression has been directly linked to leukemogenesis, with elevated HOXA9 expression consistently detected across multiple acute myeloid leukemia (AML) subtypes [100, 101, 102]. In addition, studies indicate that PBX1 functions both in early human development and in stage‐ and tissue‐specific roles later in life, with alternative splicing generating isoforms such as PBX1a in the adult brain, PBX1b in embryonic tissues, and PBX1d in CD4^+^ T cells. Its sustained expression across selected organs and immune cell subsets highlights a regulatory versatility that extends into adulthood [57], providing a conceptual bridge to other homeobox genes with persistent activity beyond embryogenesis. As a result of this functional diversity, these proteins influence a wide array of downstream targets with critical cellular roles, for example, regulators of the cell cycle and apoptosis (e.g., p53) and angiogenic factors (e.g., vascular endothelial growth factor A [VEGFA]) [21]. A broader example of representative biological processes governed by homeobox genes, alongside examples of their gene targets within various biological and cellular function, is provided in Table 3. It should be considered that some homeobox genes exhibit context‐dependent functions, extending their influence beyond a single biological process. Their regulatory effects can span multiple layers of tissue physiology, for instance, simultaneously linking developmental, regenerative, and immune pathways, as exemplified by HOXB5 and HOXA3 [103, 104]. In both in vivo and in vitro experiments, HOXB5 has been shown not only to promote revascularization and perfusion during ischemic injury by stimulating endothelial and vascular responses, but also to induce proinflammatory mediators such as MCP‐1 and IL‐6 [103]. A further example is provided by HOXA3, which has been implicated in regulating both regenerative and immune‐related aspects of wound repair. Both in vivo and in vitro studies have demonstrated that during wound healing, particularly in diabetic ulcers, HOXA3 expression facilitates skin repair by promoting migration of keratinocyte and enhancing angiogenesis, while simultaneously reducing inflammatory mediators. Maintained HOXA3 expression decreases leukocyte accumulation in these wounds and directly supports macrophage maturation. Functionally, HOXA3 suppresses M1 macrophage polarization by attenuating proinflammatory signaling, while driving an M2 phenotype through pathways such as Signal Transducer and Activator of Transcription 6 (STAT6) activation during wound healing. In parallel, HOXA3 expression contributes to wound healing by stimulating keratinocyte migration, maintaining epidermal integrity, and particularly promoting angiogenesis through upregulation of downstream effectors such as matrix metalloproteinase‐14 (MMP14), while downregulating inflammatory mediators including C–C motif ligand 2) CCL2(and CXCL12 [104, 105]. Collectively, these functions illustrate how a single homeobox gene can exert multiple, context‐dependent activities, underscoring the multidimensional and pleiotropic nature of homeobox gene activity.
Among the biological functions of homeobox TFs, notable implications have also been identified in cancer, primarily through the regulation of core processes such as apoptosis, proliferation, cell migration, and angiogenesis. By governing these processes, they assume a central role in cancer biology. Importantly, in a context‐dependent manner, these factors can exert dual effects in cancer acting as oncogenic drivers by promoting tumor growth, invasion, and angiogenesis, or functioning as tumor suppressors by various processes such as apoptosis [3, 133]. For instance, the empty spiracles homeobox 1 (EMX1) and EMX2 genes exhibit tumor‐suppressor activity by inhibiting the expression of key stemness‐related genes such as *SRY‐*box TF 2 (SOX2) and MYC, thereby reducing cancer SC (CSC) populations in sarcomas. In vivo studies have further demonstrated that loss of EMX expression is directly associated with the enhancement of tumor aggressiveness in these cancers [134]. Together, this dual context‐specific functionality contributes to a highly dynamic and intricate regulatory network. Furthermore, as mentioned, the interaction of homeodomain proteins is not restricted to embryonic stages but also occurs in later developmental and adult contexts. This underscores a deeper fundamental interface of organogenesis and tumorigenesis, mediated by homeodomains that act during embryonic development as well as those involved in cellular differentiation. Such regulatory plasticity exemplifies how homeobox TFs can function differently depending on the biological context [3, 21].
One of the most striking examples of context‐dependent functions among homeobox TFs is angiogenesis, a process in which these genes play pivotal roles across physiological and pathological contexts. During development and tissue repair, homeobox genes influence endothelial differentiation, sprouting, and vascular remodeling, contributing to placental vascularization and wound healing. Dysregulation of homeobox expression is also implicated in cancer, where aberrant angiogenesis facilitates tumor growth, metastasis, and invasion, ultimately impacting prognosis. Consequently, neovascularization represents a core biological process governed by homeobox genes Several studies have documented a close relationship between neovascularization and homeobox proteins, which modulate this process across both embryonic development and pathological conditions through dual mechanisms [133, 135, 136]. In this regulatory landscape, proangiogenic actions are achieved through upregulation of factors such as VEGFA, FGFs, TGFs, and angiopoietins, driving endothelial activation and sprouting. Conversely, antiangiogenic effects emerge via suppression of proangiogenic signals and stabilization of endothelial quiescence, exemplified by restraining components like VEGFR2 and by promoting antiangiogenic gene programs. Specific HOX members exhibit distinct roles: HOXB5, HOXA3, and HOXD3 contribute to angiogenic onset and endothelial lineage commitment, whereas HOXA5 and HOXD10 can reinforce vascular stabilization and quiescence. IRX3 has also been identified as proangiogenic, enhancing endothelial migration and influencing tip‐cell fate through VEGF–Notch integration. In cancer, the balance tilts toward proangiogenic outcomes when these genes are overexpressed, supporting tumor vascularization and progression. Notably, HOXB5, HOXB7, HOXB9, HOXC10, and DLX4 have been linked to upregulated angiogenic signaling in diverse cancers, with mechanistic examples including ANGPT2 induction and activation of extracellular signal‐regulated kinase (ERK)/AKT pathways, as well as STAT1‐mediated iNOS induction [133, 137, 138]. Angiogenesis includes a cascade of events in which endothelial cells (ECs) play a central role. During vascularization, ECs secrete various components and proteins, such as growth factors. Importantly, homeobox genes exert diverse effects on ECs, particularly during differentiation and maturation [133, 139, 140]. For example, HOXB5 has been shown to regulate angioblast differentiation into mature ECs and to upregulate ANGPT2, a key angiopoietin required for ECs sprouting [125, 140]. In addition, HOXA3 together with HOXD3 display proangiogenic functions and are expressed at early stages, correlating with endothelial invasion and sprouting, and have been reported to contribute to endometrial cancer (EC) lineage commitment [125, 140]. IRX3 has also been identified as a proangiogenic factor, directly promoting EC migration and influencing tip‐cell fate through integration of VEGF–Notch signaling [141]. By contrast, HOXA5 exerts an antiangiogenic effect by suppressing cell migration, upregulating antiangiogenic genes while downregulating proangiogenic ones (e.g., VEGFR2), thereby repressing the angiogenic process. Along with HOXD10, it is also associated with endothelial quiescence, contributing to the stabilization of the mature EC phenotype [133, 142, 143]. The relationship between homeobox gene expression and angiogenesis in disease, especially cancer, is highly complex and multifactorial. In various types of cancer, dysregulation of specific homeobox genes frequently contributes to aberrant angiogenesis, in part through their regulation of key proangiogenic related factors such as VEGF and FGFs. Therefore, overexpression of homeobox genes with proangiogenic properties leads to enhancement of tumor angiogenesis, thereby supporting tumorigenesis and its progression [133, 137]. Genes such as HOXB5, HOXB7, HOXB9, HOXC10, and DLX4 have been reported to be upregulated in different cancers, promoting vascular expansion and tumor growth [137, 144, 145, 146]. For instance, HOXB5 overexpression in esophageal cancer leads to enhance the level of ANGPT2 expression, which in turn activates the extracellular signal‐regulated kinase (ERK/AKT) pathway, thereby enhancing angiogenesis and promoting both proliferation and metastasis [144]. Similarly, DLX4 overexpression has been detected in ovarian cancer, where it promotes angiogenesis through a STAT1‐dependent mechanism that induces iNOS expression. Increased the level of iNOS is strongly linked to augmentation of tumor angiogenesis [144]. The subsequent sections will elaborate in greater detail on how alterations in the expression and function of homeobox genes contribute to cancer development and progression.
The Function of Genes Within HOX Clusters
2.2.1
Among homeobox gene families, HOX genes hold particular significance, not only because of their indispensable developmental roles but also due to their clustered genomic organization and the extensive body of research focused on them [147, 148]. As pivotal transcriptional regulators, HOX genes play a vital role in the regulation of patterning and cell fate throughout embryogenesis [3]. (Some examples are summarized in Table 3.) From a biological perspective, the function of the HOX proteins encompass numerous aspects of embryonic development, cellular physiology, and tissue homeostasis [149]. Particularly during embryonic development, the expression of HOX genes, along with intricate gene networks, determines the temporal and spatial development of human limbs [82, 150]. Within this developmental framework, HOX TFs are indispensable for the AP axis patterning across bilaterian animals through regulation of downstream target genes. Beyond the axis patterning, HOX proteins also participate in regulating diverse organogenesis, specification of individual cell types, and the coordination of morphogenetic programs throughout development. Notably, by modulating the expression of their downstream targets, HOX TFs ensure the establishment and diversification of morphological patterns along the AP axis. Such tightly regulated activity has been observed across diverse embryonic and adult tissues, highlighting the central role of HOX TFs in coordinating regional identity, developmental processes, and cell‐type specification throughout the body [11, 151, 152, 153].
As mentioned earlier, the activities of HOX genes are notably not limited to embryonic development but continue to influence key cellular functions in adult tissues throughout the human lifespan [11, 152, 153]. As mentioned earlier, these TFs contribute to the maintenance and differentiation of both embryonic and adult SCs, helping direct lineage‐specific outcomes in processes such as adipogenesis and neurogenesis. SCs are a type of cells that possess an extraordinary ability to both renew themselves and differentiate into various cell types across multiple lineages and also generate diverse cell types. These genes further support the differentiation of tissue‐specific SCs into various specialized cell types required for specific lineages within the adult body. Among their known roles, HOX genes are crucial in directing SCs differentiation across various biological pathways, such as the development of adipose tissue and neurogenesis [11, 154, 155]. A unique feature of HOX genes is their ability to reprogram the entire body regions’ identity, a process known as homeosis [11, 152, 153]. Furthermore, HOX proteins also participate in nontranscriptional activities, influencing the regulation of critical cellular processes such as DNA replication and repair, mRNA translation, and protein degradation [149]. Importantly, HOX genes extend their influence beyond developmental programs to regulate key processes in adult tissues. In cancer, their expression is often dysregulated, with specific genes being either upregulated or downregulated depending on the biological context. Such alterations have been documented across a wide spectrum of malignancies, thereby underscoring the importance of these TFs in both physiological development and pathological conditions [3, 133].
Structurally, these genes are typically divided into two exons and one intron, with the homeobox sequence located in the second exon. The functions of the HOX are reliant on a conserved 60‐amino acid HD and a hexapeptide motif (HX) that have been evolutionarily preserved. The HD is predominantly involved in binding to DNA at specific recognition sites, ultimately resulting in the transcriptional regulation of target genes through either activation or inhibition [74]. The HD acts as a DNA‐binding domain, with a preference for recognizing a specific TA‐rich core DNA sequence, such as TAAT or TTAT, among other functions [149]. Consequently, HOX genes perform key TF functions that have been preserved throughout evolution and are found in all bilaterian animals [149]. The unique structural arrangement of HOX proteins, consisting of three helices, is crucial for their DNA‐binding function. The helix–turn–helix DNA‐binding motif allows them to recognize –TAAT– motifs and facilitate binding. Both helices 2 and 3 adopt the helix–turn–helix configuration, which is a defining feature of TF binding. The HD primarily attaches to DNA by engaging helix 3, also known as the recognition helix, within the major groove of the DNA. This fundamental role of HD proteins lies in their ability to modulate the expression of various genes [156]. Interestingly, HOX proteins demonstrate nearly identical affinity in binding to these sites, underscoring the HD as a defining aspect of HOX transcriptional regulation [7, 151, 157, 158]. In addition to the HD, HOX proteins also contain an acidic C‐terminal tail (C‐tail) that allows them to bind with the three‐amino acid loop extension (TALE) and serve as a cofactor. Furthermore, a HX motif has been detected in some HOX proteins, which includes a strongly preserved YPWM motif and a flexible linker region. This motif is essential in determining TALE cofactor selectivity, which in turn regulates specific HOX target genes. The binding of HOX cofactors improves the stability of HOX‐DNA interactions [82, 150, 157].
The Involvement of Homeobox Genes in Various Signaling Pathways Under Physiological Condition
2.2.2
Across the human lifespan, a conserved set of signaling pathways, particularly Wnt/β‐catenin, Notch, Hedgehog (Hh), TGF‐β, bone morphogenetic protein (BMP), mitogen‐activated protein kinase (MAPK)/ERK, PI3K/AKT, Janus tyrosine kinase (JAK)/STAT, and NF‐κB, are frequently implicated from embryogenesis through adulthood to execute context‐specific biological processes such as tissue homeostasis, repair, and immune response regulation. Therefore, any dysregulation of these signaling pathways is intimately associated with various pathological conditions, particularly cancer [11, 159, 160, 161, 162]. Within this integrated network, homeobox TFs participate by interfacing with major pathways (e.g., WNT, Notch, and NF‐κB) to coordinate diverse biological processes such as development, cell differentiation, and immune responses. For instance, HOX clusters have been implicated in the regulation of pathways such as WNT and MAPK [11, 87]. These interactions influence both physiological stages, from embryogenesis to adulthood, as well as various pathological conditions, most notably cancer. Therefore, homeobox TFs typically serve dual roles, as downstream targets and as upstream regulators of these pathways, depending on cellular and developmental context. For example, MSX1 and MSX2 serve as downstream mediators of BMP2 signaling during woman endometrial decidualization. Moreover, during cardiogenesis, NKX2‐5 enhances canonical WNT signaling by upregulating R‐spondin3, thereby promoting this pathway activation [11, 87, 163, 164]. Other representative examples of these pathways under physiological conditions, along with selected homeobox genes implicated in each pathway, are summarized in Table 4.
Cells regulate diverse biological processes through intricate networks of receptors and signaling pathways that integrate multiple inputs and engage in extensive crosstalk, enabling context‐specific responses [181]. For instance, the WNT/β‐catenin pathway shows crosstalk with RA, BMP, Notch, NF‐κB, and Hh pathways. During osteogenesis, WNT/β‐catenin signaling potentiates BMP‐dependent target gene expression, promoting osteogenic differentiation [165, 182, 183]. In cardiac development, RA and WNT signaling play fundamental roles in morphogenesis and heart development. NF‐κB interacts with WNT, MAPK, TGF‐β, PI3K/AKT, and JAK/STAT pathways to regulate inflammation and immune responses [161]. Homeobox TFs likewise act as crucial regulators that integrate and mediate crosstalk among signaling networks. For instance, in vitro studies with murine F9 cells show that RA‐induced collinear activation of HOX cluster genes (A–D) requires PI3K/Akt signaling during gastrulation, suggesting that crosstalk helps gastrulating cells preserve positional information critical for AP axis formation [184]. In vivo and ex vivo work indicate that NKX2–5 contributes to the regulation of hemogenic and cushion endocardial cell generation through Notch activation, while RA activity is concurrently repressed by dehydrogenase/reductase 3 (Dhrs3); together, the NKX2‐5/Notch/RA signaling axis promotes the differentiation of these cells into macrophages implicated in the remodeling of cardiac valve. On the other hand, disruption of signaling crosstalk common in cancer can rewire pathway interconnections to promote tumor progression. In lung adenocarcinoma (LUAD), for example, upregulated WNT/TCF4–HOXB9 signaling facilitates metastasis to bone, augmenting aggressiveness [185]. More broadly, although WNT signaling maintains normal tissue homeostasis, its dysregulation drives oncogenesis; collectively, these findings underscore how alterations in Homeobox‐related signaling pathways can redirect developmental programs to sustain cancer [137, 165, 181].
Functional Involvement of Homeobox Genes in Human Diseases
3
Homeobox genes encode pivotal developmental TFs with precise, context‐dependent functions that influence diverse physiological processes throughout life, including maintenance of adult tissue homeostasis. Because these factors regulate broad gene networks and intersect with major signaling pathways (e.g., WNT/β‐catenin, Notch, BMP/TGF‐β), perturbations in signaling cascades can reprogram homeobox regulatory circuits and downstream outputs. As a result, alterations in homeobox activity or expression can propagate through gene‐regulatory networks, contributing to developmental anomalies or various pathological states [3, 11, 186]. Dysregulation of homeobox genes arises through multiple mechanisms, including somatic mutations, signaling perturbations, epigenetic modifications (notably DNA methylation), and noncoding RNA (ncRNA)‐mediated regulation [3, 187]. Clinically, dysregulation of homeobox genes is observed across a broad range of diseases from developmental and congenital disorders and neurodevelopmental and neurodegenerative conditions to diverse cancers [3, 187]. In cancer, homeobox genes often serve as context‐dependent modulators, acting as oncogenes or tumor suppressors depending on tissue context, and their dysregulation can drive tumor initiation, progression, and metastasis [138, 188, 189]. Emerging evidence highlights widespread DNA methylation changes affecting homeobox gene expression as a key driver of oncogenic programs [190, 191].
Furthermore, mutations within homeobox genes themselves have been identified across diverse cancer types in both germline and somatic contexts. A notable case study is HOXB13, which illustrates germline‐somatic contributions to oncogenesis, particularly in prostate cancer (PCa) [187, 192, 193]. In the germline context, a rare HOXB13 missense variant (p.Gly84Glu) has emerged as an important hereditary risk factor for PCa. Beyond PCa, evidence suggests this variant may be associated with increased risk for other cancers, including nonmelanoma skin cancer and rectosigmoid cancer, observed exclusively in male carriers. Notably, this same variant was previously reported to confer an elevated risk of colorectal cancer (CRC), particularly in a small number of mutation carriers with a family history of PCa [193, 194, 195]. In the somatic context, upregulation of HOXB13 has been observed in primary prostate tumors and is correlated with more aggressive disease, advanced tumor grade and an increased propensity for metastasis, particularly following prostatectomy. This same variant has also been reported to confer elevated CRC risk, particularly among mutation carriers with a family history of PCa. Together, these findings underscore how germline HOXB13 alterations can influence cancer susceptibility across tissues and highlight the need for integrative studies of HOX gene mutations in hereditary cancer predisposition [192]. While mutation insights are significant, this review primarily focuses on the epigenetic regulation of homeobox genes in cancer, with particular emphasis on DNA methylation. Beyond genetic alterations, epigenetic mechanisms have emerged as pivotal regulators of gene expression across a broad spectrum of human disorders, spanning cancer and noncancer conditions, with DNA methylation often serving as a principal modulator of gene activity [196]. In the sections that follow, representative noncancerous conditions and neurodevelopmental disorders linked to alterations in homeobox gene expression are outlined, supported by genetic and epigenetic evidence. Later sections address cancer biology involving homeobox dysregulation, with particular attention to the relationship between aberrant DNA methylation and malignancy, and to lung cancer, where epigenetic silencing or activation of homeobox genes is strongly implicated in tumor pathogenesis.
Noncancerous Diseases Associated With Homeobox Genes
3.1
Although homeobox genes are widely recognized for their roles in oncogenesis, mutations or dysregulation of these genes are equally central to the pathogenesis of numerous noncancerous human diseases [3, 7, 187]. Because homeobox TFs coordinate tightly regulated developmental programs, perturbations in their expression or activity can yield highly context‐specific malformations and functional impairments. Consequently, disruptions of homeobox function, or dysregulation of their expression arising from pathogenic variants or epigenetic modifications, can have far‐reaching consequences across congenital and organ‐specific disorders, including congenital malformations, metabolic syndromes, cardiac anomalies, and neurodegenerative conditions (addressed in the next section) [187, 197, 198, 199]. For instance, dysregulation of HOX genes at various stages of embryonic and postnatal development has been linked to skeletal malformations such as hand–foot–genital syndrome, syndactyly, and other limb malformations [3, 11, 186].
Congenital and Organ‐Specific Disorders Associated With Homeobox Genes
3.1.1
Because homeobox TFs regulate a broad array of developmental processes, especially morphogenesis and cell differentiation, there is strong evidence for their direct involvement in human disease. Pathogenic variants identified in affected patients often supported by animal models with corresponding gene disruptions, support the conclusion that mutations in these genes can cause serious developmental disturbances. Phenotypes frequently involve congenital defects or organ‐specific abnormalities arising from disrupted patterning and differentiation [200, 201]. For example, PBX1 deletions lead to haploinsufficiency (HI) and syndromic congenital anomalies of the kidney and urinary tract (CAKUT), with renal defects also observed in Pbx1‐null mice. Given the complexity of homeobox gene biology in noncancerous conditions, this section highlights the most frequently reported variant types in human disorders [16]. Table 5 summarizes representative examples of variants across different disease categories and outlines disorders associated with various classes of coding mutations, such as missense, nonsense, and frameshift. It should be noted that, for each gene, a wide range of mutations may be reported across different patients; the examples in Table 5 illustrate representative variant types and mechanisms rather than the full mutational repertoire. As a prominent example, around 700 distinct variants have been reported in PAX6, associated with a broad spectrum of ocular abnormalities, including aniridia, cataract, and foveal hypoplasia [202, 203]. Notably, nonsense mutations introduce premature termination codons (PTCs) and typically cause loss‐of‐function (LOF) through truncated proteins. PTCS can also arise from frameshift, splice‐site, or single‐nucleotide variants. The downstream consequences of PTC‐generating variants depend on stop‐codon position and the domain context: most truncations are functionally inactive, but in certain settings they can exert dominant‐negative effects or confer gain‐of‐function (GOF) [204, 205]. For instance, among noncancerous HOX‐related disorders, a nonsense variant in HOXA2 yields a truncated protein consistent with LOF and autosomal‐dominant bilateral microtia, while missense mutations within the CRX homeodomain can drive dominant retinopathies via GOF mechanisms [206, 207]. Quantitatively, across inherited human diseases, about one‐third of pathogenic variants are nonsense, underscoring their substantial contribution to the pathogenic‐variant spectrum; prior studies similarly estimate that nonsense and frameshift variants introducing PTCs account for roughly one‐third of characterized human genetic disorders [204, 205].
Heterozygous variants can drive disease either by a GOF or by a LOF that reduces activity below the threshold required for normal physiology. LOF in some genes leads to disease via HI, whereas others may remain clinically unaffected by half‐normal activity [241]. HI denotes a dosage‐sensitive mechanism in which a single functional allele fails to provide sufficient gene product to sustain normal physiology. Consequently, heterozygous LOF variants often manifest as dominant conditions, and HI represents a recognizable subset of rare genetic diseases [16, 241, 242, 243]. In several noncancerous homeobox disorders, diverse variant types have been linked to HI. Robust evidence of HI exists for genes such as such as PBX1, OTX2, SHOX, PAX6, and PITX2 202]. In PAX6, high dosage sensitivity is observed, with most pathogenic variants heterozygous and reducing gene dosage, thereby causing HI and resulting in aniridia with associated features such as corneal opacity, cataract, and glaucoma [16]. Likewise, PBX1 HI causes syndromic CAKUT, underscoring the critical requirement for proper gene dosage in nephrogenesis [242]. Heterozygous deletion of OTX2 within the 14q13 region has been identified as the most plausible pathogenic mechanism underlying congenital hypopituitarism and ocular malformations [242]. SHOX HI, arising from deletions, duplications, or rarer exonic mutations in PAR1, is associated with idiopathic short stature (ISS) and Léri–Weill dyschondrosteosis (LWD); in contrast, increased SHOX dosage can contribute to tall stature in sex‐chromosome polysomies (e.g., 47, XXY). Clinically, phenotypic severity in SHOX‐related conditions often correlates with hormonal context rather than the mutation class [243].
While the aforementioned mutation types illuminate pathogenic dysregulation across contexts, the phenotypic manifestations of homeobox‐gene dysfunction are often complex, shaped by tissue‐specific expression programs and the position of variants within functional domains. Genotype–phenotype correlations thus provide a critical lens for interpreting how variants in homeobox genes relate to clinical presentations. Even within the same locus, different classes or positions of mutations can yield strikingly variable phenotypes across individuals, reflecting allelic heterogeneity and variable expressivity. Across human cohorts, modest quantitative or qualitative changes can be phenotypically decisive. PAX6 serves as a well‐studied exemplar, illustrating how clinical phenotypes emerge from the interplay of gene dosage, variant position, and tissue‐specific developmental programs. Beyond its canonical role in eye development, PAX6 is expressed in neural and nonocular tissues including the forebrain, olfactory system, and endocrine pancreas consistent with its broad developmental functions. Consequently, pathogenic variants that alter PAX6 dosage or function produce clinically distinct outcomes depending on variant class, domain location, and tissue context. Given that aniridia phenotypes correlate strongly with PAX6 mutations, comprehensive variant analyses have shown that PAX6‐related disorders encompass a wide spectrum of pathogenic variant types, each contributing differently to disease severity [244, 245, 246, 247].
Among reported pathogenic variants, nonsense mutations constitute the largest fraction, followed by frameshift insertions/deletions, splice‐site alterations, and less common classes such as in‐frame indels and C‐terminal extension (CTE) mutations. These variant categories differentially affect PAX6 protein function and, consequently, the severity of related clinical phenotypes; for example, CTE and other LOF variants tend to yield more severe phenotypes, whereas most missense substitutions are associated with milder forms. Moreover, PAX6‐associated aniridia predominantly arises from heterozygous LOF variants as well as chromosomal rearrangements affecting the 11p13 locus where PAX6 resides [244, 245, 246, 247]. Together, these observations establish HI as the predominant pathogenic mechanism underlying congenital aniridia (CA) within PAX6‐related disorders. Importantly, PAX6‐related ocular disorders extend beyond isolated eye malformations. Systemic manifestations in CA frequently accompany ocular involvement, including metabolic disturbances such as thyroid dysfunction, impaired glucose regulation, and hypertension. Consistent with PAX6 expression in pancreatic tissue, this gene is essential for the development and function of pancreatic β‐cells, so pathogenic variants can contribute to metabolic disorders across life, from persistent hyperinsulinemic hypoglycemia in infancy to an elevated risk of type 2 diabetes in adulthood. Neurological involvement has also been described, including structural brain anomalies in a subset of affected individuals. Isolated foveal hypoplasia (IFVH)‐associated variants are predominantly missense and cluster within specific PAX6 regions, supporting a genotype–phenotype relationship distinct from that of classic aniridia [244, 245, 246]. Notably, IFVH has been observed in individuals carrying pathogenic PAX6 variants and typically presents with a fully formed iris. Foveal hypoplasia can co‐occur with aniridia‐associated findings such as cataract or glaucoma. In some cases, biallelic pathogenic variants of PAX6 (compound heterozygous or homozygous) have been reported and cause profound disruption of ocular development, typically resulting in anophthalmia and severe central nervous system malformations [244, 245, 246]. In addition to PAX6, a second example illustrates how biallelic inactivating variants in NKX6‐2 essential for oligodendrocyte differentiation and regulation of myelin‐associated genes abolish NKX6‐2 function and cause a severe hypomyelinating leukodystrophy, underscoring how dosage and domain context across homeobox genes shape distinct neurodevelopmental outcomes [208].
Beyond biallelic pathogenic mutations that correlate with severity, polyalanine extension mutations represent a distinct mutational mechanism shaping homeobox‐related phenotypes in a length‐dependent manner. The correlation between polyalanine repeat length and disease severity provides a particularly instructive paradigm for understanding genotype–phenotype relationships. Two instructive examples illustrate how these mutation classes converge on clinical expressivity: HOXD13 and PHOX2B. Pathogenic expansions of polyalanine tracts can perturb protein folding and subcellular trafficking in a length‐dependent manner, promoting misfolding and intracellular aggregation of the mutant protein [248, 249]. HOXD13 harbors diverse mutations causing synpolydactyly (SPD). Comprehensive clinical and molecular analyses have identified three main classes of HOXD13 mutations associated with SPD: polyalanine expansions, missense mutations, and truncating variants, each linked to distinct molecular mechanisms and phenotypic variability. Polyalanine expansions are the most frequent and are generally associated with the classic, more severe SPD forms compared with missense or LOF variants. Mechanistically, expanded polyalanine tracts promote protein misfolding, cytoplasmic aggregation, and disruption of HOXD13’s normal nuclear functions. Missense variants within the homeodomain primarily impair transcriptional activation without causing aggregation. Truncating mutations (nonsense or frameshift) can occur inside or outside the homeobox region; truncations involving the homeodomain abolish DNA binding and transcriptional activity [248]. In congenital central hypoventilation syndrome (CCHS), the vast majority of cases arise from PARMs in PHOX2B. In healthy individuals, each allele contains about 20 alanines; disease‐associated alleles typically harbor expansions from 24 to 33 alanines. Importantly, individuals with longer polyalanine tracts tend to exhibit severe forms of clinical manifestations of CCHS [249].
Epigenetic Regulation of Homeobox Genes in Noncancer Human Disease
3.1.1.1
Epigenetic modifications have been implicated in a range of noncancerous diseases, highlighting that dysregulation of homeobox gene expression via epigenetic mechanisms can contribute to pathology beyond coding mutations. Epigenetic regulation encompassing DNA methylation, histone modifications, higher‐order chromatin structure, and ncRNAs mediated processes has emerged as a central determinant of gene expression, extending beyond traditional sequence variants [196]. While the interface between homeobox genes and epigenetic regulation has been most extensively explored in oncology, growing evidence demonstrates that similar epigenetic interactions influence noncancerous conditions.
Recent studies across disorders such as endometriosis, type 2 diabetes, and chronic kidney disease show that promoter DNA hypermethylation can repress expression of homeobox genes critical for maintaining tissue‐specific function. Consequently, DNA methylation‐mediated downregulation of homeobox TFs has emerged as a recurring pathogenic mechanism in various noncancerous diseases, whereby epigenetic repression directly impairs tissue physiology. To illustrate this, five representative examples HOXA10, HOXA11, HOXD10, HOXA5, and PDX1 are discussed below to show how promoter hypermethylation of homeobox genes contributes to distinct pathological processes in noncancerous conditions [197, 250, 251, 252]. Among noncancer conditions, endometriosis offers a well‐characterized case where epigenetic regulation of homeobox genes intersects with disease. Across the menstrual cycle, HOXA10 and HOXA11 expression is phase dependent: low in the proliferative phase, rising through the secretory phase to a mid‐secretory peak, and remaining elevated in the early decidua after successful implantation. Attenuation of secretory‐phase upregulation of HOXA10 and HOXA11 correlates with reduced implantation rates [250]. In contrast, women with endometriosis fail to exhibit the typical cyclic modulation of HOXA10 and HOXA11 across menstrual phases, maintaining persistently dysregulated expression throughout the cycle. Multiple independent studies have shown that this disruption of cyclic gene activity is primarily driven by aberrant promoter hypermethylation affecting both genes. Elevated HOXA10 promoter methylation has been consistently observed in eutopic endometrial tissue of affected women, particularly during the secretory phase, where it directly correlates with marked reductions in gene expression. Moreover, abnormal promoter methylation of HOXA10 and HOXA11 has also been reported in other infertility‐associated conditions, such as chronic endometritis and polycystic ovary syndrome, suggesting a shared epigenetic mechanism contributing to endometrial dysfunction across multiple reproductive pathologies [199, 250]. Beyond the reproductive system, similar epigenetic repression of homeobox genes has been observed in other organs. In vitro and in vivo studies have demonstrated that TGF‐β1 stimulation is associated with promoter hypermethylation of HOXD10, resulting in suppressed of its expression. Under physiological conditions, HOXD10 functions as a transcriptional repressor of NADPH oxidase 4 (NOX4) by directly binding to its promoter; decreased HOXD10 expression leads to NOX4 upregulation, increased ROS production, and contributes to renal tubular injury and fibrosis in chronic kidney disease [251]. Another example is HOXA5, which becomes hypermethylated at its promoter during kidney fibrogenesis, leading to loss of expression. HOXA5 normally represses JAG1, thereby restraining NOTCH signaling; loss of HOXA5 expression derepresses JAG1, activates NOTCH signaling, and promotes fibrogenesis in the kidney [252]. Epigenetic regulation also features in metabolic disease, such as type 2 diabetes. In pancreatic islets from individuals with T2D, hypermethylation of distal HOXA5‐regulated regions within PDX1 has been detected and is associated with suppressed PDX1 expression and attenuated glucose‐stimulated insulin secretion [197].
Neurodegenerative Diseases Associated With Homeobox Gene Dysregulation
3.1.2
Homeobox genes are increasingly implicated as key contributors to NDDs and related disease states, through both genetic variants and epigenetic dysregulation. Under normal physiology, many homeobox genes not only guide early development but also support adult neuronal function, including preservation of neuronal terminal identity, maintenance of neurotransmitter identity, and maintenance of synaptic structure [253, 254]. For example, the LMX1A/B‐driven transcriptional program is crucial for adult midbrain dopaminergic neurons, regulating mitochondrial metabolism and sustaining mitochondrial function [255]. Evidence regarding homeobox gene expression in the adult brain is nuanced: while some studies report that HOX genes are undetectable in healthy adult brain tissue, context‐dependent HOX activity has been observed in restricted cell populations and disease models, where it appears to help maintain cellular identity. Certain HOX TFs have also been implicated in modulating synaptogenesis and supporting neurotransmitter identity [254]. Collectively, dysregulation of homeobox gene expression emerges as a potential pathogenic mechanism linking these genes to neurodegenerative processes. A concrete example is the HOXB6 locus, where a differentially methylated region containing cg17179862 is hypermethylated in Alzheimer's disease (AD), with elevated methylation in hippocampal tissue correlating positively with tau burden. Table 6 summarizes representative examples of homeobox genes implicated in various neurodegenerative disease related syndromes (NDDS), illustrating the diversity of mechanisms by which their dysregulation can influence neuronal vulnerability and degeneration [256].
A Central Role of Homeobox Genes in Cancer Development
4
Homeobox genes orchestrate a wide spectrum of biological programs across the lifespan, from embryonic development to maintenance of tissue identity in adulthood [7]. Consequently, precise transcriptional regulation of these genes is essential for tissue homeostasis; even subtle deviations in expression can profoundly reshape cellular behavior and promote tumor progression. Dysregulation of homeobox gene expression whether upregulation or downregulation within the HOX clusters has been consistently detected across multiple malignancies, underscoring their fundamental contribution to tumor initiation and progression [3, 7, 191]. Aberrant expression of homeobox genes in cancer cells arises from multiple layers of genetic and epigenetic modifications that disrupt normal transcriptional patterns. These regulatory disturbances may involve genetic alterations such as loss of heterozygosity or gene amplification, alongside epigenetic modification. Across human cancers, and most notably within HOX clusters, epigenetic dysregulation rather than recurrent coding mutations constitutes the dominant axis of homeobox gene alteration. Consequently, these mechanisms act as crucial drivers of tumor initiation, progression, and metastasis through persistent misexpression of key homeobox regulators [3, 191, 270].
In cancer cells, the dysregulation of homeobox genes, especially those within the HOX clusters, has far‐reaching biological consequences, as these TFs rewire core developmental and lineage programs that underlie malignant behaviors [147]. Homeobox genes, particularly those clustered within HOX loci, influence cancer progression by modulating diverse signaling networks, revealing substantial functional plasticity [18]. A prominent example is the EMT, a developmental program hijacked in oncogenesis to enhance tumor cell plasticity. In malignant contexts, EMT promotes dissemination by bestowing epithelial cells with migratory and invasive capabilities, a process closely associated with therapeutic resistance across multiple cancers, including lung carcinoma [271, 272, 273]. Mechanistically, dysregulated homeobox factors intersect with major oncogenic pathways such as Notch, Wnt/β‐catenin, TGF‐β/SMAD, and PI3K/AKT/mTOR, with extensive crosstalk shaping hallmark tumor phenotypes proliferation, invasion, EMT, and angiogenic remodeling. The integrated influence of HOX genes on these signaling cascades underscores their context‐dependent roles in tumor biology and supports their potential as targets for therapeutic intervention [71, 274].
Another critical aspect underscoring the centrality of homeobox genes in cancer is their remarkable dual functionality as both oncogenes and tumor suppressors. Across cancer biology, dysregulation of oncogenes or tumor suppressor genes driven by diverse mutation types or epigenetic mechanisms leading to upregulation or downregulation constitutes a fundamental driver of tumorigenesis [188]. In this landscape, homeobox genes exemplify this dualistic paradigm, displaying either oncogenic or tumor‐suppressive effects depending on cellular context, tissue origin, and developmental stage. Consequently, misexpression of these genes is consistently associated with tumorigenesis in a tissue‐ and stage‐dependent manner. During embryogenesis, those homeobox genes with oncogenic potential may be transiently expressed and contribute to malignant traits when aberrantly re‐expressed in cancer cells, while in fully differentiated adult tissues their expression is often reduced or silenced. Conversely, a subset of homeobox genes remains physiologically expressed in adult tissues but becomes downregulated across various cancers [275]. This dynamic adds a layer of complexity to cancer biology, underscoring that the functional outcome of homeobox genes’ dysregulation is not context‐invariant but contingent on tissue type and developmental state. Illustrative examples of homeobox genes functioning as either oncogenes or tumor suppressors, depending on expression patterns across different tumor contexts, are discussed below. For instance, oncogenic activity is exemplified by HOXA1, which is overexpressed in breast cancer and consistently associated with tumor progression and poor prognosis. HOXA1 is likewise upregulated with tumor‐promoting effects in other malignancies, including PCa, glioblastoma, and head‐and‐neck squamous cell carcinoma (HNSCC) [276]. Similarly, HOXB5 exhibits oncogenic activity in breast cancer, where its overexpression correlates with increased cellular invasiveness [277]. HOXB7 is another oncogenic HOX member overexpressed across several cancer types; in breast cancer, high HOXB7 expression associates with tumor progression and adverse outcomes, and in HNSCC its upregulation correlates with advanced disease stage and poor prognosis [278]. In contrast, certain homeobox genes display tumor‐suppressive functions and are frequently downregulated across cancers. EMX2 functions as a tumor suppressor, with marked downregulation in lung cancer and esophageal adenocarcinoma; loss of EMX2 enhances cellular proliferation and invasiveness [278, 279]. Similarly, reduced PITX1 expression has been linked to tumor progression in esophageal squamous cell carcinoma (ESCC) [280].
In addition, some homeobox genes exhibit distinct expression patterns across cancers, being upregulated in certain tumor types and downregulated in others. The most striking examples of their functional duality are those that exert opposite roles depending on cancer type and cellular context. For instance, HOXA9 displays dual functionality across human cancers: its oncogenic activity is evident in both hematologic and solid malignancies. HOXA9 overexpression has been reported in AML; ∼70% of cases and in acute lymphoblastic leukemia (ALL), where it is strongly associated with disease aggressiveness and poor prognosis. On the other hand, HOXA9 tumor‐suppressive activity has been observed in cancers such as cutaneous SCC and breast cancer, where its expression is downregulated. Collectively, HOXA9 can function as either an oncogene or a tumor suppressor, depending on tumor type and cellular context [281, 282, 283]. Among the most striking examples of dual‐function homeobox genes is NKX3.1, which plays a pivotal role in prostate development and tumorigenesis. Under physiological conditions, NKX3.1 promotes epithelial differentiation and acts as a cofactor for the androgen receptor. During PCa progression, its role shifts from tumor suppressor in early stages to exhibiting oncogenic potential in advanced stages [284]. The functional duality of homeobox genes largely depends on multiple factors, including tumor type, the developmental stage of the malignancy, and the surrounding tumor microenvironment (TME). These context‐dependent parameters collectively shape whether a given homeobox gene acts as an oncogene or a tumor suppressor.
In addition to dysregulation of homeobox genes caused by genetic and epigenetic alterations, multiple extrinsic factors influence their expression and function. Among these, the tumor microenvironment (TME) exerts a profound regulatory impact on how these genes contribute to tumor initiation and progression. The TME is a highly complex and dynamic milieu whose composition varies across tumor types, yet consistently includes cancer‐associated fibroblasts, diverse immune cells, vasculature, and extracellular matrix (ECM). Each component can modulate the expression and function of homeobox genes through a range of integrated mechanisms [285, 286]. Among these, the ECM plays a particularly critical role in tumor progression. ECM remodeling not only supports angiogenesis but also promotes tumor cell motility and the induction of EMT, thereby increasing malignant potential and the likelihood of metastasis in solid tumors [286, 287, 288]. EMT is well known for its critical role in the progression of various cancers, driving metastasis and resistance to conventional therapies, particularly in solid tumors. Distinct signaling molecules, such as Wnt, TGF‐β, and BMP, also orchestrate various types of EMT, with each contributing to specific EMT types during processes such as embryonic development, tissue remodeling, and cancer progression. EMT is a central driver of cancer progression, metastasis, and therapeutic resistance, particularly in solid tumors. Distinct signaling pathways such as Wnt, TGF‐β, and BMP orchestrate various EMT types that operate during embryogenesis, tissue remodeling, and cancer progression. EMT entails extensive reprogramming of gene expression, as well as remodeling of cellular morphology and metabolism, enabling epithelial cancer cells to acquire migratory capabilities and more resilient phenotypes. A diverse set of TFs orchestrates EMT, with several core EMT‐TFs acting as central regulators that influence both their own expression and that of other EMT‐TFs. Notably, the ZEB1 and ZEB2 zinc‐finger E‐box‐binding homeobox proteins are regarded as important core EMT regulators [289, 290, 291]. Consequently, EMT shapes the expression and regulatory dynamics of various homeobox genes across multiple cancer types, contributing to tumor progression and metastasis. For instance, in esophageal cancer*, forkhead box C1* (FOXC1) modulates ZEB2 expression through its association with the pioneer factor PBX1, thereby promoting EMT [292]. In addition, HOX cluster‐linked lncRNAs (HOX‐lncRNAs) have emerged as key regulators of EMT, modulating transcriptional networks that drive tumor progression and metastatic potential [293].
Beyond their involvement in EMT, homeobox genes significantly influence other TME‐associated hallmarks of cancer, particularly angiogenesis. Angiogenesis is a fundamental mechanism by which the TME promotes malignant progression, and multiple HOX family members act as pivotal regulators of this process. HOX genes have long been implicated in tissue patterning and vascular development, in part by regulating ECs function. Their expression has been associated with proliferation and migration of vascular smooth muscle cells, atherosclerotic plaque formation, and remodeling of cardiac tissue after injury [294, 295, 296]. Emerging evidence also indicates that dysregulation of specific HOX genes contributes to tumorigenesis by promoting angiogenesis within tumors. Conversely, HOX gene dysregulation has been linked to cardiovascular diseases such as atherosclerosis, heart failure, and arrhythmias, highlighting the broader relevance of HOX pathways to tissue remodeling and pathology [294, 295]. In cancer, angiogenesis is tightly coordinated by a balance of pro‐ and antiangiogenic factors, and HOX genes can influence this balance through context‐dependent regulation of endothelial and stromal cell behavior as well as interactions with other TME components. Notably, angiogenesis is a critical process in tumor progression as it involves in cancer invasion, and metastasis. This process is modulated by a range of pro‐ and antiangiogenic factors [137, 296, 297]. One of the well‐established HOX regulators of angiogenesis is HOXA9. HOXA9 promotes angiogenesis by interfacing with multiple signaling pathways and TFs. Moreover, accumulating evidence shows that HOXA9 overexpression enhances angiogenic activity while HOXA9 downregulation markedly impairs these processes. This dual role physiological in some contexts and pathological in cancer highlights the importance of HOXA9 in cancer biology. Dysregulation of HOXA9, whether by aberrant DNA methylation or other mechanisms, can contribute to angiogenesis deregulation, a hallmark of cancer progression and metastasis [281, 298]. In humans, HOXA9 also drives EC migration, a key step in vasculature formation, in part by upregulating EphB4, a receptor tyrosine kinase involved in blood vessel development and maturation. EphB4 is frequently overexpressed in various cancers and supports tumor growth by enhancing angiogenesis; its expression levels correlate with tumor growth and differentiate status in cancers such as lung cancer, underscoring its oncogenic potential [296, 297, 299, 300]. Other HOX family members linked to angiogenesis and dysregulation in cancer include HOXB3, HOXB9, and HOXD3, which have been implicated in regulating angiogenic processes. HOXB9, in particular, is often overexpressed across solid tumors (e.g., lung adenocarcinoma) and is associated with increased proangiogenic factor expression and activation of proangiogenic signaling cascades. This mechanistic link is clinically relevant because angiogenesis is a major route through which tumors resist antiangiogenic therapies, suggesting that HOXB9 angiogenic programs can contribute to therapeutic resistance [137].
Cancer‐Associated Epigenetic Modulation of Homeobox Genes
4.1
As described, extensive research over recent decades has established epigenetic modifications as critical contributors to the onset and progression of numerous diseases. These alterations extend beyond cancer to a range of noncancerous and NDDs, underscoring their broad impact on human health. Epigenetic mechanisms regulate gene expression without altering the DNA sequence, thereby influencing disease initiation and progression, including cancer. The major modalities include DNA methylation, histone acetylation and methylation, and ncRNA‐mediated regulation, all of which can silence tumor‐suppressor programs or activate oncogenic circuits. Consequently, these modifications can drive tumorigenesis and shape cancer development and metastasis. Aberrant DNA methylation patterns at CpG sites can silence tumor suppressor genes or aberrantly activate oncogenes. A particularly relevant area in cancer is the dysregulated expression of ncRNAs within the HOX cluster networks [71, 301, 302]. Abnormal methylation of homeobox genes constitutes a crucial area of study in epigenetics, given its impact on regulatory roles and cancer prognosis.
Methylation‐Mediated Silencing and Activation of Homeobox Genes in Cancer
4.1.1
A large body of evidence indicates that aberrant DNA methylation and sequence mutations can dysregulate multiple homeobox genes, promoting cancer initiation and progression by disturbing control of cell proliferation, apoptosis, differentiation, and angiogenesis. Across many cancers, particularly solid tumors, methylation abnormalities are a major contributor to HOX genes’ misexpression. Within the four HOX clusters, numerous genes show recurrent dysregulation with functional consequences for tumorigenesis [149, 191]. As discussed, homeobox gene misexpression especially within HOX clusters drives tumorigenesis in a context‐dependent fashion, with individual genes acting as oncogenes or tumor suppressors depending on context. Aberrant DNA methylation is a key mechanism governing this regulation [74, 147]. DNA methylation, mediated by DNA methyltransferases (DNMTs; notably DNMT1 and DNMT3A/3B), involves adding a methyl group to cytosine, influencing gene expression by recruiting repressive proteins or hindering TF binding. This process is dynamic, involving de novo methylation and demethylation that contribute to tissue‐specific gene regulation and the establishment of stable methylation patterns in differentiated cells [283, 303]. In mammals, DNA methylation at the C5 position of cytosine can impact gene regulation by recruiting repressive proteins or inhibiting the binding of TFs. This process is highly dynamic, encompassing both de novo methylation and demethylation events, and contributes to tissue‐specific gene regulation and the establishment of stable methylation patterns in differentiated cells [301, 302]. Ongoing research is dedicated to unraveling the intricate mechanisms underlying DNA methylation and its profound impact on gene regulation, development, and the manifestation of various disease states [301, 304, 305]. Notably, aberrant DNA methylation patterns have important roles in cancer onset, progression, and metastasis [306].
Although aberrant DNA methylation is widespread in cancer, its impact on transcription is region and context dependent, not uniformly silencing [71]. In general, CpG‐island promoter hypermethylation represses transcription by recruiting methyl‐CpG‐binding proteins (e.g., MeCP2), thereby reducing transcription‐factor and RNA polymerase II (RNAP II) access [307]. For instance, HOXD10 is frequently hypermethylated at its promoter and transcriptionally silenced in endometrial cancer (EC), where its loss correlates with tumorigenic progression, particularly in endometrial adenocarcinoma [308]. Similarly, promoter hypermethylation of HOXA5 has been reported in several malignancies, including CRC, non‐small‐cell lung cancer (NSCLC), and chronic myeloid leukemia (CML); notably, hypermethylation of HOXA4/HOXA5 is strongly associated with imatinib resistance in CML patients [309, 310]. In breast cancer, HOXD13 promoter methylation is a common event and predicts worse overall survival (OS), supporting its value as a prognostic [311]. Collectively, aberrant promoter methylation of homeobox genes is linked to disease progression, prognosis, and patient outcomes across diverse cancer types. In line with this, promoter CpG island hypermethylation is a well‐established driver of neoplastic transformation, enforcing stable transcriptional silencing of tumor suppressor genes and enabling oncogenic programs to proceed [306]. HOXC10 serves as a clear example of hypomethylation‐mediated activation within two different types of cancer, with promoter CpG‐island hypomethylation reported in NSCLC and hypomethylation at its CpG sites within the first intron in gastric cancer, accompanying HOXC10 overexpression and tumor progression [312, 313].
Although aberrant promoter CpG‐island hypermethylation typically represses gene expression, methylation within gene‐bodies often tracks with active transcription and has been linked to tumor initiation and progression an observation widely referred to as the “DNA methylation paradox” [307]. This paradox is prominently observed at homeobox genes; the pan‐cancer integration of analyses has shown that “DNA methylation canyons,” large under‐methylated tracts, tend to become hypermethylated in various types of cancer, and this kind of hypermethylation is strongly associated with upregulated expression in ≈43% of homeobox genes, which are significantly enriched for oncogenes. For illustration at homeobox loci, gene‐body “canyon” hypermethylation at DLX1 coincides with aberrant overexpression in bladder urothelial carcinoma (BLCA), uterine corpus endometrial carcinoma (UCEC), lung SCC (LUSC), and LUAD; similarly, POU3F3 harbors a hypermethylated gene‐body canyon with overexpression in LUSC, BLCA, UCEC, supporting that gene‐body hypermethylation can increase expression at these loci [314]. As a whole, the complexity of these methylation patterns underscores the potential for tissue‐specific therapeutic strategies targeting the epigenetic dysregulation of homeobox genes in cancer [312].
The Interplay Between Homeobox Genes’ Expression, lncRNAs, and miRNAs in Cancer
4.1.2
The human genome predominantly comprises ncRNAs, which account for approximately 90% of its sequence, while only about 2% of the genome is dedicated to protein‐coding. Notably, ncRNAs are broadly categorized into small ncRNAs (sncRNAs) and lncRNAs [315]. In various biological sources, sncRNAs are also defined as ncRNAs with fewer than 200 nucleotides in length and are categorized into different classes, including miRNAs, tRNA‐derived stress‐induced small RNAs (tiRNAs), small interfering RNA (siRNA), small nucleolar RNAs (snoRNAs), and PIWI‐interacting RNAs. Collectively, these different classes of sncRNAs are involved in modulating gene expression through various mechanisms at multiple levels. Moreover, they are known to contribute to epigenetic regulation [316]. Notably, miRNAs, the most prevalent class of ncRNAs, are compromised approximately 19–25 nucleotides in length. They are participated in posttranscriptional gene regulation by binding to the 3′‐untranslated regions (3′‐UTRs) of their specific target mRNAs. This interaction typically leads to either mRNA degradation or translational repression. HOX genes are remarkable targets of miRNAs. A notable connection exists between miRNAs and these genes regulation, as some miRNAs are located within HOX gene clusters [317, 318]. Notably, miRNAs regulate approximately 30–50% of human protein‐coding genes, underscoring their extensive involvement in biological and pathological processes, including carcinogenesis. Significantly, their expression levels are dynamically regulated across various cell types and developmental stages, and aberrations in their expression have been strongly linked to disease progression. Therefore, dysregulation of miRNAs function or expression, whether upregulation or downregulation, are particularly associated with various cancers, including lung cancer. In lung cancer, several miRNAs have been identified as key regulators [317, 319, 320]. For example, miR‐21, often overexpressed in NSCLC, is associated with tumor progression, whereas miR‐128, which acts as a tumor suppressor, is frequently downregulated in NSCLC. These findings underscore the critical roles miRNAs play in cancer development and their potential as therapeutic targets or biomarkers [321]. In contrast, lncRNAs are characterized by having more than 200 nucleotides in length. They are well known for their lack of protein‐coding potential and exhibit diverse characteristics and functionalities, allowing further classification into subclasses such as sense lncRNAs, promoter‐upstream lncRNAs, intergenic lncRNAs, and bidirectional lncRNAs [315, 322]. LncRNAs employ diverse and complex mechanisms to influence cellular processes, such as transcriptional regulation, regulation of chromatin structure, RNA splicing, gene imprinting, and cell proliferation. While the precise functions of many lncRNAs remain elusive, their roles in gene expression and transcriptional regulation are widely reported. These functions occur through interactions with DNA, mRNA, miRNA, and proteins. For instance, lncRNAs can either activate or suppress transcription through various mechanisms. Interestingly, lncRNAs share structural analogies with mRNAs, as both are transcribed by RNAP II [322, 323, 324]. Despite their inability to encode proteins, certain lncRNAs possess features such as an N7‐methylguanosine (m^7^G) at the 5′ cap and a 3′ polyadenylated (polyA) tail. Notably, nearly half of all lncRNAs contain polyA tails, and almost all undergo splicing and accumulate in the cytoplasm, further enhancing their functional versatility. These attributes underscore the intricate mechanisms through which lncRNAs regulate transcription [323]. Specifically, one of the primary mechanisms by which lncRNAs regulate target gene expression is through their interaction with miRNAs, a process also widely reported in various types of cancer. Functionally, lncRNAs interact with miRNAs via multiple mechanisms, significantly influencing gene expression. A key role of lncRNAs is to act as competitive endogenous RNAs (ceRNAs), often described as exhibiting “miRNA sponge” activity by binding to miRNAs at specific complementary sites. As ceRNAs, by binding to miRNAs at specific complementary sites, lncRNAs limit miRNA availability for interaction with target mRNAs. This reduction in miRNA regulatory influence ultimately leads to the upregulation of the target genes. Consequently, this regulatory interaction establishes a complex network involving lncRNAs, miRNAs, and their targets, emphasizing the multifaceted role of lncRNAs in gene expression regulation and their potential implications in tumorigenesis [324]. In addition, lncRNAs play integral roles in epigenetic modifications, particularly by recruiting chromatin‐modifying complexes through their interactions with DNA, RNA, and related proteins [322, 325].
In various types of cancer, epigenetic modifications, particularly DNA methylation, play a significant role in regulating the expression of ncRNAs, including both miRNAs and lncRNAs. Moreover, interactions between epigenetic mechanisms and miRNA regulation critically contribute to the initiation and progression of human cancers. Such alterations in their expression can substantially affect the development and progression of lung and other cancers. For example, the methylation status of CpG islands within promoter regions determines miRNA transcription, with hypomethylation enhancing and hypermethylation suppressing miRNA expression, respectively. Similarly, DNA methylation is one of the primary factors regulating lncRNA expression; abnormal hypermethylation of lncRNA promoters can lead to their silencing. Moreover, lncRNAs themselves can modulate methylation patterns by interacting with DNMTs and demethylases, directing these enzymes to specific promoter regions to regulate gene expression. Therefore, methylation‐related lncRNAs play a key role in shaping tumor biology, with certain lncRNAs modulating DNA methylation to regulate gene expression. For instance, aberrant expression of lncRNA ELF3‐AS1 is associated with hypermethylation of miR‐212, which contributes to increased NSCLC cells invasion. Similar methylation‐dependent regulatory mechanisms have also been reported in other malignancies. For instance, in gastric cancer, loss of TFF1 expression has been linked to promoter hypomethylation and subsequent activation of the HOXA10/miR‐196b‐5p axis, thereby enhancing cell proliferation and invasion. It should be noted that, the bidirectional interplay between lncRNAs and DNA methylation underscores their significant role in epigenetic regulation and their contribution to tumor progression, growth, and patient outcomes in lung cancer [326, 327, 328]. As motioned above, in lung cancer, DNA methylation regulates the expression of specific lncRNAs, either silencing or activating them. For instance, hypomethylation of the MIR503HG promoter in LUAD leads to its upregulation, promoting tumor proliferation through the lncRNA MIR503HG/SNHG17/miR‐330–3p axis. Conversely, hypermethylation‐induced downregulation of lncRNA lung cancer immune cell infiltration associated RNA (LCIIAR) suppresses LUAD metastasis, highlighting the dual role of methylation in lncRNA‐mediated cancer regulation. In lung cancer metastasis, evidence also suggests that exosomal lncRNAs, such as Ubiquitin‐Fusion Protein 1 (UFC1), have been identified as contributors to these processes. Consequently, lncRNAs are known to exhibit oncogenic and tumor‐suppressive functions, highlighting their diverse roles in cancer biology [327, 329, 330]. In recent years, several lncRNAs have been identified as key players in lung cancer, including cancer‐associated LncRNA‐1 (SCAL1), antisense ncRNA in the INK4 Locus (ANRIL), UFC1, metastasis‐associated lung adenocarcinoma transcript 1 (MALAT1), and HOXA transcript antisense RNA, myeloid‐specific 1 (HOTAIRM1). Notably, a subset of lncRNAs is closely associated with HOX gene clusters such as HOTAIR, HOTAIRM1, HOXA11 antisense RNA (HOXA11‐AS), HOXA Cluster Antisense RNA 3 (HOXA‐AS3), and HOXD cluster antisense RNA 1 (HOXD‐AS1). For instance, HOTAIR, transcribed from the antisense strand of the HOXC gene cluster on chromosome 12, is particularly well known for its significant upregulation in lung cancers, especially NSCLC. Its strong oncogenic properties make HOTAIR a critical regulator of lung cancer progression and metastasis [322, 329, 330]. While HOX‐linked lncRNAs such as HOTAIR act predominantly as oncogenic drivers in lung cancer, their functions are not uniformly protumorigenic across tissues. This context dependence is exemplified in breast cancer, where diminished HOTAIRM1 expression associates with more aggressive phenotypes, indicating a tumor‐suppressive role. Loss of HOTAIRM1 drives breast‐cancer cells to proliferate, form more colonies, and invade, whereas restoring its expression suppresses these malignant traits. Consistently, reduced HOTAIRM1 levels predict worse outcomes, suggesting it as a potential therapeutic lever in breast invasive carcinoma [331]. Therefore, HOX‐cluster associated lncRNAs constitute a context‐dependent epigenetic axis in cancer.
The Interplay of Homeobox Genes and the Wnt Signaling Pathway Across Cancers With Emphasis on Lung Cancer
4.2
Homeobox genes participate in a wide array of signaling networks that regulate developmental physiology, and under pathological conditions particularly in human cancers—they engage in multiple oncogenic cascades. A broad set of homeobox TFs functionally interacts with core oncogenic pathways, including, Wnt/β‐catenin, TGF‐β/Smad, PI3K/Akt, MAPK/ERK, NF‐κB, and JAK/STAT, in development and tumorigenesis across contexts [87]. Among these, Wnt signaling stands out as central to many fundamental cellular processes, and its dysregulation is a common driver of tumorigenesis. In CRC, aberrant Wnt activity was early linked to disease pathogenesis, and dysregulated Wnt/β‐catenin signaling has since been recognized as a hallmark in a broad range of malignancies. Mutations in pathway components such as adenomatous polyposis coli (APC) or β‐catenin can sustain constitutive activation, promoting unchecked proliferation, survival, and metastatic potential. Beyond CRC, aberrant Wnt signaling has been implicated in liver, breast, gastric, and other cancers, underscoring its wide oncogenic relevance. Consequently, therapeutic strategies targeting Wnt receptors, core mediators, or downstream effectors are actively explored as potential anticancer approaches. The Wnt network does not operate in isolation; it engages in extensive crosstalk with pathways such as Hh, Not Hh, Notch, Hippo, TGF‐β/Smad, NF‐κB, and PI3K/AKT, shaping both normal development and tumor behavior. Understanding these interconnections is crucial for designing effective interventions and for interpreting cancer biology in translational contexts [165].
Accordingly, the Wnt signaling pathway remains one of the most extensively studied to date, though its full functionality is not yet fully understood. This critical pathway was first identified in 1982 with the discovery of the int1 (Wnt1) gene's role in tumor growth [332]. The Wnt/β‐catenin signaling cascade plays a pivotal part in a wide range of physiological processes. It regulates key functions like embryonic development, immune responses, cell growth, and death, as well as maintaining homeostasis in vital organs such as the lungs, intestines, and liver. This pathway is involved in the broadest array of biological activities, including cell proliferation, differentiation, organogenesis, regeneration, and is associated with conditions like neurodevelopmental disorders and cancer [159, 332, 333, 334, 335]. In the lungs, the Wnt/β‐catenin signaling pathway plays a pivotal role that extends beyond maintaining homeostasis. It is essential for regulating lung epithelial SCs and facilitating their differentiation into specialized epithelial cell types. For example, research indicates that this pathway is probably associated with the regulation of epithelial differentiation in lung resident MSCs (LR‐MSCs). During lung development, Wnt/β‐catenin signaling governs critical processes, particularly the precise fate determination of progenitor cells. Beyond its developmental roles, the pathway is also instrumental in tissue remodeling and regeneration under stress or damage. However, dysregulation of this pathway through various mechanisms can impair these functions, leading to pathological conditions in the lungs, including lung cancer [301, 336]. Therefore, given the paramount significance of this pathway in the lung cells, it is evident that this pathway's abnormal activation could potentially be the underlying cause of various lung diseases, particularly in lung carcinogenesis [333]. Hence, recent research on lung cancer has demonstrated a significant correlation between the activation of the Wnt/β‐catenin pathway and an increased tumor mutational burden in NSCLC. This finding underscores the strong association between Wnt/β‐catenin pathway dysregulation and NSCLC tumor development, highlighting its potential impact on the progression of this cancer type [301, 337, 338]. Given the crucial role of the Wnt/β‐catenin signaling pathway in cancer development and progression, particularly through its interaction with the TME, it is vital to explore its regulation at the molecular level [339]. A comprehensive understanding of the various proteins involved in regulating the Wnt signaling pathway is essential for elucidating its impact on cancer progression, including lung cancer. This pathway comprises various proteins and is primarily subdivided into canonical and noncanonical pathways. The Wnt/β‐catenin pathway, commonly referred to as the canonical pathway, is characterized by the translocation of cytoplasmic β‐catenin into the nucleus. Key proteins involved in the Wnt signaling pathway include Wnt ligands, Frizzled receptors, lipoprotein receptor‐related protein 5 and 6 (LRP5/6), Dishevelled 1 (Dvl1), Glycogen Synthase Kinase 3 Beta (GSK3β), APC, Axin, β‐catenin, and T‐cell factor/lymphoid enhancer factor (TCF/LEF) family transcriptional regulators [301].
Various genes within HOX family exhibit significant involvement in the Wnt signaling cascade. For instance, HOXB5 has been shown to upregulate the expression of β‐catenin, a key effector in the pathway. Conversely, HOXA4 appears to decrease β‐catenin levels while increasing the protein and mRNA abundance of GSK3β, a negative regulator of the pathway. Interestingly, the stability of β‐catenin has been linked to the expression of HOXB9, which is itself a target gene of the Wnt/TCF signaling axis. Additionally, HOXB7 has been found to form a physical complex with β‐catenin, potentially modulating its activity. Conversely, HOXB13 has been reported to downregulate the expression of TF 4 (TCF4), a crucial TF in canonical Wnt signaling [87]. Furthermore, GSK3β has been shown to promote the conditional association of the transcriptional factor cyclic amp‐responsive element‐binding protein (CREB) and its coactivators with the HOX cofactor MEIS1. This interplay is thought to facilitate HOX‐mediated transcriptional regulation and potentially contribute to tumorigenesis. It should be highlighted that the role of HOX proteins in modulating Wnt signaling can be complex, with some HOX factors promoting cancer progression while others may inhibit it. Further research is needed to fully elucidate the intricate crosstalk between these two pivotal developmental pathways (shown in Figure 3) [87]. Thus, HOX proteins, play essential roles in mediating the Wnt signaling pathway [71, 87].
The Wnt/β‐catenin signaling pathway and its bidirectional crosstalk with HOX genes. In the active state (left), Wnt ligand binding disrupts the β‐catenin destruction complex, leading to β‐catenin stabilization and nuclear translocation. Nuclear β‐catenin then associates with TCF/LEF factors to activate transcription of target genes, including HOX genes. HOX proteins, in turn, modulate the pathway through feedback regulation: HOXA4 downregulates β‐catenin expression, while HOXB5 upregulates it. In the inactive state (right), the destruction complex remains active, leading to β‐catenin phosphorylation, ubiquitination by β‐TrCP, and proteasomal degradation. This schematic highlights the key regulatory loop between Wnt signaling and HOX‐mediated transcriptional control.
Among the members involve in Wnt signaling pathway, it is crucial to highlight that β‐catenin acts as a pivotal effector. Once inside the nucleus, β‐catenin can interact with T‐cell factor/lymphoid enhancing factor (TCFs/LEF) TFs, leading to the initiation of the TCFs/LEF transcription complex. Therefore, this pathway ultimately involves the regulation of the expression of target genes. On the other hand, the noncanonical pathways, such as the Wnt/calcium pathway, occur independently of β‐catenin and are more closely related to cell differentiation [159, 333]. As a consequence, the Wnt signaling pathway is crucial in governing gene expression regulation, notably including the HOX genes’ expression. In contrast, elevated levels of β‐catenin are directly correlated with the enhanced activation of transcription processes, leading to the overexpression of key genes such as cyclin D1 and c‐MYC [159, 333]. Hence, the overexpression of Cyclin D1 and c‐Myc has been detected in various cancers, including lung cancer, and they are deeply implicated in the process of carcinogenesis [340]. Cyclin D1 plays a critical role in cell cycle regulation, particularly in facilitating the progression from the G1 phase to the S phase. By activating cyclin‐dependent kinases 4 and 6 (CDK4/6), Cyclin D1 ensures proper cell cycle advancement. Due to its critical role in cell cycle regulation, the overexpression of Cyclin D1 is directly associated with neoplastic growth. Such dysregulation has been observed in various types of cancer, especially lung cancer. Existing evidences have consistently detected Cyclin D1 overexpression in a significant proportion of NSCLC cases, with reported frequencies ranging from approximately 60 and 76% [341, 342]. Similarly, c‐Myc, a prominent proto‐oncogene, encodes a TF that plays a pivotal role in regulating the expression of genes essential for fundamental cellular functions. By selectively amplifying the expression of its target genes, c‐Myc influences diverse processes such as cell growth, proliferation, differentiation, and division [341, 343, 344]. Additionally, it is a key regulator of apoptosis and angiogenesis. The overexpression of c‐Myc has been observed in multiple types of cancers, including lung cancer, where it is strongly associated with tumorigenesis and cancer progression. The involvement of c‐Myc in relationship with aggressiveness is reported across numerous human malignancies. In NSCLC, c‐Myc overexpression has been reported with significant variability, ranging from 18 to 91% across different studies. For example, a study analysis of 30 NSCLC samples found that 37% exhibited Myc overexpression, reflecting inconsistencies in the existing data [343, 345, 346]. These relationships underscore the critical importance of β‐catenin in regulating gene expression linked to cancer progression. As a consequence, dysregulation of the Wnt/β‐catenin signaling pathway is directly associated with a range of cancers. Notably, in various tumor types, it has been observed that the aberrant and excessive activation of the Wnt/β‐catenin signaling pathway is associated with the promotion of CSCs, which is highly associated with tumorigenesis, tumor metastasis, and chemotherapy resistance such as cisplatin, docetaxel, and radiotherapy, and Wnt inhibitors may restore sensitivity [333, 347].
Some HOX genes appear to impact lung cancer development by modulating the Wnt signaling pathway, particularly the Wnt/β‐catenin axis, which is commonly disrupted in various types of cancer. Research indicates that HOX proteins interact with β‐catenin and play a crucial role in regulating this pathway, underscoring their significance in cancer progression. Notably, specific HOX proteins, such as HOXA4, HOXA13, and HOXB5, have been directly implicated in the regulation of the Wnt/β‐catenin pathway, particularly in lung cancer [87, 348]. These proteins play crucial roles in cellular differentiation, proliferation, and tumor suppression. A brief overview of their functions and interactions with this pathway is provided below. HOXA4 exhibits dual roles, functioning either as an oncogene or a tumor suppressor, depending on the cancer type. Abnormal HOXA4 expression has been observed in various cancers, particularly in lung and cervical cancers. In lung cancer tissues, research shows that HOXA4 levels are significantly reduced, a change associated with more aggressive tumor characteristics, such as larger tumor size and also poorer patient outcomes. This reduction indicates a critical tumor‐suppressive role for HOXA4. Specifically, HOXA4 primarily acts as a tumor suppressor, largely through its interaction with the Wnt/β‐catenin signaling pathway in lung cancer. Notably, HOXA4 exerts its tumor‐suppressive effects by enhancing the transcription of GSK3β, a key regulator of the Wnt pathway. GSK3β facilitates the phosphorylation and subsequent degradation of β‐catenin, thereby reducing its levels within the pathway. Additionally, HOXA4 overexpression directly inhibits β‐catenin expression by binding to the promoter of its gene (CTNNB1). This suppression further diminishes the activity of key downstream target genes, including Cyclin D1 and c‐Myc. Through these mechanisms, HOXA4 effectively disrupts the Wnt/β‐catenin pathway, leading to reduced cell proliferation and metastasis in lung cancer [349, 350]. In contrast, studies have demonstrated that HOXA13 plays an oncogenic role in lung cancer. Experimental evidence from both cell line studies and patient samples confirms that HOXA13 is significantly overexpressed in NSCLC tissues compared with healthy samples, suggesting its involvement in cancer progression. In lung cancer, a key mechanism underlying HOXA13's oncogenic activity involves the transcriptional regulation of both the Wnt/β‐catenin and p53 signaling pathways. HOXA13 activates the Wnt/β‐catenin while inhibiting p53 signaling pathways, thereby promoting the tumor progression [351]. Consequently, the overexpression of HOXA13 expression not only reduces the expression of p53 itself but also directly leads to downregulation of p53 downstream target genes, such as P21 and protein Bcl‐2‐associated X protein (Bax). As a result, HOXA13 disrupts essential processes such as apoptosis. It should be noted that in normal condition, the p53 protein is one of the most important TF that has serious responsibility in regulation of various process, particularly regulating cell cycle arrest and promoting apoptosis. It should be noted that the p53 pathway encompasses a network of genes and interacts with several other pathways such as the Wnt pathway, the cyclin‐CDK pathway, and the p38 MAP kinase pathway [351, 352]. Specifically, p53 also involvement in two critical pathways, namely, p53/p21/p27 and p53/antiapoptotic protein B‐cell lymphoma‐2 (BCL‐2)/Bax, are essential for regulating apoptosis as well as cell cycle progression, especially during the G2/M phase. As a consequence, disrupting the p53 pathway can directly correlated with upregulation of cell proliferation in cancer [351, 352]. As mentioned earlier, HOXA13 activates the Wnt/β‐catenin signaling pathway, which in turn increases the expression of genes such as CTNNB1, MYC, and Cyclin D1. These genes are associated with enhance tumorigenesis and cancer progression. Consequently, HOXA13 overexpression significantly contributes to promoting cancer cell metastasis [351, 353]. Findings also have demonstrated a strong association between the expression of HOXB5 and tumor development in multiple cancer types [348]. HOXB5 plays a crucial role in modulating the Wnt/β‐catenin signaling pathway, and its association with β‐catenin has been extensively documented in various cancers, including lung, breast, HNSCC, as well as gastric cancers. HOXB5 significantly impacts cancer progression by directly interacting with the CTNNB1 promoter, which encodes β‐catenin. This interaction stimulates β‐catenin transcription and upregulates downstream targets such as cyclin D1 and c‐Myc, enhancing the invasive potential in gastric cancer, breast cancer, and particularly NSCLC [71, 354]. Experimental studies on NSCLC cell lines also show that knocking down HOXB5 reduces β‐catenin levels, which in turn decreases the expression of cyclin D1 and c‐Myc. This downregulation impairs NSCLC cell proliferation and invasion, emphasizing the crucial role of HOXB5 in regulating the Wnt/β‐catenin pathway during lung cancer progression [277, 354, 355]. Similarly, the Wnt/TCF signaling pathway plays a crucial role in the progression and metastasis of LUAD, particularly through its target genes HOXB9 and lymphoid enhancer binding factor 1 (LEF1). Therefore, aberrant activity of this pathway enhances the metastatic potential of tumor cells, enabling colonization of distant organs such as the brain and bones. HOXB9, a key target gene of TCF4, is notably upregulated in lung cancer and has been strongly linked to enhanced invasive and metastatic potential in malignant cells [356, 357]. Likewise, LEF1, one of the key transcriptional effectors of the canonical Wnt pathway, is regulated by Wnt3a and TCF4, strengthens Wnt signaling during malignancy Notably, LEF1 can interact with β‐catenin, driving increased transcriptional activity even in the absence of changes to β‐catenin levels, and is implicated in increasing tumor colony outgrowth [356]. Moreover, studies have shown that HOXB9 expression is induced by N‐acetylgalactosaminyltransferase (GalNAc‐T14). GalNAc‐T14 significantly contributes to enhancing the responsiveness of the Wnt pathway and to elevating the stabilization of β‐catenin. These molecular occurrences are probably linked to the development of an invasive characteristic in LUAD [87, 356, 358]. In addition, PITX2, as a TF, is also implicated in modulating the Wnt/β‐catenin signaling pathway. Within this context, PITX2 activates certain genes’ expression such as cyclin D1 and c‐Myc. Studies have indicated PITX2's association with LUAD, suggesting that it functions as an upregulated oncogene in this cancer type. Findings also have been reported that PITX2 is overexpressed in LUAD and correlates with poor patient prognosis. Furthermore, research demonstrates that PITX2 enhances the transcription of Wnt3a, thereby promoting oncogenic effects through activation of the Wnt/β‐catenin pathway [359]. Wnt3a, a well‐known activator of this pathway, is implicated in various cancers due to its critical role in tumorigenesis. Its involvement in this pathway allows Wnt3a to influence tumor progression in a complex manner, either suppressing or promoting activity. Correspondingly, this dual role highlights the complexity of Wnt3a's function in cancer biology, suggesting that its effects may vary based on the specific cancer type. Studies have specifically shown that Wnt3a enhances the development and progression of solid tumors, particularly lung cancers. The mechanism by which Wnt3a exerts its effects involves promoting cancer cell proliferation and self‐renewal [359, 360].
As mentioned earlier, dysregulation of the Wnt signaling pathway is closely implicated in the tumorigenesis of lung cancer and other cancer types. The transcriptional landscape of Wnt genes is also markedly altered in LUAD and LUSC, revealing potential biomarkers for these malignancies [359]. Therefore, alterations in the expression levels of Wnt genes and their inhibitors, often driven by genetic mutations and epigenetic factors such as abnormal DNA methylation, can substantially contribute to cancer development. These alterations can lead to key oncogenic processes such as increased cell proliferation, and metastasis. Hence, DNA hypermethylation plays a crucial role in disrupting Wnt signaling pathways by suppressing the expression of essential regulatory genes. In lung cancer, studies have identified abnormal methylation patterns in multiple Wnt inhibitors, correlating with the aberrant activity of the Wnt signaling pathway. Notably, key Wnt inhibitors such as APC, wingless‐type protein 7a (Wnt7a), Wnt inhibitory factor‐1 (WIF1), and axis inhibition protein (AXIN) are frequently found to be hypermethylated in lung cancer, particularly NSCLC [347]. This epigenetic silencing leads to the disruption of normal Wnt signaling regulation, thereby promoting oncogenic processes [301]. Among Wnt inhibitors genes, two homeobox genes, CDX2 and EMX2, have been identified as Wnt inhibitors. The hypermethylation of the promoters for CDX2 and EMX2 is associated with their downregulation, leading to transcriptional silencing in lung cancer [347, 361, 362, 363]. Both CDX2 and EMX2 are pivotal in regulating Wnt signaling pathways through their roles as inhibitors. Their promoter hypermethylation leads to decreased expression, facilitating enhanced Wnt signaling that promotes lung carcinogenesis. Understanding these mechanisms highlights potential therapeutic targets for restoring the function of Wnt inhibitors in lung cancer treatment strategies. Importantly, individual homeobox genes frequently converge on multiple oncogenic signaling cascades, with engagement patterns that are highly context‐dependent and shaped by tumor type, intrinsic genetic and epigenetic alterations, cell state, and the TME. For example, HOXA9 cooperates with cofactors such as PBX and MEIS to orchestrate a broad gene network and to interface with multiple signaling pathways, including Wnt, TGF‐β, PI3K/AKT, and NF‐κB, collectively regulating oncogenic processes such as EMT, autophagy, cell‐cycle progression, and cellular metabolism [281].
A Central Role of Homeobox Genes in Five Highest Incidence Cancer
4.3
As outlined earlier, dysregulation of homeobox genes is widespread across human malignancies, spanning solid tumors and hematologic cancers, and includes recurrent alterations in breast, colorectal, prostate, gastric, and notably lung cancers. These patterns provide a mechanistic basis for tumor‐type‐specific expression programs and clinical behavior. In particular, the dysregulation of HOX gene clusters has been reported in various solid tumors, with distinct expression patterns observed across different cancer types. For instance, genes in the HOXA cluster exhibit altered expression predominantly in breast and ovarian cancers. Additionally, genes in the HOXB cluster are commonly associated with colon cancer. Similarly, genes in the HOXC cluster are frequently upregulated in several malignancies, including colon and PCa. Moreover, genes in the HOXD cluster show aberrant expression in colon and breast cancers. Likewise, most genes across all four clusters of HOX have been reported to be dysregulated in lung cancer. Notably, recent studies suggest that HOX genes are also regulated at the level of nuclear‐cytoplasmic transport in carcinomas, adding another layer of complexity to their role in tumorigenesis. This suggests that the any dysregulation of HOX genes’ expression and localization may play an important role in the development and progression of various solid tumors. Furthermore, it appears that tumors arising from tissues with similar embryonic origins exhibit relatively similar patterns of HOXA and HOXB family gene expression. For example, tumors originating from endodermal tissues, such as the colon, prostate, and lung, show more comparable HOXA and HOXB genes expression profiles compared with breast tumors, which arise from the ectodermal mammary tissue [1, 3]. Importantly, HOXA11‐AS antisense RNA (HOXA11‐AS) has been found to be upregulated in NSCLC, suggesting its potential involvement in the development and progression of this cancer type. In contrast, the downregulation of the PITX1 gene has been significantly associated with more advanced tumor stages across various cancer types. This highlights its potential role as a tumor suppressor and suggests its possible application as a prognostic marker [149, 364].
Beyond solid tumors, HOX genes are also involved in later developmental processes that establish cell identity, such as hematopoiesis. Expression of HOX gene clusters A, B, and C has been detected in HSCs, underscoring their critical regulatory roles in blood cell development [365]. Deregulation of specific HOX genes has been linked to leukemia, with aberrant expression patterns contributing to the pathogenesis of ALL and AML. This evidence highlights the important role HOX genes play in maintaining normal hematopoietic function and how their misexpression can drive malignancies in blood‐forming tissues [148]. Because homeobox dysregulation is widespread across lineages, it is informative to consider the five most common cancers as global exemplars, illustrating how HOX networks extend across solid tumors. According to GLOBOCAN 2022, lung cancer has the highest number of new cases and remains the leading cause of cancer death. The five cancers with the greatest global incidence in 2022 were lung (∼2.5 million cases; 12.4%), female breast (∼2.3 million; 11.6%), colorectal (∼1.9 million; 9.6%), prostate (∼1.5 million; 7.3%), and stomach (∼0.97 million; 4.9%). These figures provide the current worldwide benchmark for statements written for 2025 [366].
Aberrant Expression of Homeobox Genes in Lung Cancer
4.3.1
In 2024, approximately 2,001,140 new cancer cases and 611,720 cancer‐related deaths were reported in the united states [367]. Lung cancer remains the first most common malignancy and the leading cause of cancer‐related mortality worldwide in 2024, resulting in the loss of about 350 lives each day [368, 369]. Lung cancer is classified into two main histological groups: NSCLC and small cell lung cancer (SCLC), which account for approximately 85% and 15% of all lung cancer cases, respectively. The NSCLC group can be further divided into various subtypes, with LUAD being the most common, representing around 40% of all lung cancer cases. The second most prevalent NSCLC subtype is LUSC, making up approximately 25% of lung cancer diagnoses. Additionally, large cell carcinoma with or without neuroendocrine features accounts for roughly 10% of lung cancer cases [370]. In lung cancer patients, tobacco use is significantly associated with 35% to % 50 of cases in men and around 17% in women. Additionally, the rising incidence of lung cancer cases can also be attributed to factors such as sedentary lifestyle, urban pollution, obesity, and increased use of cigarettes and alcohol. Moreover, the unregulated expression of the HOX genes in cancer can be influenced by factors such as temporospatial heterogeneity, gene dominance, genetic or epigenetic mechanisms, or a combination of these variables, as indicated by research studies [369].
As mentioned previously, homeobox genes are involved in the complex dynamics of cancer development, employing diverse mechanisms that contribute to the process of carcinogenesis. Their dual function as transcriptional regulators acting as both transcriptional activators and repressors is modulated by various factors, which renders their contribution to tumorigenesis multifaceted [3]. Therefore, any alterations in the expression of homeobox genes, whether through upregulation or downregulation, have been observed across different cancers, particularly in solid tumors such as lung cancer, especially in NSCLC. Importantly, NSCLC, which accounts for approximately 85% of all primary pulmonary carcinomas, remains the leading cause of cancer‐related mortality worldwide. Lung cancer is a complex biological process that arises from the intricate dysregulation of various oncogenes associated with cancer development. Despite advances in multimodal therapies including surgery, radiation, and chemotherapy, the 5‐year OS rate for NSCLC patients remains suboptimal. This underscores the critical need to identify precise and specific biomarkers that can enhance survival outcomes for NSCLC patients [371]. Research studies have demonstrated that numerous HOX genes, such as those from the HOXC and HOXD clusters, exhibit overexpression in primary NSCLC tissues and cell lines. For example, genes like HOXC4 and HOXC8 show significantly elevated levels in malignant cells compared with normal lung tissues. This dysregulation of HOX genes’ expression suggests that these TFs may contribute to the oncogenic processes involved in the development and progression of lung cancer [372]. In contrast, the downregulation of certain HOX gene clusters, such as HOXC9 and HOXD10, has been linked to increased cell proliferation, while overexpression of HOXA1 has also been observed in lung carcinoma. Furthermore, genes associated with four clusters of HOX gene exhibit distinct and complex expression patterns in lung cancer. Collectively, these alterations in HOX genes’ expression have been reported to negatively impact lung development, contributing to congenital abnormalities and the progression of lung carcinomas [1, 373].
In solid tumors, angiogenesis plays a significant role in the progression of them, recognized as an initial step in tumorigenesis. Moreover, it is a primary driving force behind tumor recurrence [3]. Research has reported the crucial role of specific HOX genes in angiogenesis associated with solid tumors. In particular, genes such as HOXB7 and HOXA11‐AS have been discovered to actively promote angiogenesis in lung cancer [149, 374, 375]. In LUAD, HOXB7 has been identified as a key factor involved in tumorigenicity and metastasis. Elevated levels of HOXB7 expression are associated with poor clinical outcomes and reduced survival rates in patients. Notably, HOXB7 is strongly linked to cell proliferation and metastasis in LUAD. Previous studies have shown that its knockdown leads to a reduction in the expression of certain genes such as VEGFA and matrix metalloproteinase‐2 (MMP‐2) [376, 377]. In addition, aberrant expression of HOXA11‐AS has garnered attention for its potential involvement in the development and progression of NSCLC through the regulation of certain genes and various signaling pathways, such as the TGF‐β signaling pathway. A significant overexpression of HOXA11‐AS has been observed in NSCLC tissues, including LUAD and SCC. This implies that HOXA11‐AS likely plays a direct role in tumorigenesis, angiogenesis, and invasion in NSCLC [149, 374, 375].
Homeobox Genes Dysregulation in Lung Cancer Pathogenesis
4.3.1.1
HOXA Cluster's Role in Lung Cancer
4.3.1.1.1
HOXA cluster genes play an integral role in several types of cancer, including lung cancer, leukemia, oral SSC, CRC, and pancreatic cancer [378, 379]. Notably, there is an abundance of CGIs within the HOXA gene cluster, which are believed to facilitate transcription initiation. In normal tissues, these CGIs are generally nonmethylated, allowing for active transcription of the HOXA genes. However, aberrant methylation patterns within the HOXA cluster can lead to gene silencing, which is often observed in various cancers, including lung cancer. According to an earlier report, the HOXA loci in the human LUAD was methylated at several CpG islands [380], although the methylation of the HOXA cluster genes is predominantly found in NSCLC [381]. In lung primary SCC, frequent methylation patterns in HOXA‐associated CpG islands were identified in the first stage of tumors, particularly involving HOXA1 and HOXA7 [2].
HOXA1 is known for its role in regulating various biological processes such as cell proliferation, differentiation, and apoptosis [190, 382]. The elevated levels of HOXA1 expression has been consistently linked to various malignancies, including various types of lung cancer, where they play a significant role in tumorigenesis [383]. Accordingly, research has demonstrated that HOXA1 expression is markedly elevated in NSCLC, suggesting its potential utility as a reliable biomarker for the diagnosis of this malignancy. Moreover, its expression correlates with advanced stages of the disease and higher stage tumor node metastasis (TNM) of in LUAD and LUSC. Additionally, its upregulation is correlated with advanced disease stages and higher TNM classifications in both LUAD and LUSC [384] [385]. Consequently, HOXA1 appears to be intricately linked to various biological processes associated with lung cancer. Furthermore, functional analyses, incorporating gene ontology and Kyoto encyclopedia of genes and genomes (KEGG) pathway assessments, indicating that HOXA1 plays a pivotal role in essential biological processes such as centromeres’ DNA binding, and demonstrates a significant linkage to the P53 signaling pathway. This connection suggests that HOXA1 may influence the progression of NSCLC by regulating pathways critical for cell cycle control and programmed cell death [385]. However, a previous study has reported that hypermethylation of the HOXA1 CpG island is present in LUAD. Subsequent studies have revealed that HOXA1 expression is detectable in 46% of SCLC patient samples. Notably, reduced HOXA1 expression is strongly associated with poorer prognosis and decreased survival rates in SCLC patients. Building on this, research has shown that HOTAIR regulates HOXA1 expression through methylation. These findings underscore the close relationship between HOTAIR and HOXA1 methylation [382, 384, 386]. Therefore, research indicates that depletion of HOTAIR leads to a significant decrease in the expression of the DNA methyltransferase enzymes DNMT1 and DNMT3b, which in turn correlates with decreased methylation of the HOXA1 gene [382]. This suggests a direct regulatory interaction between the HOTAIR and the HOXA1 gene, where in HOTAIR modulates the methylation status of HOXA1 through its influence on DNMT expression [387]. The mechanism by which HOTAIR influences HOXA1 further involves its interaction with epigenetic regulators, particularly the polycomb repressive complex 2 (PRC2). HOTAIR has been shown to recruit PRC2 to target genes, facilitating the trimethylation of histone H3 at lysine 27 (H3K27me3), a mark associated with gene silencing. This process is crucial for maintaining the transcriptional repression of genes like HOXA1, thereby influencing cellular processes such as proliferation and invasiveness in cancer cells, while the mechanism is unclear [388, 389]. As mention earlier, HOTAIR significantly influences the expression of HOXA1, demonstrating a multifaceted regulatory role in its downstream target gene. This interaction is intricately linked to the regulation of the NF‐κB signaling pathway, which plays a crucial role in cancer progression and drug resistance. The NF‐κB pathway plays a crucial role in various cellular processes, such as cell survival, and proliferation, as well as innate and adaptive immune response. However, when this pathway is abnormally activated, it is strongly associated with the transcription of genes that promote tumorigenesis, contributing to the process such as enhanced cancer cell proliferation, inhibition of apoptosis, and the enhancement of angiogenesis in various types of cancer [390]. In addition to HOTAIR role in lung cancer, HOTAIR has also been shown to significantly influence the regulation of the NF‐κB pathway across various cancer types. This underscores the critical nature of HOTAIR’s interaction with this pathway, demonstrating its potential as a key regulatory element in tumor biology. In vitro studies on SCLC cell lines have shown that silencing HOTAIR leads to inhibition of the NF‐κB pathway, while the overexpression of HOXA1 also diminishes this pathway's activity. In contrast, reducing HOXA1 expression reverses these effects, resulting in the activation of the NF‐κB pathway. This implies a dynamic regulatory relationship between HOTAIR, HOXA1, and in the context of SCLC. Besides, the NF‐κB pathway is significantly activated in multidrug‐resistant SCLC cell lines compared with their parental counterparts, leading to a reduced sensitivity of these cells to chemotherapy drugs. Consequently, research suggests that inhibiting this pathway with specific agents has been shown to improve the responsiveness of these resistant cells to various chemotherapeutic treatments. Notably, blocking NF‐κB not only facilitates apoptosis but also induces cell cycle arrest. Therefore, NF‐κB inhibitors could represent a potential therapeutic strategy for overcoming drug resistance in patients with elevated HOTAIR expression [382, 386, 387]. Another key factor involved in the regulation of HOXA1 expression is the influence of miRNAs in lung cancer. Several miRNAs exhibit a significant relationship with HOXA1 expression, emphasizing their regulatory roles [320]. Specifically, miR‐100, miR‐181b‐5p, miR‐181d‐5p, and miR‑577 have been identified as key regulators of HOXA1 expression in various types of lung cancer [385, 391, 392]. For instance, miR‐100 has been implicated in regulating HOXA1 expression in both SCLC and NSCLC, highlighting its complex role in lung cancer progression [392, 393]. In SCLC, HOXA1 expression is inversely correlated with miR‐100 levels, as miR‐100 directly targets the 3′‐UTR of the HOXA1 gene, leading to reduced HOXA1 expression. In vitro studies on multidrug‐resistant SCLC cell lines have demonstrated that restoring HOXA1 expression enhances sensitivity to chemotherapy agents. Conversely, silencing HOXA1 further exacerbates drug resistance in these cells. Additionally, elevated miR‐100 levels in drug‐resistant cell lines are inversely correlated with HOXA1 expression, underscoring the regulatory role of miR‐100 in chemoresistance [392]. In NSCLC, studies have revealed another aspect of miR‐100's influence on HOXA1 expression, underscoring its antitumor role. Notably, miR‐100 is frequently downregulated, leading to the upregulation of HOXA1 expression, which is associated with aggressive clinical features such as advanced TNM stage, lymph node metastasis, and poor OS. Functional studies confirm that restoring miR‐100 expression suppresses NSCLC cell proliferation, invasion, and migration by downregulating the expression of HOXA1 alongside with key pathways, including EMT process and Wnt/β‐catenin signaling pathway. Moreover, miR‐100 overexpression leads to decreased levels of key Wnt/β‐catenin components, such as cyclin D1 and c‐Myc, further limiting tumor progression. These findings underscore the pivotal role of miR‐100 in suppressing the invasive and aggressive properties of NSCLC cells [393]. Another miRNA that targets HOXA1 expression in NSCLC is miR‐577. Functionally, miR‐577 directly binds to the 3′‐UTR of HOXA1. The expression of miR‐577 has been found to be significantly reduced in NSCLC tissue samples and related cell lines compared with their nontumorous counterparts. In contrast, HOXA1 was notably upregulated at both the mRNA and protein levels in NSCLC, with its overexpression inversely correlated with miR‐577 expression, emphasizing its oncogenic role in the tumorigenesis of NSCLC. Experimental analyses also revealed that restoring miR‐577 expression led to a downregulation of HOXA1 and suppressed both the proliferation and invasion of NSCLC cells, suggesting its potential tumor‐suppressive role. These findings collectively imply that miR‐577 acts as a tumor suppressor in NSCLC by downregulating HOXA1 [320]. In LUSC, miR‐181b‐5p and miR‐181d‐5p have explored to significantly reduce compared with normal lung tissues, showing an inverse relationship with increased HOXA1 expression. This interaction is believed to influence several signaling pathways associated with lung cancer progression, particularly P53 pathway [385].
Expanding on the involvement of other genes within the HOXA cluster, HOXA2 has been identified as another member whose expression is altered through hypermethylation in both SCC and NSCLC. Evidence suggests that aberrant HOXA2 methylation patterns could serve as a potential biomarker for lung cancer, as these patterns stratify lung SCC into distinct transcriptional phenotypes, which are associated with varying prognoses [303, 394]. Previous research suggested that HOXA2 is more likely to be methylated in patients with recurrent SCC compared with those without recurrence, although no significant link was found between them [394]. The methylation profiles of HOXA2, frequently hypermethylated in SCC and linked to its prognosis, can provide prognostic information for SCC patients and also may assist in identifying biomarkers associated with the prognosis of NSCLC patients [303, 394, 395]. In NSCLC, HOXA2 is often downregulated and acts as a tumor suppressor, contributing to lung cancer progression. Additionally, several factors influence HOXA2 expression in NSCLC, including LINC00472 (an intergenic lncRNA) and miR‐1275. In NSCLC, both LINC00472 and HOXA2 expressions are significantly reduced, while miR‐1275 levels are elevated. The increased expression of miR‐1275 is likely associated with a malignant cell phenotype in NSCLC, as it targets HOXA2 expression. Experimental studies have shown that the re‐expression of LINC00472 can directly interact with miR‐1275, inhibiting its expression, which subsequently restores HOXA2 levels and mitigates the miR‐1275‐related malignant phenotype. Consequently, restoring HOXA2 and overexpressing LINC00472 lead to a reduction in malignant behaviors such as proliferation and EMT, while promoting apoptosis; however, these effects can be reversed by elevated miR‐1275 levels or silencing of HOXA2. The interplay between the LINC00472/miR‐1275/HOXA2 axis is crucial in the progression of NSCLC and accentuates potential therapeutic targets for intervention [395].
Another member of the HOXA cluster with a recognized role in various cancers is HOXA3. Extensive studies have reported its potential involvement in cancers such as colon cancer and nasopharyngeal carcinoma. Interestingly, the expression of HOXA3 shows cancer‐specific variation. For instance, HOXA3 expression is elevated in colon cancer, which is associated with poor survival rates. In contrast, HOXA3 expression is reduced in lung cancer, particularly NSCLC, due to elevated methylation levels. Notably, the methylation levels of HOXA3 are significantly elevated in LUAD samples, suggesting lower HOXA3 expression may be an independent protective factor. These findings suggest that lower HOXA3 expression, driven by hypermethylation, may act as an independent protective factor in LUAD, correlating with improved prognostic outcomes [378, 396]. In vitro studies on NSCLC cell lines indicate that HOXA3 is downregulated, which correlates with enhanced chemotherapy resistance and directly influences EMT‐related markers, such as a decrease in E‐cadherin expression. Research has also demonstrated that, both HOXA3 mRNA and its protein can interact with the lncRNA HOXA‐AS3, located on chromosome 7p15.2, suggesting that HOXA‐AS3 may contribute to the downregulation of HOXA3 in NSCLC. Experimental evidence further indicates that HOXA‐AS3 is upregulated in response to drug resistance, including cisplatin treatment in NSCLC. Notably, this elevated expression has been observed both in vivo and in vitro. Moreover, silencing HOXA‐AS3 enhances cisplatin efficacy by upregulating HOXA3 expression, which in turn promotes cell apoptosis. Furthermore, silencing HOXA‐AS3 improved the efficacy of cisplatin and upregulated HOXA3 expression, which subsequently enhanced cell apoptosis [273].
HOXA5 is a critical TF involved in both normal lung development and the progression of lung cancer, particularly in NSCLC. Its dual role indicates its importance in both physiological and pathological contexts [397]. HOXA5 plays a critical role in the proper development of the lung. Research on mouse models has shown that a lack of HOXA5 leads to severe respiratory distress at birth, due to significant structural defects in the lung, including impaired innervation of the diaphragm and abnormal tracheal formation. This underscores the pivotal role of HOXA5 in regulating the development of lung epithelial cells and associated structures, such as alveoli and airway cell. Specifically, HOXA5 influences the differentiation of various lung cell types and is involved in the signaling pathways that govern lung morphogenesis [398]. HOXA5 also plays a critical role in regulating cell proliferation through its interaction with the CDK inhibitor p21. This interaction is essential for controlling the cell cycle and promoting apoptosis in cancer cells. Additionally, it inhibits tumor growth, highlighting the tumor‐suppressive function of HOXA5 [399]. Research has established a direct link between the downregulation of HOXA5 expression and increased cell proliferation and migration in lung cancer, particularly in NSCLC samples. Its expression is significantly correlated with clinicopathological features, including tumor size and lymph node metastasis. These findings indicate the critical role of HOXA5 as a tumor suppressor, with its reduced expression strongly associated with lung tumorigenesis. So far, the lower level of HOXA5 expression is correspondingly linked to various adverse clinicopathological features, emphasizing its importance in cancer progression. Experimental evidence supports this notion, showing that ectopic HOXA5 overexpression in invasive lung cancer cell lines suppresses cell migration and invasion. Functionally, HOXA5 also suppresses cancer invasiveness partly by regulating cytoskeletal remodeling. Emerging research has revealed a correlation between the downregulation of HOXA5 expression and miRNA regulation, specifically, miR‐196a. Therefore, HOXA5 expression is influenced by miR‐196a, a miRNA that directly binds to its 3′‐UTR regions, leading to reduced HOXA5 levels at both mRNA and protein levels. This interaction promotes NSCLC cell proliferation, migration, and invasion, creating an inverse relationship between miR‐196a and HOXA5 expression in NSCLC tissues. While miR‐196a has been detected to implicate in multiple types of cancers, its complete role in lung cancer remains under investigation. However, these findings suggest that targeting miR‐196a could be a promising therapeutic strategy to restore HOXA5 expression and inhibit tumor progression [400, 401]. The abnormal methylation of HOXA5 is another identified epigenetic factor that downregulates its expression in lung cancer, a key epigenetic mechanism contributing to lung cancer tumorigenesis. In lung cancer, the expression of HOXA5 has detected to often absent due to the abnormal methylation of its promoter region, particularly in NSCLC's samples. This methylation‐induced silencing of the HOXA5 gene contributes to the development and progression of NSCLC's samples, and it can consider as a significant factor in the pathogenesis of this lung cancer subtype. Remarkably, patients with Stage I lung cancer are more likely to have methylated HOXA5 than never‐smokers, with methylated HOXA5 being detected in an average of 81.3% of NSCLCs and 51.8% of nonmalignant lung tissues [301, 397, 399, 402]. In NSCLCs, the level of HOXA5 expression also has been reported to dramatically reduce due to promoter hypermethylation. Consequently, the epigenetic downregulation of HOXA5 in NSCLC is associated with several clinically relevant factors, including increased tumor size, metastasis to both the primary tumor and lymph nodes, as well as higher TNM staging. Furthermore, the suppression of HOXA5 appears to be linked with the promotion of LUAD, suggesting that HOXA5 functions as a tumor suppressor in NSCLC. This downregulation of the HOXA5 gene also likely contributes to the progression and development of the cancer, particularly LUAD [394, 399].
HOXA7 has been found to exhibit promoter hypermethylation in lung cancer, with a higher incidence in NSCLC, especially the LUAD subtype. Numerous studies have demonstrated that HOXA7 promoter hypermethylation is significantly more detectable in both plasma and tumor samples of lung cancer patients compared with benign or healthy controls, underscoring its potential as a reliable diagnostic biomarker for lung cancer. For instance, HOXA7 hypermethylation was highly detectable in plasma samples from Stage IV patients. Notably, elevated HOXA7 methylation levels are associated with advanced TNM stages, particularly stage IV disease, further emphasizing its potential as a prognostic biomarker [403, 404]. In addition, studies on the methylation profiles of certain genes in plasma samples suggest that combining the detection of HOXA7 promoter hypermethylation with the promoter methylation of other genes, such as cysteine dioxygenase type 1 (CDO1), tachykinin precursor 1 (TAC1), and SRY‐box TF 17 (SOX17), demonstrates promising diagnostic performance for early‐stage NSCLC, achieving sensitivity and specificity values of up to 90% and 71%, respectively [403]. Beyond its epigenetic regulation, the reduction of HOXA7 expression in lung cancer is further influenced by its involvement in a ceRNA network, particularly with miR‐17‐5p and lncRNA histocompatibility leukocyte antigen complex P5 (HCP5). Studies have highlighted the role of the HCP5/miR‐17‐5p/HOXA7 axis in LUAD progression, with implications for ferroptosis a type of regulated cell death metastasis, and EMT. In LUAD samples, lncRNA HCP5 is significantly upregulated and strongly associated with EMT, promoting tumor growth and metastasis. In vitro studies using LUAD cell lines have demonstrated that HCP5 overexpression increases HOXA7 levels by binding to and suppressing miR‐17‐5p. This upregulation of HOXA7 by HCP5 promotes ferroptosis. Conversely, silencing HCP5 increases miR‐17‐5p levels, leading to the inhibition of HOXA7 expression and suppression of ferroptosis. Additionally, HOXA7 overexpression has been shown to significantly enhance cellular growth, proliferation, and migration in LUAD cells. Overall, this regulatory axis underscores the oncogenic role of HCP5/miR‐17‐5p/HOXA7 in LUAD and highlights its potential as a promising therapeutic target to inhibit tumor progression and metastasis [405].
HOXA9 functions as a key transcriptional regulator, binding to DNA and modulating the expression of numerous genes. This activity is essential during embryonic development and in preserving the characteristics of HSCs. The protein collaborates with other TFs, enhancing its specificity in binding to target genes, many of which are involved in cell division and cell survival processes [283, 406]. HOXA9 has been reported to involve in the pathogenesis of various cancers, particularly AML and solid tumors such as lung cancer [407, 408]. Methylation of the HOXA9 promoter region is a crucial mechanism by which its expression is repressed in cancer. In lung cancer, for instance, excessive methylation of the HOXA9 promoter results in diminished transcriptional activity, thereby contributing to tumorigenesis. This epigenetic alteration disrupts the normal regulatory pathways, enabling cancer cells to avoid differentiation and maintain uncontrolled proliferation [283, 381]. As noted earlier, the NF‐κB signaling pathway is involved in the regulation of various cellular processes, including inflammation, proliferation, and apoptosis. Therefore, deregulation of this pathway is implicated in the development and progression of multiple types of cancer, such as lung, gastric, and hepatocellular carcinoma. Remarkably, HOXA9 as a TF has been found to be associated with the NF‐κB signaling pathway [283]. HOXA9 has been reported to promote apoptosis and inhibit autophagy by transcriptionally regulating the p65 subunit of NF‐κB (RELA) in cutaneous SCC. Further studies have shown that the responsive region of NF‐κB is located within the first 400 base pairs of the HOXA9 promoter. In NSCLC, HOXA9 expression has detected to downregulate in tumor tissues compared with matched nontumor tissues, and overexpression of HOXA9 inhibited cell migration and invasion, which is linked to the regulation of NF‐κB activity. However, there are conflicting reports regarding the effects of HOXA9 on the activation of the NF‐κB signaling pathway. This could be attributed to the potential existence of cell heterogeneity, and further in‐depth investigations are warranted to elucidate the precise mechanisms by which HOXA9 regulates the NF‐κB signaling pathway [283]. The frequent hypermethylation of the HOXA9 gene plays a significant role in ovarian carcinogenesis, particularly in the early stages (FIGO stage I‐II) [408]. In breast cancer, HOXA9 and HOXA10 function as tumor suppressor genes, and methylation at their promoter CpGs islands has been shown to provide valuable prognostic information in breast cancer through interactions at the HOXA10–HOXA9 promoter CpG sites [407, 409]. A HOXA9 promoter hypermethylation was detected in 42.6% of NSCLC tumors (23 out of 54) [410]. Previous studies on NSCLC have observed that HOXA9 methylation rates increase with disease progression and cancer stages, with the rate of methylation increasing from Stage I (38.5%) to Stages III–IV (66.7%). However, no significant correlation was found with the TNM stage, and low detection rates were observed with the methylation frequency [410]. The study results indicated that among Stage IA LUAD samples, 42.9% of the HOXA9 gene was methylated, which is consistent with the primary tumor DNA. Additionally, a prior study also found hypermethylated HOXA9 in 80% of lung cancers during their early stages [381, 411]. Furthermore, HOXA9 hypermethylation in its promoter region has been detected in six lung cancer cell lines, whereas it is not observed in normal epithelial cells. Poor recurrence‐free survival (RFS) was observed in HOXA9 hypermethylation in never‐smokers, appearing that HOXA9 hypermethylation can serve as an independent prognostic factor for RFS in patients with nonsmoking NSCLC [412]. Functionally, the HOXA9 protein appears to prevent lung cancer cell migration, whereas its downregulation enhances cell invasiveness and promotes migration [406, 412]. Accordingly, HOXA9 overexpression in NSCLC cell lines has been shown to prevents invasion [410]. Moreover, HOXA9 methylation is highly prevalent in SCC compared with adenocarcinoma in lung cancer [413], and dysregulated HOXA9 is implicated in a variety of solid tumors, particularly in lung cancer [406, 412]. Additionally, HOXA9 promoter methylation can be considered a potential early lung cancer diagnostic biomarker and a tool for prognosis prediction. However, a deeper understanding of the mechanisms underlying the relationship between HOXA9 and the signaling pathways is still needed such as NF‐κB [381, 411].
Based on a cohort study, HOXA9 gene methylation along with two other methylated genes (CDO1 and TAC1) were considered as valuable biomarkers for diagnostic tests for NSCLC due to their sensitivity and specificity. Therefore, characterized methylation biomarkers might be useful for accurately detecting the early diagnosis and stages of NSCLC [414]. Subsequent experiments have revealed that hypermethylated HOXA9 has been detected in bronchial lavage fluids, indicating its potential as a biomarker in lung cancer [406]. HOXA2 and HOXA9 hypermethylation have been associated with high‐methylation and low‐methylation epigenotypes in NSCLC [415]. Within the HOXA clusters, both HOXA7 and HOXA9 promoters contain a high proportion of methylated DNA in the initial stage of SCCs, with HOXA9 being prominently methylated in 80% of the Stage I tumors, suggesting its potential as a promising marker for lung cancer [2]. In primary lung carcinomas, particularly LUSC, the CpG islands of HOXA7 and HOXA9 are methylated in tumors by 45 and 68%, respectively, despite being unmethylated in normal lung tissue [2]. These epigenetic modifications, which result in reduced expression of HOXA7 and HOXA9, may contribute to tumor recurrence and could be useful for detecting NSCLC [2, 303, 412]. In sputum and plasma, a high level of diagnostic accuracy can be identified for early‐stage lung cancer by detecting hypermethylated promoters of HOXA7 and HOXA9 [406, 416]. Furthermore, since HOXA9 exhibits higher methylation levels in SCLC compared with NSCLC, both genes may serve as valid biomarkers for SCLC. Data from lung cancer liquid biopsies support that HOXA9 methylation levels in circulating cell‐free DNA (cfDNA) can be effectively assessed in lung cancer subtypes, demonstrating *HOXA9’*s high sensitivity (63.8%) [303, 413]. In LUAD, current evidence suggests that HOXA9 promoter methylation serves as a potential biomarker for early‐stage diagnosis and risk stratification. Studies on Brazilian patients have shown that lower HOXA9 methylation levels are associated with improved cancer‐specific survival (CSS); however, this marker was not identified as an independent prognostic factor in this study [417].
HOXA10 expression in lung cancer has been reported inconsistently across various studies, with conflicting findings regarding its role. HOXA10 is also referred to by several alternative names, including PL, HOX1, HOX1H, and HOX1.8. Studies have reported methylation of HOXA10 in different lung cancer subtypes, such SCC, NSCLC, and LUSC [303, 394, 418]. These findings suggest that the methylation profiles of HOXA10 may provide valuable prognostic information for patients with SCC and NSCLC [394]. Notably, HOXA10 methylation has been highly detected in invasive peripheral pulmonary adenocarcinoma (ADC) compared with atypical adenomatous hyperplasia (AAH) or adenocarcinoma in situ (AIS). This indicates that HOXA10 methylation could be involved in tumor progression in the ADC. However, other studies have reported increased HOXA10 expression in lung cancer, highlighting the complexity and potential dual role of HOXA10 in tumor development [418, 419], HOXA10 overexpression is probably involved in the tumorigenesis and progression of NSCLC, particularly impacting LUSC more than LUAD. Analysis of extensive datasets indicates that HOXA10 mRNA expression is significantly elevated in NSCLC, with levels markedly higher in LUAD and even more upregulated in LUSC compared with noncancerous tissues—highlighting its importance in LUSC. Accordingly, studies have demonstrated that HOXA10 plays a significant role in the tumorigenesis and progression of NSCLC by influencing various signaling pathways. For instance, HOXA10 overexpression in NSCLC has been reported to associate with dysregulation of the Wnt and FGF pathways, both critical for tumor progression. Elevated HOXA10 levels are associated with increased FGF10 and FGF17 are correlated with the marked upregulation of HOXA10 in NSCLC, further supporting its role in tumor progression [418, 420].
Furthermore, the regulation of HOXA10 expression in lung cancer is influenced by lncRNAs and miRNAs. Research has identified several lncRNAs that play crucial roles in regulating HOXA10 expression, including HOXA11‐AS (also known as ENSG00000240990), LINC00461, LINC00466, and LINC00483. Accordingly, miRNAs also play a crucial role in modulating the level of HOXA10 expression, including miR‐144, miR‐195, and miR‐588, further emphasizing the complexity of its regulation and its importance in NSCLC pathogenesis [421, 422, 423]. A computational study on a ceRNA network has demonstrated that lncRNA HOXA11‐AS regulates the expression of HOXA10. The interaction between the HOXA11‐AS–HOXA10 pair and four miRNAs, specifically hsa‐let‐7a/b/f/g‐5p, is significantly associated with survival outcomes in lung cancer patients [421, 424]. Notably, lncRNA HOXA11‐AS can function either as a tumor suppressor or promoter depending on the cell type, and its expression has been explored to be aberrantly altered in various cancers. Therefore, its involvement in complex molecular networks highlights its critical role in tumorigenesis and cancer progression [423, 425]. For instance, lncRNA HOXA11‐AS is significantly upregulated in LUAD, alongside the overexpression of HOXA10. This suggests that HOXA11‐AS plays a crucial role in the progression of NSCLC, potentially through its influence on target genes, particularly HOXA10. Further research is essential to elucidate the precise mechanisms by which HOXA11‐ AS influences different malignancies, and emerging studies suggest that it may serve as a valuable biomarker for lung cancer prognosis [418, 421, 424]. LINC00466 and LINC00483 also have detected to play crucial roles in LUAD progression by regulating HOXA10 expression through interactions with their specific related miRNAs, particularly miR‐144. It should be noted that in LUAD, LINC00483 and LINC00466 are significantly upregulated, while miR‐144 is downregulated, resulting in the enhanced expression of HOXA10 [423, 426]. Studies on LUAD tissues have also demonstrated that elevated expression of LINC00466 and HOXA10, combined with reduced miR‐144 expression, is strongly associated with cancer progression. Notably, silencing LINC00466 or increasing miR‐144 expression leads to a significant reduction in HOXA10 levels. Silencing LINC00466 reduces its binding to miR‐144, thereby increasing miR‐144 levels and effectively suppressing processes that related to cancer development such as cell proliferation and migration, while promoting apoptosis. These findings collectively underscore the critical role of the LINC00466–miR‐144–HOXA10 axis in driving LUAD progression and suggest promising therapeutic targets within this pathway [426]. Similarly, LINC00483 binds competitively with miR‐144, effectively impacting the enhancement of HOXA10 in LUAD. This lncRNA contributes to tumor progression by promoting EMT, cell invasion, migration, and reducing radiosensitivity. Therefore, investigation had indicated that knockdown of LINC00483 reduces its binding to miR‐144, thereby increasing miR‐144 levels, which suppress HOXA10 expression. This suppression is directly associated with reduced EMT, cell proliferation, and invasion and increased radiosensitivity. Additionally, silencing LINC00483 decreases the expression of MMP‐2 and MMP‐9 (mesenchymal markers) while increasing the expression of E‐cadherin (an epithelial marker). These findings highlight the oncogenic role of LINC00483 and its potential as a therapeutic target in LUAD [423, 426]. In addition, the LINC00461/miR‐195/HOXA10 axis has also been implicated in the progression of LUAD. Overexpression of LINC00461 suppresses miR‐195, leading to increase the levels of HOXA10 expression. Additionally, upregulation of LINC00461 is associated with elevated the level of expression of critical genes, including MMP‐2, MMP‐9, and BCL‐2, while simultaneously reducing the expression of the BAX proapoptotic protein. As a result of upregulation of LINC00461, this axis promotes cell proliferation, migration, and reducing radiosensitivity in LUAD. Conversely, silencing LINC00461 exerts suppressive effects on LUAD by modulating the interaction between miR‐195 and HOXA10, highlighting its potential as a therapeutic target for LUAD [427]. Circular RNAs (circRNAs), a stable subclass of ncRNAs, have emerged as critical regulators in cancer biology, often functioning as miRNAs sponges. Under normal physiological conditions, circRNAs participate in various biological processes such as gene regulation. Although their role in lung cancer remains relatively underexplored, growing evidence suggests that circRNAs contribute significantly to cancer progression and malignancy, including in lung cancers such as NSCLC and LUAD. Recent studies have identified circRNAs, particularly circCSNK1G3 and circ_0010235, as key players in regulating HOXA10 expression [422]. In primary LUAD tissues and related cell lines, circCSNK1G3 has been shown to be aberrantly expressed, promoting critical processes such as cell proliferation and migration by sponging miR‐143‐3p. This interaction suppresses miR‐143‐3p expression, leading to HOXA10 upregulation and facilitating tumor growth and metastasis [422]. Similarly, circ_0010235 is overexpressed in NSCLC, where it regulates HOXA10 by sponging miR‐588, thereby driving malignancy and reducing radiosensitivity. Overexpression of circ_0010235 facilitates tumor progression by increasing HOXA10 levels, while its downregulation, coupled with miR‐588 upregulation, suppresses malignancy and enhances radiosensitivity in NSCLC. These findings underscore the potential therapeutic value of targeting the circCSNK1G3/miR‐143‐3p/HOXA10 and circ_0010235/miR‐588/HOXA10 axes in lung cancer [422, 428].
HOXA11 as an important TF plays a critical role in the proliferation, differentiation, and embryonic development of various tissues, particularly the endometrium [190, 283]. A prior study has indicated that the methylation of the promoter regions of HOXA5 and HOXA11 leads to transcriptional silencing, resulting in the loss of their tumor suppressor activities [381]. Evidence suggests that the HOXA11 gene is hypermethylated at CpG sites in adenocarcinomas compared with normal lung tissue; however, later studies revealed that HOXA11 was hypermethylated in SCC (74%) and adenocarcinoma (63%), indicating a higher prevalence in SCC [429, 430]. There are instances when LUADs originate from preneoplastic AAH lesions, which progress into AIS and eventually lead to invasive lung cancer. Hypermethylation of HOXA1 and HOXA11 at CGIs has been detected in AIS, suggesting that it might be possible to create specific biomarkers for the stages of development of LUAD based on the methylation level of these genes in lesions [431]. HOXA11 has been found hypermethylated in six lung cancer cell lines, including 69% of primary NSCLCs. HOXA11 hypermethylation is accompanied by downregulation. It is possible that hypermethylation of HOXA11 at its promoter in NSCLC drives progression by promoting cell proliferation and migration. In LUAD patients, HOXA11 hypermethylation may be used as a diagnostic and prognostic marker [429, 432].
Even‐Skipped Homeobox Genes’ Role in Lung Cancer
4.3.1.1.2
Previous studies have shown that hypermethylation of EVX1 is epigenetically associated with PCa [26]. Similarly, in NSCLC, particularly LUAD, EVX1 promoter promoter hypermethylation at CpG sites has been reported to correlate with lack of gene expression in [394, 433]. However, within some normal lung tissues have also detected to be methylated with EVX1 [434]. Additionally, it has been indicated that methylated EVX1 causes downregulation in lung primary tumors [394]. According to the study, a significant relationship has been identified between clinicopathological parameters and EVX1 hypermethylation. Furthermore, hypermethylated EVX1 may be associated with its silencing and determination of precancerous stages that influence tumor aggressiveness [433]. It was reported that the even‐skipped homeobox 2 (EVX2) gene has been detected hypermethylated at CGIs in SCCs’ samples [434, 435]. Moreover, methylated EVX2 was more prevalent in SCCs than in adenocarcinomas, whereas methylated Ras‐association domain family member 1A (RASSF1A) was more frequently observed in adenocarcinomas compared with SCCs in clinical samples from NSCLC patients. A high level of EVX2 promoter methylation has also been observed in NSCLC compared with noncancerous lung tissues [436]. In NSCLC, methylated EVX2 due to epigenetic alteration might be caused by the over‐activation of the PI3K/Akt pathway [434].
HOXB Cluster's Role in Lung Cancer
4.3.1.1.3
The expression levels of HOXB cluster genes exhibit notable variation across different cancer types, including lung cancer, and are regulated by a complex interplay of factors. Among these, HOXB5 plays a pivotal role in the Wnt signaling pathway, as previously described [3, 18]. Therefore, alterations in the expression of HOXB5 in lung cancer have been shown to disrupt this essential pathway, thereby implicating in the tumor progression and malignancy. Similarly, HOXB9 emerges as a significant player in cancer biology, particularly during lung cancer metastasis, where it acts as a target gene in the Wnt/TCF signaling pathway. Furthermore, epigenetic mechanisms, such as aberrant methylation of specific HOXB cluster genes, have been identified as contributors to dysregulation of genes within HOXB cluster, ultimately influencing the onset and progression of various types of cancer, including lung cancer [3, 18, 356]. For instance, hypermethylation of genes in the HOXB cluster, including HOXB2, HOXB3, HOXB4, HOXB9, and HOXB1, has been reported in several types of cancers. Specifically, HOXB3 hypermethylation has been linked to epithelial ovarian cancer [303]. In lung cancer, particularly LUAD, hypermethylation of HOXB3 and HOXB4 has been identified, suggesting their potential as diagnostic biomarkers [303, 396, 437]. Consequently, alterations in the expression of some genes within the HOXB cluster have been observed in lung cancer studies, as discussed in greater detail below [438].
HOXB2, similar to other HOX proteins, acts as a TF that governs gene expression critical for cellular processes such as differentiation and development. Dysregulation of HOXB2 expression has been linked to tumorigenesis and the progression of lung cancer, including NSCLC. This gene is regulated by miR‐139‐5p, a miRNA with potent tumor‐suppressive effects. By selectively binding to the 3′‐UTR region of HOXB2, miR‐139‐5p downregulates its expression, thereby inhibiting tumor growth through enhanced apoptosis and reduced cellular proliferation. Experimental studies on the lung cancer cell lines have demonstrated that the interaction between miR‐139‐5p and HOXB2 plays a pivotal role in modulating sensitivity to chemotherapy drugs, including cisplatin. Moreover, upregulating miR‐139‐5p has been shown to reverse resistance to chemotherapeutic agents such as cisplatin and paclitaxel in these cell lines. This effect is mediated through the suppression of HOXB2 and concurrent modulation of key signaling pathways, particularly the PI3K/AKT pathway and the activation of caspase‐3. The PI3K/AKT pathway, known for regulating cell survival and proliferation, is downregulated following the overexpression of miR‐139‐5p. Simultaneously, this upregulation increases the expression of caspase‐3, a critical enzyme involved in apoptosis. Together, these changes not only suppress tumor cell proliferation but also enhance apoptosis, providing a potential therapeutic approach to overcome drug resistance in lung cancer. Thus, targeting the miR‐139‐5p/HOXB2 axis holds promise for improving the effectiveness of chemotherapy and reducing tumor progression in lung cancer patients [439, 440].
The involvement of the HOXB3 gene in the prenatal lung during developmental stages has been linked to cellular differentiation processes. For instance, studies have demonstrated that HOXB3 promotes the expression of clara cell marker genes, facilitating the differentiation of M3E3/C3 cells into clara‐like cells, thereby underscoring its importance in normal lung development. However, the precise mechanisms underlying its function during these stages remain unclear [441]. Beyond its developmental role, HOXB3 has emerged as a significant factor in cancer biology, particularly in lung cancer. While aberrant methylation of HOXB3 has been observed, other studies have identified elevated HOXB3 expression levels in various malignancies, with a notable emphasis on lung cancer [303, 396, 437]. In lung cancer, particularly LUAD, HOXB3 and HOXB4 are epigenetically implicated through abnormal methylation patterns. Methylation analysis revealed that the promoter regions of the HOXB3 and HOXB4 genes are highly methylated in 75% of tumor samples, whereas no methylation is detected in healthy lung tissues. Additionally, the elevated methylation of the HOXB3/HOXB4 region is particularly evident in metastatic tumors, such as those in the brain and adrenal glands. This aberrant methylation is more pronounced in tumors with metastatic potential compared with nonmetastasizing tumors, underscoring its potential involvement in cancer progression. As a result, HOXB3/HOXB4 methylation has demonstrated high sensitivity and specificity in detecting lung cancer, underscoring its potential as a diagnostic biomarker [396]. On the other hand, high HOXB3 expression has been reported in lung cancer, particularly in LUAD, where it is consistently associated with poor clinical outcomes and reduced OS. Additionally, in vitro studies have demonstrated that HOXB3 downregulation inhibits LUAD cell proliferation and enhances their susceptibility to apoptosis. Computational analyses using tools like tumor immune estimation resource (TIMER) and tumor immune single‐cell database (TISIDB) further reveal a strong association between HOXB3 expression and tumor immunity such as immune checkpoint molecules (ICMs). These findings highlight HOXB3's oncogenic role and its link to tumor immunity in LUAD [438]. In addition, recent studies have also shown that HOXB4 promoter hypermethylation is frequent in LUAD, associates with early‐stage disease and poorer survival, and carries diagnostic/prognostic value [442].
Dysregulation of HOXB7 expression has been implicated in tumorigenesis and metastasis across various cancers. While its role in facilitating cancer progression is documented, the specific mechanisms by which HOXB7 expression levels contribute to malignancy remain largely unclear. Notably, HOXB7 overexpression has been observed in multiple cancer types, particularly lung and breast cancers biology [376, 443]. For example, in breast cancer cells, HOXB7 overexpression has been shown to drive the EMT, thereby enhancing tumor progression and metastasis, particularly to the lungs. In vivo and in vitro experiments have shown that this metastatic process may involve the regulation of the TGF‐β/SMAD3 signaling pathways [443]. Remarkably, TGF‐β signaling plays a complex role in cellular processes such as cell proliferation and differentiation, acting through both canonical SMAD‐dependent and noncanonical pathways. This signaling is initiated when one of the three TGF‐β isoforms (TGF‐β1, TGF‐β2, or TGF‐β3) binds to transmembrane serine/threonine kinase receptors. Additionally, dysregulation of the TGF‐β pathway is frequently observed in various cancers [160, 444]. The results of a study suggest that HOXB7 overexpression is directly linked to elevated TGFβ2 expression, indicating a potential role for HOXB7 upregulation in modulating TGFβ signaling. HOXB7 overexpression has been shown to attract tumor‐associated macrophages and drive an M2 phenotype via TGFβ2 signaling, which may contribute to metastatic progression. Notably, silencing TGFβ2 in the breast cancer cell lines overexpressing HOXB7 significantly reduces lung metastasis, emphasizing the critical interplay between HOXB7 and TGFβ2 in regulating tumor metastasis, particularly to lung. In LUAD, current evidence indicates that the higher level of HOXB7 expression is associated with increased inferred fractions of M0/M1 macrophages and decreased levels of resting mast cells and dendritic cells [17, 443]. Therefore, HOXB7 is significantly overexpressed in NSCLC and LUAD, suggesting its role as a key oncogene involved in carcinogenesis. Particularly, HOXB7 expression is significantly elevated in LUAD tissues compared with normal lung epithelial tissues. This upregulation is linked to increased cell proliferation and enhanced metastatic potential, contributing to poor clinical outcomes, advanced tumor stages, and reduced patient survival rates. Mechanistically, HOXB7 promotes cell growth and tumor progression by activating the MAPK and PI3K/Akt signaling pathways. Conversely, studies have demonstrated that silencing HOXB7 effectively suppresses associated metastatic processes in LUAD [443]. Moreover, the regulation of HOXB7 expression is intricately connected to taurine‐upregulated gene 1 (TUG1), an lncRNA that binds to the PRC2, facilitating H3K27 trimethylation at the HOXB7 promoter locus. In contrast, TUG1 downregulation disrupts PRC2 binding, leading to the loss of H3K27 trimethylation and increased HOXB7 expression. The expression of TUG1 is also directly regulated by the p53 protein, as TUG1 serves as a transcriptional target of p53. This regulatory relationship between p53 and TUG1 also suggests that p53 plays a role in influencing the expression of HOXB7. In NSCLC, TUG1 had been detected to be downregulated, leading to HOXB7 overexpression and promoting tumor proliferation through the MAPK and PI3K/Akt pathways. Correspondingly, reduced TUG1 expression is associated with advanced tumor stage and lower OS. These results indicate the critical role of the p53/TUG1/PRC2/HOXB7 axis in NSCLC tumorigenesis [445].
HOXB8 overexpression has been documented in several cancers, including CRC, and more recently, it has been found to be significantly upregulated in NSCLC tissues compared with adjacent normal tissues. This overexpression is strongly correlated with tumorigenesis and lymph node metastasis, which are linked to advanced tumor stages and poor survival rates. Functionally, HOXB8 drives NSCLC progression by promoting EMT. In vitro studies on NSCLC cell lines have demonstrated that silencing HOXB8 via siRNA suppresses EMT, evidenced by an increase in E‐cadherin expression and downregulation of N‐cadherin, MMP‐2, and vimentin. These findings highlight the potential of HOXB8 knockdown as a therapeutic strategy to inhibit EMT and reduce NSCLC progression [69, 446].
As described earlier, HOXB9 is consistently overexpressed in various cancers, including breast and lung cancer, where it functions as a key oncogene. Studies on lung cancer tissue samples demonstrate significantly elevated HOXB9 expression in NSCLC tissues compared with the noncancerous tissues. Specifically, HOXB9 overexpression has been detected in 21.3% of LUAD cases and is strongly associated with tumor progression, advanced TNM stages, and poor clinical outcomes, including reduced OS. Notably, its overexpression correlates with lower 5‐year survival rates, underscoring its role in aggressive tumor phenotypes [447, 448, 449]. In lung cancer, HOXB9 drives tumor‐promoting processes through mechanisms such as the Wnt pathway, the AMP kinase (AMPK)–HOXB9–Kirsten rat sarcoma viral oncogene homolog (KRAS) axis, and the EMT. By promoting EMT, HOXB9 overexpression is closely linked to larger tumor sizes and enhances the migratory and invasive abilities of NSCLC cells, directly contributing to metastases, including lymph node involvement and distant organ metastases, particularly to the brain and bones [356, 357, 447, 448]. In brain metastases, the role of HOXB9 is closely tied to its ability to activate the EMT. This process enables tumor cells to acquire greater mobility and invasive properties, allowing them to disseminate into the bloodstream and colonize other organs. Interestingly, elevated levels of HOXB9 have been linked to compromised blood–brain barrier (BBB) integrity through the disruption of ECs adhesion junctions via the upregulation of MMP‐9 expression. As a positive regulator of MMP‐9, its overexpression enhances the metastatic potential of NSCLC cells. The endopeptidase activity of MMP‐9 facilitates the degradation of ECM proteins and diminishes the expression of junctional proteins, ultimately weakening the BBB and allowing cancer cells to pass into the brain. This dual mechanism enhancing EMT and disrupting BBB integrity highlights the pivotal role of HOXB9 in driving metastasis of NSCLC to the brain. Clinical and experimental evidence consistently demonstrates that HOXB9 is significantly upregulated in the majority of primary NSCLC tumor samples. Moreover, patients with elevated HOXB9 expression in these tumors are more likely to have shorter brain metastasis‐free survival compared with those with lower HOXB9 expression, underscoring its potential role in NSCLC progression and metastasis [447]. Similarly, in addition to its role in lung cancer metastasis, HOXC9 has been reported to involve in breast cancer metastasis, particularly to the lungs. In vivo studies reveal that HOXC9 upregulation is associated with high tumor grade and promotes tumor growth and angiogenesis by upregulating the expression of angiogenic factors such as VEGF and TGF‐β. These factors drive cell motility, mesenchymal transitions, and alter the TME, enhancing neovascularization and facilitating distal metastasis to the lungs [449].
HOXC Cluster's Role in Lung Cancer
4.3.1.1.4
Several genes within the HOXC cluster have been implicated in the development and progression of various cancers, including lung cancer, and have attracted significant research interest. Among these, members such as HOXC8 and HOXC13 are notably overexpressed in primary lung tumors and lung cancer cell lines, underscoring their potential roles in tumorigenesis [372]. Therefore, overexpression of certain genes in this cluster have been observed in lung cancer studies, contributing to its pathogenesis, as detailed below. HOXC4 has been identified as an oncogene involved in tumorigenesis, contributing to the development and progression of multiple cancers, including lung cancer. Studies integrating data from the cancer genome atlas (TCGA) and genotype tissue‐expression (GTEx) datasets have demonstrated that HOXC4 is significantly upregulated in at least 21 different cancer types, highlighting its potential role in carcinogenesis. Notably, its abnormal overexpression has been observed in lung cancer, particularly in LUAD and LUSC, as well as in other malignancies such as colorectal adenocarcinoma and HNSCC [450, 451]. In lung cancer, evidence suggests that HOXC4 overexpression is associated with poor prognosis, including reduced OS, disease‐specific survival (DSS), disease‐free interval, and progression‐free interval (PFI). This correlation is particularly strong in early‐stage squamous cell lung cancer (SqCLC), where HOXC4 has one of the most pronounced impacts on poor prognosis and survival outcomes, especially OS. Additionally, in LUAD, studies indicate that aberrant HOXC4 expression correlates with mutations in mismatch repair (MMR) genes such as mutL homolog 1 (MLH1), mutS homolog 2 (MSH2), MSH6, and postmeiotic segregation increased 2 (PMS2). This suggests that the elevated level of HOXC4 expression may influence DNA repair pathways by regulating MMR genes, which are critical for repairing DNA mismatch errors, thereby enhancing the survival of related malignant cells [450, 451].
Aberrant HOXC6 expression has been explored to participate in the progression of various cancers, including BLCA and lung cancer. In lung cancer, HOXC6 overexpression has been observed across multiple histological subtypes, including NSCLC, LUAD, and LUSC, with approximately 66.6% of NSCLC tumors exhibiting elevated HOXC6 levels compared with healthy controls, suggesting its potential role as a biomarker. This overexpression is associated with enhanced cell proliferation, invasion, migration, lower immune scores, and poor clinical outcomes, including poor OS, advanced tumor size (T), and lymph node involvement (N) based on the TNM staging system. In cancer cells, prior studies have indicated that HOXC6 upregulation contributes to chemotherapy resistance by activating the multidrug resistance1 (MDR1) gene promoter and influencing other ATP‐binding cassette (ABC) transporters, specifically ABCG2 and MRP1, which expel chemotherapeutic agents from cancer cells [452]. Moreover, current evidence indicates that higher HOXC6 expression is associated with an increased inferred fraction of M0 macrophages, alongside decreased levels of resting mast cells and dendritic cells resting in LUAD [17]. It should be noted that HOXC6 also plays a crucial role in determining the effectiveness of targeted therapies, particularly gefitinib (Gef), which is an epidermal growth factor receptor‐tyrosine kinase inhibitor (EGFR‐TKI) commonly prescribed for the treatment of NSCLC. Correspondingly, in vitro studies on Gef‐resistant NSCLC cells demonstrate that knocking down HOXC6 expression not only enhances Gef sensitivity but also leads to increased apoptosis, G2/M phase cell cycle arrest, and suppression of cell migration [453]. Interestingly, it should be noted that patients with LUAD exhibiting upregulation in the expression of HOXC6 have been found to be more responsive to chemotherapeutic agents such as camptothecin and vorinostat. These results demonstrate the importance of considering HOXC6 expression as a potential therapeutic target for improving treatment strategies in lung cancer [453, 454]. In addition to its role in modulating the expression of various genes associated with lung cancer, HOXC6 has been identified as a crucial regulator of several genes that contribute to tumorigenesis. It affects the expression of key tumorigenesis‐related genes such as WNT6, MMP‐2, and secreted protein acidic and rich in cysteine (SPARC). Furthermore, HOXC6 is regulated by miR‐27 a, which directly influences its expression levels [453, 454]. In lung cancer, research indicates that while HOXC6 is often upregulated, miR‐27a is frequently downregulated in NSCLC, particularly LUAD; this imbalance is linked to aggressive malignancy and poor clinical outcomes in these patients. In contrast, in vitro and in vivo studies on Gef‐resistant NSCLC cells have indicated that the upregulation of miR‐27a can reverse Gef resistance by suppressing HOXC6 expression. Correspondingly, miR‐27a downregulates HOXC6, which subsequently influences downstream drug resistance‐related markers such as ABCG2, Bcl‐2, and N‐cadherin, all of which are implicated in drug resistance [453].
HOXC8 overexpression has been observed in various cancers, including breast and lung cancer, where its elevated levels are associated with disease progression and metastasis. In lung cancer, HOXC8 is prominently expressed in NSCLC, particularly in LUAD and LUSC, with significantly higher mRNA levels compared with healthy lung tissues. Studies in NSCLC cell lines have shown that HOXC8 is likely involved in regulating gene expression; its overexpression in these cell lines leads to the upregulation of genes such as TGFβ1 and vimentin, while downregulating E‐cadherin, thereby indicating its role as a transcriptional activator [379]. TGFβ1 is a well‐known cytokine with a critical role in cancer cell proliferation, migration, and metastasis across various cancer types. Notably, TGFβ1 expression is nearly doubled in NSCLC compared with normal tissues and is strongly correlated with tumorigenesis, including advanced TNM stages and lymph node metastases. In NSCLC, HOXC8‐mediated TGFβ1 upregulation directly contributes to enhancement cancer cell proliferation and anchorage‐independent growth [455, 456]. HOXC8 upregulation is also implicated in the EMT process in lung cancer by downregulating E‐cadherin and upregulating TGFβ1 and vimentin. The downregulation of E‐cadherin via HOXC8 is particularly associated with increased tumor growth, migration, and metastasis in NSCLC, indicating its significant role in cancer progression and poor clinical outcomes, including worse RFS. Moreover, HOXC8 upregulation contributes to increased chemoresistance in NSCLC. Conversely, in vitro studies have shown that silencing HOXC8 reduces chemoresistance and enhances NSCLC cell sensitivity to chemotherapy drugs such as cisplatin [379, 457]. Overexpression of HOXC9 has been observed in various cancers, including lung cancer, although its expression levels in lung cancer remain controversial. While many studies report elevated HOXC9 expression, some findings suggest that HOXC9 may exhibit aberrant methylation in lung cancer samples, particularly when analyzed alongside other genes such as RASSF1A and APC. These genes often display aberrantly methylated in their promoter regions, indicating a complex regulatory role for HOXC9 in lung cancer progression. Interestingly, the methylation status of HOXC9 and associated genes appears to be more characteristic of advanced stages of NSCLC compared with early stages [458, 459]. In LUSC, hypermethylation of the HOXC9 gene is observed at a frequency of 66%, whereas in Stage I NSCLC samples, HOXC9 methylation occurs in 13.71% of cases. These findings suggest that HOXC9 hypermethylation may serve as a potential biomarker for specific lung cancer subtypes and stages [459, 460]. On the other hand, HOXC9 overexpression in lung cancer, particularly in NSCLC subtypes such as LUAD, is widely reported and associated with poor clinical outcomes. HOXC9 overexpression has been confirmed in LUAD tumor tissues compared with adjacent noncancerous tissues, underscoring its role as a prognostic biomarker. In LUAD, upregulated HOXC9 expression correlates with advanced tumor stages and reduced OS, highlighting its oncogenic potential. However, the association between HOXC9 expression levels and OS is absent in LUSC. As an oncogene, HOXC9 overexpression contributes to carcinogenesis in lung cancer by promoting cell proliferation, migration, and invasion, particularly in LUAD [458, 461, 462]. Moreover, HOXC9 overexpression in LUAD is associated with immune suppression, as evidenced by reduced CD8+ T cell activation and diminished IFN‐γ production, whereas its knockdown enhances both, demonstrating its suppressive role in the TME. In LUAD, combining HOXC9 knockdown with immune checkpoint inhibitor therapies, such as PD‐1 blocking therapies, has demonstrated synergistic effects in reducing tumor growth and enhancing antitumor immune responses through pronounced activation of CD3+/CD8+/IFN‐γ+ T cells, which are critical for targeting and killing lung cancer cells [461]. In lung cancer, HOXC9 expression is also tightly regulated by specific circRNAs‐miRNA interactions [458, 462]. Notably, hsa_circ_0020123 functions as a miR‐495 sponge, preventing miR‐495 from binding to the HOXC9 3′‐UTR and thereby promoting HOXC9 expression. This regulatory mechanism enhances tumor cell migration and proliferation, as demonstrated by the reversal of miR‐495‐induced suppression upon HOXC9 overexpression. Elevated hsa_circ_0020123 expression is consequently associated with poor prognoses in NSCLC patients [458, 462]. Similarly, circCENPF functions as a hsa‐miR‐184 sponge, increasing HOXC9 expression and driving cell proliferation, migration, and invasion in NSCLC, particularly in LUAD. These findings underscore the significance of circRNA–miRNA–HOXC9 axis in NSCLC progression and their potential for therapeutic intervention [462, 463].
Aberrant expression of HOXC10 has been reported in various cancers, including lung cancer, where its elevated levels are strongly linked to tumorigenesis. Studies indicate that HOXC10 expression is significantly higher in tumor tissues compared with noncancerous tissues in NSCLC, particularly in LUAD patients. Overexpression of HOXC10 in LUAD is strongly associated with aggressive disease features, including advanced clinical stages and reduced distant metastasis‐free survival, underscoring its role in tumor progression and metastasis [464]. As an oncogene, elevated HOXC10 drives tumor progression in LUAD by promoting angiogenesis through VEGFA upregulation and increased microvessel density, which correlates with aggression, metastasis, advanced stages, and poor outcomes. Beyond its role in angiogenesis, HOXC10 upregulation has been reported to interact with the TME and immune regulatory pathway [313, 465, 466]. As mention earlier, research indicates a direct correlation between HOXC10 expression and metastasis in lung cancer patients. Notably, in cases of bone metastasis, HOXC10 expression is strongly associated with the presence of KRAS mutations, further emphasizing its role in lung cancer metastasis [464, 465]. KRAS, part of the sarcoma viral oncogene homolog (RAS) gene family, is the most frequently mutated oncogene in NSCLC, particularly in cases with bone metastases. These mutations, particularly in patients suffering bone metastases from LUAD, are linked to poor outcomes and have proven resistant to related‐targeted therapies such as mitogen‐activated protein kinase (MEK) inhibitors [464, 467, 468]. Experimental studies have shown that in NSCLC with KRAS mutations, around 50% of cases display abnormal expression of HOXC10. This overexpression is mainly linked to defects in PRC2, a vital histone methyltransferase that plays a significant role in regulating HOX gene expression. PRC2 is frequently associated with various cancers, functioning both as a tumor suppressor and as an oncogene [464, 469, 470]. Consequently, defects in PRC2 genes play a pivotal role in the dysregulation of HOXC10. Specifically, the downregulation of PRC2 results in the upregulation of HOXC10 in NSCLC, contributing to enhancement HOXC10’s role in tumor progression. HOXC10 interacts with the promoter region of nucleotide‐binding oligomerization domain 1 (NOD1) at its specific binding‐site, leading to enhanced expression of NOD1. This interaction plays a crucial role in the progression of KRAS‐mutant lung cancer bone metastasis by activating the NOD1/ ERK signaling pathway. Studies have demonstrated that suppressing HOXC10, particularly in combination with a STAT3 inhibitor, significantly inhibits the proliferation and migration of lung cancer cells with KRAS mutations in both in vivo and in vitro models. This suppression effectively reduces the metastatic spread of these cells to osteolytic bone sites by disrupting key mechanisms driving tumor progression. Specifically, HOXC10 inhibition has been shown to impair the NOD1/ERK signaling pathway, which plays a critical role in reprogramming the EMT and modulating the bone microenvironment [466].
HOXC11 has been found to be involved in the progression of various cancers, including renal cell carcinoma and CRC, where its overexpression correlates with poor clinical outcomes. In lung cancer, particularly in LUAD and LUSC, recent studies emphasize its significant role. Both of experimental and bioinformatics analyses demonstrate that HOXC11 expression is significantly elevated in lung cancer tissues compared with the noncancerous tissues, with its overexpression especially notable in LUAD and LUSC. This upregulation is strongly associated with tumorigenesis, aggressive tumor phenotype, and poor clinical outcomes, including shorter OS in LUAD, highlighting its potential as a prognostic biomarker [471]. In particular, upregulation of HOXC11 in LUAD cells has been reported to promote tumorigenesis and enhance malignant phenotypes, significantly affecting key characteristics such as cell proliferation, migration, and invasion. Elevated levels of HOXC11 are also linked to increased colony formation, as well as promotion of subcutaneous and lung metastasis, thereby contributing to the overall aggressiveness of the tumor. Moreover, HOXC11 expression is regulated through several mechanisms, including IκB kinase α (IKKα), and miR‐1197 [471, 472]. In LUAD, HOXC11 expression has been detected to be tightly regulated by IKKα. IKKα has been detected in various types of cancer and performs multiple functions through both NF‐κB‐dependent and independent mechanisms. In canonical NF‐κB signaling, IKKα, as part of IKK complex, phosphorylates IκBα, leading to its degradation via the ubiquitin‐proteasome pathway and subsequent nuclear translocation of NF‐κB dimers. Additionally, IKKα can be influenced in the regulation of cell proliferation by stabilizing β‐catenin to enhance cyclin D1 expression or by phosphorylating cyclin D1 to induce its degradation [161, 473]. Elevated levels of IKKα are directly associated with the posttranscriptional regulation of HOXC11, enhancing the levels of HOXC11 protein and promoting its stability by preventing degradation through the ubiquitin‐proteasome pathway. These results suggest the critical role of IKKα in promoting HOXC11 expression and driving tumor progression in lung cancer [471, 473]. HOXC11 also regulates the expression of sphingosine kinase 1 (SPHK1) (HOXC11 downstream target gene) by binding to its promoter region. SPHK1 is notably upregulated in NSCLC, particularly in LUAD, where its elevated expression correlates with a more malignant phenotype. Experimental studies indicate that inhibiting SPHK1 in LUAD cells with stable HOXC11 overexpression reduces migration and invasion, emphasizing the importance of the HOXC11‐SPHK1 axis in lung cancer progression. Moreover, the elevated levels of HOXC11 and SPHK1 are strongly associated with poor prognosis in LUAD patients [473]. Interestingly, previous studies have revealed a distinct regulation and function for HOXC11 in lung cancer, where it is downregulated by miR‐1197. In vitro studies have demonstrated that miR‐1197 is upregulated in NSCLC, and its expression inversely regulates HOXC11 expression in NSCLC, leading to HOXC11 downregulation. Conversely, silencing miR‐1197 results in the upregulation of HOXC11, which, in turn, inhibits NSCLC cell proliferation and migration. These studies indicate that miR‐1197 plays a tumor‐suppressing role when downregulated and provides a novel epigenetic regulatory axis involving miR‐1197 and HOXC11 in lung cancer [472].
HOXC13 has been identified as a potential oncogene in lung cancer, with experimental and bioinformatics analyses confirming its significant upregulation in LUAD and LUSC compared with normal tissues. In vitro studies show that HOXC13 mRNA and protein levels are notably higher in LUAD cell lines than the control cell lines. By analyzing data from the UCSC Xena and GEPIA databases on HOXC13 expression, it was shown that HOXC13 is consistently overexpressed across all stages of lung cancer. Clinically, this elevated expression is strongly associated with poor prognosis and adverse outcomes, suggesting HOXC13 as a promising prognostic biomarker [474, 475]. HOXC13 exerts its oncogenic effects in lung cancer by regulating the expression of key oncogenes, CCND1 (Cyclin D1) and CCNE1 (cyclin E1), which are crucial for tumor cell proliferation and are associated with poor clinical outcomes. Both genes are upregulated in lung cancer and correlate with enhanced metastasis and poor prognosis, highlighting their role as critical prognostic biomarkers. Specifically, the upregulation of CCND1 promotes cell proliferation, invasion, and migration [476, 477, 478]. Moreover, elevated CCNE1 expression has been observed in NSCLC, particularly LUAD, and is strongly associated with tumor growth [478, 479]. Thus, HOXC13‐mediated regulation of CCND1 and CCNE1 significantly contributes to lung cancer progression [476]. In lung cancer, upregulation of CCND1 leads to promote cell cycle progression into the S phase, while CCNE1 facilitates the transition from the G1 phase to the S phase. In LUAD, evidence indicates that elevated HOXC13 expression strongly correlates with the upregulation of both genes, enhancing cell proliferation and reducing G1‐phase arrest. This promotes continuous cell cycle progression and tumor growth, particularly in LUAD. Conversely, silencing HOXC13 downregulates both CCND1 and CCNE1, inducing G1‐phase arrest and inhibiting LUAD progression [476, 477].
In lung cancer, the expression of HOXC13 has also been demonstrated to be modulated by miR‐141 and the LncRNA HOXC‐AS2. In LUAD, miR‐141 suppresses the expression of HOXC13 by directly binding to its 3′‐UTR region, which subsequently leads to the downregulation of CCND1 and CCNE1. This regulation enables miR‐141 to effectively inhibit cell proliferation in LUAD. Notably, studies have shown that the upregulation of HOXC13 can counteract the inhibitory effects of miR‐141, underscoring the critical role of elevated HOXC13 levels in promoting cell growth by mitigating miR‐141‐mediated suppression. The lncRNA HOXC‐AS2 is located in close proximity to its target gene, HOXC13. Both experimental and bioinformatics analyses have confirmed a positive regulatory relationship between LncRNA HOXC‐AS2 and HOXC13, leading to elevated HOXC13 at both the mRNA and protein levels, particularly in NSCLC. Importantly, HOXC13 has been identified as a coexpressed target of LncRNA HOXC‐AS2, demonstrating the interdependence of their activities and emphasizing the critical role of the LncRNA HOXC‐AS2/HOXC13 regulatory axis in NSCLC pathogenesis [475, 476].
HOXD Cluster's Role in Lung Cancer
4.3.1.1.5
Several genes within the HOXD cluster have been implicated in the development and progression of various cancers, including lung cancer. This subfamily exhibits context‐dependent functions, acting as either oncogenes or tumor suppressors depending on the type of cancer. Therefore, dysregulated HOXD expression has been associated with key clinical and pathological features, emphasizing its contribution to tumor growth and malignancy [480, 481]. Many genes in this subfamily have been reported to exhibit altered regulation in lung cancer, as discussed in greater detail below.
HOXD1 is significantly downregulated in LUAD tissues and cell lines compared with their normal counterparts, and this reduced expression is associated with poor prognosis and lower OS in patients. DNA methylation of the HOXD1 promoter region has been identified as a key mechanism driving its downregulation. However, studies in LUAD have demonstrated that upregulation of HOXD1 can function as a tumor suppressor. Its overexpression has been shown to inhibit cell proliferation, migration, and invasion in both in vitro and in vivo models, particularly suppressing tumor growth in mouse models. Furthermore, elevated levels of HOXD1 act as a TF, promoting the expression of BMP2 and BMP6, which are implicated in the inhibition of LUAD progression. Although HOXD1 downregulation in LUAD has been reported, the key pathways and mechanisms associated with its aberrant methylation remain poorly understood [481].
HOXD3 has been the focus of conflicting reports regarding its expression levels in lung cancer. Studies on SCLC tissue samples report that HOXD3 is overexpressed in tumors originating from primary or metastatic sites, but not in noncancerous lung tissues, emphasizing its role in lung metastasis. However, other studies have been found that HOXD3 undergoes abnormal promoter methylation in lung cancer, particularly LUAD. These findings underscore the pivotal role of HOXD3 in lung cancer progression [481, 482, 483]. In vitro studies on lung cancer cell lines have demonstrated a direct association between elevated HOXD3 expression and increased invasion and metastasis, identifying HOXD3 as a potential metastatic gene. Therefore, the expression of the HOXD3 gene in lung cancer cell lines has been closely linked to alterations in cell adhesion, which directly contribute to promoting metastasis. Evidence suggests that elevated HOXD3 levels drive lung cancer metastasis through the regulation of genes’ expression involved in cell adhesion and metastasis. Specifically, HOXD3 overexpression is associated with the reduced expression of E‐cadherin and plakoglobin, both critical for maintaining epithelial integrity and cell–cell adhesion, while concurrently upregulating integrins and also N‐cadherin [484, 485]. E‐cadherin, a crucial cell–cell adhesion protein, plays an essential role in maintaining epithelial integrity. However, it is significantly downregulated in lung cancer, particularly in NSCLC, which can disrupt this important function [486, 487]. Similarly, plakoglobin, a key component of adherens junctions and desmosomes, is vital for the regulation of cell–cell adhesion. Studies indicate that plakoglobin expression is significantly downregulated in NSCLC cell lines, which contributes to their increased metastatic potential. Conversely, restoring plakoglobin levels has been shown to effectively suppress proliferation, invasion, and migration in lung cancer cells, indicating a tumor suppressor role of plakoglobin in lung cancer [488]. In contrast, aberrant expression of N‐cadherin is tightly associated with enhanced motility and malignancy of cancer cells. Its upregulation facilitates cancer progression by driving metastasis and invasion while promoting interactions between cancer cells and the extracellular microenvironment [489, 490]. Remarkably, this dual regulatory effect of HOXD3 underscores its role in enhancing the invasive and metastasis potential of lung cancer cells [485].
On the other hand, aberrant methylation of the HOXD3 gene has been specifically reported in various studies. Analyses of samples from patients with LUAD have demonstrated that the aberrant methylation of HOXD3 is specific to cancerous tissues and is not present in healthy lung tissues, highlighting its potential as a cancer‐specific epigenetic alteration. Similarly, research utilizing blood‐based liquid biopsy samples from cancer patients, particularly those with lung cancer, has identified HOXD3 as one of the genes undergoing abnormal methylation in its promoter region. Moreover, studies have shown that circulating methylated HOXD3 levels, along with other genes such as RASSF1A, are significantly elevated in SCLC compared with NSCLC. Panels incorporating methylated HOXD3 and RASSF1A achieve high sensitivity (75%) and specificity (88%) in distinguishing between lung cancer subtypes, from SCLC to NSCLC. Furthermore, elevated levels of methylated HOXD3 are associated with node‐positive disease, metastatic dissemination, and an increased disease‐specific mortality rate. Consequently, methylated HOXD3 has emerged as a potential biomarker for both diagnosis and prognosis in lung cancer [481, 482, 483].
The expression of HOXD8 in lung cancer has demonstrated variability across studies. While certain investigations have reported upregulated HOXD8 expression, others have highlighted abnormal methylation of its promoter region. These findings suggest a complex regulatory mechanism influencing HOXD8’s role in lung cancer pathogenesis [436, 491]. Notably, HOXD8 is implicated in processes such as cell migration and has been found to be methylated in LUADs. This methylation is associated with its downregulation, which is observed more frequently in the metastatic tumors than in primary ones, underscoring its critical involvement in metastasis [436]. On the other hand, HOXD8 overexpression has been observed in various cancers, including ovarian and lung cancers, where it plays a significant role in tumor progression. In lung cancer, HOXD8 expression is markedly elevated in tumor tissues compared with adjacent noncancerous tissues, indicating its potential oncogenic role. Studies have demonstrated that HOXD8 protein levels are particularly increased in NSCLC. Consequently, in NSCLC, this upregulation is correlated with increased cancer cell proliferation, the formation of CSCs, and enhanced migratory capacity. These findings underscore the oncogenic role of HOXD8 in driving the aggressive behavior of lung cancer cells. HOXD8 expression in lung cancer has been demonstrated to be modulated by miRNAs. Specifically, HOXD8 has been identified as a direct target gene of miR‐142‐5p and miR‐520a‐3p, suggesting that these miRNAs play a crucial role in the regulating of HOXD8 expression levels in lung cancer cells. Studies have further demonstrated a direct correlation between the expression levels of miR‐142‐5p and miR‐520a‐3p and the regulation of HOXD8, which significantly contributes to the resistance of lung cancer cell lines to Gef [491, 492]. Therefore, HOXD8 and miR‐142‐5p play critical roles in modulating Gef sensitivity in NSCLC cell lines. Studies have shown that miR‐142‐5p binds to the 3′‐UTR of HOXD8 mRNA, resulting in the downregulation of HOXD8 expression. Consequently, overexpression of miR‐142‐5p significantly reduces protein expression level of HOXD8, whereas its downregulation results in HOXD8 upregulation, demonstrating a clear negative correlation between their expressions. Notably, miR‐142‐5p enhances Gef‐induced apoptosis in lung cancer cells by activating the mitochondria‐dependent apoptosis pathway, a process modulated through HOXD8 regulation. Therefore, studies indicate that the coexpression of miR‐142‐5p and HOXD8 significantly reduces the levels of apoptosis‐related proteins such as Bax, cleaved‐caspase3, and cleaved‐PARP, potentially influencing the cells response to Gef [492]. Studies on NSCLC and SCLC tissues and the related‐cell lines have demonstrated distinct roles of miR‐520a‐3p expression in NSCLC. While miR‐520a‐3p expression is decreased in NSCLC, its upregulation acts as a tumor suppressor by directly targeting HOXD8. Overexpression of miR‐520a‐3p not only degrades HOXD8 mRNA but also inhibits cancer cell proliferation and CSC phenotypes specifically in NSCLC, with no notable effect in SCLC. Moreover, overexpression of miR‐520a‐3p leads to downregulate the level of the mesenchymal–epithelial transition (MET), while HOXD8 has an adverse effect on its expression and leads to an increase in its expression [491]. MET, a proto‐oncogene expressed on epithelial cells across various organs, plays a pivotal role in biological processes such as embryonic development and tissue regeneration. Notably, MET–hepatocyte growth factor pathway is one of the well‐known pathways involved in several types of cancer, particularly lung cancer. Therefore, dysregulation of the MET pathway, often associated with mutations or overexpression, contributes to tumorigenesis and cancer progression through pathways such as Wnt/β‐catenin signaling, which drives proliferation, metastasis, and invasion in cancer cells [493, 494]. In NSCLC, aberrant MET dysregulation is frequently reported and is probably linked to resistance against Gef. Moreover, HOXD8‐mediated MET upregulation correlates with Gef resistance, whereas suppression of the MET has been shown to reverse this resistance in NSCLC cell lines [491, 493].
The aberrant expression of HOXD9 has been detected in various cancers, including glioblastomas, gastric cancer, and lung cancer. In NSCLC, HOXD9 is significantly upregulated in tissues and cell lines compared with normal counterparts, correlating with poor prognosis and lower OS rate. Therefore, overexpression of HOXD9 in NSCLC directly contributes to tumorigenesis such as cell proliferation, migration, invasion, and apoptosis inhibition. In addition, its upregulation is correlated with metastasis in NSCLC [494, 495, 496]. Research suggests that immunoglobulin TF 2 (ITF2), also known as TCF4, plays a crucial role in the regulation of HOXD9 expression. ITF2, known as a downstream target of the Wnt/β‐catenin signaling pathway, is a key TF whose reduced expression is frequently observed in lung cancer. ITF2 expression is often reduced in lung cancer, but its overexpression inversely correlates with HOXD9 levels, improving patient outcomes by inhibiting tumor progression. This regulation appears to be mediated through an alternative mechanism involving Wnt pathway activation. Consequently, upregulation of ITF2 is associated with lower HOXD9 expression and improved OS in NSCLC patients. In vivo and in vitro studies further support the idea that silencing HOXD9 suppresses metastasis, invasion, cell proliferation, and cell migration, while inducing apoptosis in NSCLC [495, 497]. Moreover, evidence indicates that the silence of HOXD9 in NSCLC cell lines suppresses proliferation by inducing G1 cell cycle arrest. This process is characterized by reduced expression of cyclin E and cyclin B1, along with increased expression of p53, a critical regulator of DNA repair and cell cycle progression. Cyclin E, a key regulator of the G1/S transition in the cell cycle, is frequently overexpressed in tumors, contributing to dysregulated cell cycle progression in lung cancer. Similarly, cyclin B1 (also known as CCNB1), critical for the G2‐M phase transition, is typically low under normal conditions but is upregulated during the transition and associated with malignancy in various cancers, including NSCLC, particularly in LUAD. Consequently, the aberrant expression of both of them drives uncontrolled cell cycle progression [495, 498, 499]. HOXD9 also contributes to the regulation of angiogenesis and immune evasion in NSCLC. Specifically, it regulates the expression of ANGPT2 and programmed death ligand‐1 (PD‐L1), both of which are closely associated with tumor progression. ANGPT2, a proangiogenic factor, works alongside VEGF to significantly contribute to tumor vascularization and is upregulated in several cancers, including lung cancer. Elevated plasma levels of ANGPT2 have been observed in NSCLC patients, with further increases noted following tumor resection [495, 500]. The overexpression of HOXD9 is positively associated with elevated levels of ANGPT2, thereby promoting metastasis and angiogenesis in lung cancer. In contrast, downregulation of HOXD9 leads to a reduction in ANGPT2 expression. ANGPT2, a proangiogenic factor, besides VEGF plays a significant role in tumor vascularization and is upregulated in several cancers, including lung cancer. Elevated plasma levels of ANGPT2 have been observed in NSCLC patients, with further increases noted after tumor resection. Additionally, HOXD9 inhibition promotes apoptosis by activating caspase‐3 and polymerase 1 (PARP1) cleavage, disrupting DNA repair and inducing apoptosis [495, 500]. In addition, both in vivo and in vitro studies on lung cancer cell lines have demonstrated that HOXD9 upregulates PD‐L1, a critical ICM. This upregulation significantly aids tumor cells in evading recognition by the immune system, particularly T cells, thereby contributing to unfavorable clinical outcomes in these patients. In contrast, silencing HOXD9 markedly reduces PD‐L1 expression, underscoring its role in immune evasion mechanisms in NSCLC [495]. As previously noted, HOXD9 plays a significant role in lung cancer metastasis. Among the mechanisms linking HOXD9 to this process is its ability to promote increased glycolysis. The metabolic reprogramming observed NSCLC, characterized by the Warburg effect, is closely tied to the transcriptional activation of 6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase 3 (PFKFB3) by HOXD9. By directly binding to the promoter region of PFKFB3, a critical enzyme in glycolysis, HOXD9 enhances its expression, establishing a positive relationship between HOXD9 overexpression and PFKFB3 upregulation in lung cancer. Notably, PFKFB3 upregulation has been reported across various cancer types, including NSCLC, where it plays a central role in enhancing glycolysis, a hallmark of the Warburg effect [496, 501]. Prior evidence highlights a strong correlation between the Warburg effect and metastasis in NSCLC. In tumor cells, including those from NSCLC, glycolysis is the preferred pathway for glucose metabolism, rather than relying on mitochondrial oxidative phosphorylation. Consequently, elevated levels of HOXD9 in NSCLC tissues enhance glycolysis and significantly contribute to tumor metastasis. Importantly, suppressing PFKFB3 has been shown to markedly reduce the oncogenic effects of HOXD9, including its ability to promote metastasis [496].
The expression of HOXD10 is notably reduced in lung cancer, demonstrating its role as a tumor suppressor in this disease. Furthermore, HOXD10 methylation has been identified in plasma samples from patients with lung cancer. Consequently, the methylation of HOXD10, along with other genes such as PAX9, STAG3, and PTPRN2, suggests its potential utility as a diagnostic biomarker [303, 502, 503]. HOXD10 expression is markedly reduced in NSCLC tissues and cell lines when compared with normal, noncancerous tissues. This downregulation is further influenced by miR‐224, a miRNA that is highly expressed in metastatic NSCLC tissues and the cell lines. MiR‐224 directly targets the 3′‐UTR of HOXD10, leading to its suppression. Overexpression of miR‐224 significantly reduces HOXD10 levels, impairing its tumor‐suppressive functions and promoting cell migration and invasion in NSCLC. These findings highlight the critical interplay between HOXD10 and miR‐224 in lung cancer progression, although further research is needed to elucidate the precise mechanisms underlying this relationship [503]. Studies have shown that HOXD11 exhibits inconsistent expression patterns in lung cancer, with evidence of both upregulation and downregulation depending on the lung cancer subtype. This dual profile underscores its complex role in lung cancer pathogenesis [474, 504, 505]. On the CpG island, the HOXD11 gene has been found to be methylated in lung tumors in four out of eight cases. In contrast, DNA methylation in normal lung tissue was also observed in three out of eight cases for HOXD11 [380]. cfDNA methylation analyses in plasma samples from lung cancer patients have identified HOXD11, along with genes like BCAR1 and HOPX, as hypermethylated [504]. Methylation of HOXD11 is frequently observed in the oral epithelium of lung cancer patients, particularly among smokers and individuals over 50 years of age. Combined analyses of methylation profiles have demonstrated high specificity and predictive value for assessing lung cancer risk, especially in relation to smoking and age. Aberrant HOXD11 methylation in NSCLC is also detected to associate with resectable tumors, emphasizing its potential for early detection and risk stratification. Therefore, DNA methylation studies further emphasize the methylation of HOXD11 as a promising biomarker [505]. Conversely, evidence has revealed significant overexpression of HOXD11, along with other HOX genes, in LUAD and LUSC tissues compared with the healthy tissue. This elevated expression is strongly correlated with poor clinical outcomes, including reduced OS and postprogression survival (PPS), further highlighting the complexity and dual role of HOXD11 in lung cancer progression and prognosis [474].
The reported expression patterns of HOXD13 in lung cancer vary significantly across studies, reflecting its complex role in tumor biology. Some research highlights hypermethylation of the HOXD13 promoter, which may lead to transcriptional silencing. Conversely, other studies report increased expression of HOXD13 in lung cancer [474, 483]. Methylation of the HOXD13 promoter CpG island has been observed in lung tumors, reported in seven out of eight cases studied, highlighting context‐specific epigenetic silencing within the HOXD cluster. However, DNA methylation in normal lung tissue was observed in only one case [380]. Moreover, aberrant hypermethylation of HOXD13 has been particularly observed in LUAD, suggesting its potential as a biomarker [483]. Bioinformatics analyses, including GEPIA, have demonstrated that HOXD13, along with other HOX genes such as HOXA13, HOXB13, HOXC13, and HOXD11, shows significantly higher expression in LUAD and LUSC tissues compared with the healthy tissues. Notably, this overexpression appears to persist across all stages of lung cancer and is strongly associated with a worse prognosis. The elevated expression of HOXD13 and related HOX genes underscores their significant diagnostic and prognostic value, correlating with reduced OS and PPS. This makes them promising targets for further research and clinical applications in lung cancer management [474].
Paired‐Like Homeodomain Family's Role in Lung Cancer
4.3.1.1.6
However, the role of PITX1 in lung development remains not fully understood, dysregulation of genes within PITX family has been observed in LUAD patient samples, and methylation of PITX1 and PITX2 has also been reported in lung cancer [45]. Despite of this, the expression profile of the PITX gene family has not been thoroughly explored in lung cancer, and further investigation will be required. Additionally, accumulating evidence suggests that PITX1 functions as a tumor suppressor in various cancers, including lung cancer, melanoma, and ESCC [45, 364] [506, 507]. Hypermethylation of PITX1 gene in its promoter region has been detected in primary lung tumors and lung cancer, leading in downregulated expression. This suggesting that PITX1 downregulation is associated with tumor progression, development, and higher stages of the tumor [364]. Studies have demonstrated that higher levels of PITX1 are associated with decreased cell proliferation and enhanced apoptosis in melanoma cells. In ESCC, hypermethylation of the PITX1 promoter was linked to increased tumor depth and advanced stages, indicating that reduced PITX1 expression may contribute to aggressive cancer behavior [280]. PITX1 has been reported to inhibit the RAS signaling pathway and promote p53 activity, which are crucial in regulating cell proliferation and survival. This suggests that PITX1 may play a pivotal role in tumor suppression by modulating these key cellular pathways [507]. In 62% of the lung cancer samples, the lack of PITX1 expression was identified, and lower levels of expression were associated with advanced tumor stages. Despite this fact, the mechanism behind the downregulation of PITX1 in lung cancer may not be due to its promoter hypermethylation, and further investigation will be required to determine the exact cause of downregulation [364]. Therefore, restoring PITX1 function or targeting its regulatory pathways may offer therapeutic avenues for treating cancers where PITX1 is silenced.
PITX2 is responsible for lungs left–right asymmetry but may not be necessary for other organs [364]. Although PITX2 involvement in carcinogenesis has yet to be precisely elucidated, the role of PITX2 hypermethylation has been implicated in a variety of cancer types such as AML, prostate, and breast carcinoma [508, 509]. Hypermethylation of PITX2 gene has been detected in lung SCC, with higher levels in tumor tissue compared with adjacent nontumor lung tissue [460]. Subsequent studies have revealed that methylation of PITX2 occurred in both hypo‐ and hypermethylation in NSCLC. It is possible that hypomethylation of PITX2 could lead to PITX2 expression and subsequently cause the expression of GTP‐binding protein Di‐Ras3 (DIRAS3) [508]. DIRAS3, also known as aplysia ras homology member I (ARHI), is an imprinted gene that is predominantly expressed from the paternal allele. Its downregulation has been implicated in various cancers, including lung cancer, where the loss of DIRAS3 expression can occur through different mechanisms. Studies have shown that the re‐expression of DIRAS3 can inhibit multiple oncogenic signaling pathways, such as PI3K/AKT and RAS/MAPK, thereby blocking malignant transformation and promoting autophagy in cancer cells [510]. The evidence indicates that the DNA methylation status of PITX2 is associated with the risk of disease. Thus, in patients with low PITX2 methylation, the risk of disease progression was significantly higher than in patients with high methylation. Hypermethylation of PITX2, but not hypomethylation, has been reported to be associated with prolonged survival. As a biomarker in patients with NSCLC, the methylation of PITX2 has been demonstrated to provide independent prognostic information regarding disease progression [508].
Short Stature Homeobox Family's Role in Lung Cancer
4.3.1.1.7
Studies confirm that SHOX2 has important roles in lung carcinogenesis. SHOX2 hypo‐ and hypermethylation has been detected in patients with NSCLC. Hypermethylation of SHOX2, but not hypomethylation, has been reported to be associated with indicative of prolonged survival [508]. In 96% of tumor samples from lung cancer patients, hypermethylation of the SHOX2 locus was found compared with that of normal adjacent tissues [47]. Accordingly, methylation of SHOX2 was present in 70% of NSCLC, 97% of SCLC, 82% in squamous carcinoma, and 47% of adenocarcinoma [511]. The later study revealed SHOX2 as a biomarker has been detected in 80% of SCLC and 63% of SCC, along with high sensitivity in advanced stages of cancer in comparison with the first stage [512]. In patients with low SHOX2 methylation, the risk of disease progression was significantly higher than in patients with high SHOX2 methylation [508]. The detection of hypermethylated SHOX2 in bronchial aspirates could potentially provide a helpful biomarker for diagnosing lung carcinoma patients, particularly in cases with unclear results from cytological and histological examinations [511]. Despite the absence of visible tumors in the bronchoscopy, the methylation of SHOX2 as a potential biomarker allowed the detection of malignant lung disease through blood plasma [512]. The methylation of SHOX2 has been demonstrated to provide independent prognostic biomarkers for the progression of cancer in patients with NSCLC [508].
The 3q region of the chromosome is considered a critical region because it is frequently amplified in lung cancer. The differences between adenocarcinomas and SCC of the lungs are largely attributed to differential gene expression and 3q chromosomal copy number alterations. According to the study, SHOX2 (located at the 3q locus) exhibits higher methylation in lung SCC compared with LUAD. Therefore, it is possible to justify the connection between SHOX2 hypermethylation and the locus amplification [508]. Accordingly, hypermethylation of the SHOX2 gene locus has been found to be associated with frequent gene amplification, despite no differences in the expression of the SHOX2 gene [47]. SHOX2 hypermethylation shows promise as a biomarker in two key areas: risk stratification for the development of secondary primary lung cancer and as a prognostic indicator. It may serve as a surrogate marker for prolonged survival, particularly linked to the amplification of the 3q locus. Additionally, studies have been revealed that SHOX2 and PITX2 DNA methylation levels are significantly higher in SCC compared with LUAD. Hypomethylation of PITX2 and SHOX2 has been detected in patients with NSCLC with a high risk for tumor progression, while hypermethylation of PITX2 and SHOX2 has been observed in patients with low risk and is related to poor prognosis [508]. Both SHOX2 and PITX2 methylation could be potential predictors for tumor progression. Additionally, SHOX2 and PITX2 hypermethylation has been observed in relation with Thyroid TF‐1 (TTF‐1) downregulation, which has been identified as an independent biomarker associated with an adverse prognosis in LUADs [508]. Studies have demonstrated that methylation of RASSF1A and SHOX2 plays crucial roles in the tumorigenesis, progression, and metastasis in lung cancer. This makes these markers highly relevant as diagnostic biomarkers, offering high sensitivity and specificity for precise lung cancer screening [46]. In bronchoalveolar lavage fluid (BALF), both methylated SHOX2 and RASSF1A were found to have high specificity and sensitivity (97.4 and 81% respectively), which can be utilized as a noninvasive method for lung cancer detection, particularly during the early stage [513]. The aberrant hypermethylation of the RASSF1A promoter region has been widely documented in lung cancer, occurring in approximately 63% of NSCLC cases. This distinct methylation pattern, absent in normal epithelial cells, highlights RASSF1A as a promising biomarker for lung cancer research. Notably, the combined analysis of SHOX2 and RASSF1A methylation has demonstrated significant diagnostic potential, with sensitivity ranging from 71.5 to 83.2% in BALF samples. This combined methylation analysis correlates strongly with clinical parameters such as tumor size and TNM stage, providing valuable insights into disease progression. Importantly, the methylation status of SHOX2 and RASSF1A can serve as an early indicator of tumor invasiveness, suggesting that patients testing positive for these markers may face a more aggressive clinical types of cancer [514].
Iroquois Homeobox Family's Role in Lung Cancer
4.3.1.1.8
The IRX genes family participate in both development and differentiation as TFs, and they have been suggested to both suppress and promote several types of cancer. Specifically, IRX1 promoter hypermethylation leads to the lack of its expression in lung cancer. Furthermore, IRX1 promoter hypermethylation is more frequent in NSCLC than in SCLC [54]. In normal lung tissue, IRX1 has a lesser degree of methylation in the promoter region, whereas in primary adenocarcinoma and SCC, the lack of IRX1 expression is caused by hypermethylation of the IRX1 gene promoter CpG sites. It is worth noting that in lung cancer, IRX1 promoter hypermethylation occurs more often in NSCLC samples as compared with SCLC samples. It is likely that hypermethylation and downregulation of IRX1 can be used to determine the prognosis and diagnosis of patients with adenocarcinoma [515]. IRX2 is involved in cell migration and apoptosis, and in mouse embryos, it plays an important role in early lung development. Studies have found that the IRX2 gene is hypermethylated at CpG sites within the promoter region in LUADs compared with normal lung tissue [516]. Furthermore, the IRX2 gene has been detected as hypermethylated at CpG islands in lung SCC samples, compared with normal lung tissue. In Stage I lung cancer cases, IRX2 may serve as useful biomarkers for early lung cancer diagnosis, particularly in SCC [435].
Caudal‐Related Homeobox Family's Role in Lung Cancer
4.3.1.1.9
This multifaceted role of CDX2, due to its involvement in cell proliferation, differentiation, and apoptosis, makes it a key player in tumorigenesis across various cancer types. The expression and function of CDX2 have been extensively studied, particularly in the context of CRC and lung cancer. However, the role of CDX2 can vary significantly depending on the cancer type and cellular context, acting either as a tumor suppressor or an oncogene [92, 362, 517, 518]. In the context of lung cancer, emerging evidence suggests that CDX2 may act as a tumor suppressor by suppressing Wnt signaling, thereby preventing uncontrolled cell proliferation [362]. Interestingly, promoter hypermethylation of Wnt signaling antagonistic components has been observed to disrupt the Wnt signaling pathway in lung cancer. The re‐expression of CDX2 has been shown to suppress cell proliferation and block cells in the G1 phase of the cell cycle by inhibiting β‐catenin/TCF activity and downstream target genes such as c‐Myc and Cyclin D1. Importantly, CDX2 was found to be silenced in lung cancer due to its own promoter hypermethylation. In a study involving 110 primary lung cancer samples, CDX2 was methylated in approximately 55% of cases, while it was not observed in normal lung tissue samples [362]. This suggests that CDX2 functions as a tumor suppressor by negatively regulating Wnt signaling, thereby preventing the proliferation of lung cancer cells. Furthermore, a recent study indicated that the hypermethylation of CDX2 in CpG islands was detected in 100% of LUAD samples, with a median level of methylation 10 times higher than that of adjacent nontumor lung tissue. This suggests that the hypermethylation of HOXA1 and CDX2 in CpG islands in LUAD could serve as a promising biomarker for the detection of lung cancer, particularly in early‐stage (Stage IA) tumor samples, using plasma or sputum samples [384].
LMX Homeobox Family's Role in Lung Cancer
4.3.1.1.10
Recent studies have also uncovered tumor‐suppressive roles for several other LHX genes. Specifically, LHX6, LHX9, ISL2, and LMX1A have been found to exhibit anticancer functions in depends on the cancer types [519]. Research has uncovered that several members of the LMX family, including LHX2, LHX3, LHX4, LHX4, LHX5, LHX6, and LHX9, frequently display hypermethylation in different types of cancer tissues [90]. In lung cancer, some members of the LHX family have been reported to exhibit either aberrant expression or abnormal methylation, as detailed in the sections below. LHX2 can exhibit either hypermethylation or overexpression, depending on the type of cancer. In NSCLC, LHX2 is predominantly reported to function as an oncogene, with studies indicating its upregulation in 70% of NSCLC tissues compared with paired noncancerous samples. Therefore, evidence indicates that elevated LHX2 expression promotes NSCLC progression by enhancing cell proliferation, migration, and invasion, largely through its regulation of the cell cycle [90, 319, 519]. Notably, miR‐1238 levels are significantly reduced in NSCLC tissues and cells, with 62% of NSCLC tissue samples exhibiting this reduction. Analysis of patient tissue samples further reveals an inverse correlation between miR‐1238 and LHX2 expression, with simultaneous miR‐1238 downregulation and LHX2 overexpression observed in 77.4% of cases. By targeting the 3′‐UTR of LHX2, miR‐1238 directly reduces LHX2 mRNA and protein levels. Conversely, in vitro studies on lung cancer cell lines have shown that upregulation of miR‐1238 suppresses cell viability, proliferation, migration, and invasion through downregulation of LHX2, highlighting its tumor‐suppressive potential. These research suggests that restoring miR‐1238 expression may provide a promising therapeutic strategy for managing NSCLC by reducing *LHX2‐*driven oncogenic activity [319]. Similarly, low levels of miR‐124 are frequently detected in NSCLC tissues and show an inverse correlation with elevated LHX2 expression. This upregulation of LHX2 is associated with enhanced migratory and invasive abilities, increased metastasis, and poorer prognosis in NSCLC. Consequently, miR‐124 downregulation exacerbates these tumorigenic phenotypes, while enhanced miR‐124 expression has been shown to suppress LHX2 levels, reducing NSCLC cell migration and invasion. As a result, these findings underscore the tumor‐suppressive role of miR‐124 in NSCLC progression [520]. On the other hand, aberrant methylation of the LHX2 gene has been prominently observed in lung cancer tissues. Although low levels of methylation were detected in some normal tissues excised during tumor surgery, methylation was significantly more pronounced in tumor samples. Specifically, LHX2 is found to be methylated in 58% of primary lung tumor cases, a frequency markedly higher than that observed in adjacent normal lung tissues. These results indicate distinct methylation patterns associated with LHX2 in lung cancer, suggesting that its aberrant methylation may serve as an epigenetic mechanism driving tumor progression [90, 519, 521]. Similarly, LHX4, located on chromosome 1q25, shows significantly higher methylation levels in lung tumor tissues compared with noncancerous lung tissues. Studies indicate that 75% of primary lung tumors exhibit LHX4 methylation, highlighting its potential role in lung cancer progression through epigenetic modifications. Specifically, low expression of LHX4 has been correlated with an undifferentiated state of lung tumors [90, 519, 521].
LHX3 exhibits significantly elevated expression levels in lung cancer tissues compared with adjacent noncancerous tissues. This overexpression suggests its role as a potential oncogene, particularly in NSCLC and more prominently in LUAD. Its elevated expression is closely associated with advanced clinical stages, tumor metastasis, and reduced OS, particularly in LUAD patients. Functionally, LHX3 promotes cancer cell proliferation and invasion while inhibiting apoptosis, underscoring its oncogenic role and its designation as an unfavorable independent prognostic factor. Furthermore, LHX3 acts as a radiosensitivity prognostic biomarker, especially in early‐stage LUAD, with higher expression levels observed in patients undergoing radiotherapy. Nevertheless, the precise molecular mechanisms underlying LHX3’s oncogenic activity remain poorly understood, necessitating further research [522].
LHX6 is frequently hypermethylated at its CpG islands in lung cancer, a phenomenon observed in 56% of primary lung cancer cases. This epigenetic silencing results in a significant downregulation of LHX6 expression in lung cancer tissues, highlighting its role as a tumor suppressor in lung carcinogenesis [523, 524]. In vitro and in vivo studies, including experiments on lung cancer cell lines and mouse models, demonstrate that enforced expression of LHX6 markedly inhibits cell viability and tumor growth. Notably, LHX6 expression is closely associated with inducing apoptosis, promoting G1/S cell cycle arrest, and suppressing migratory and invasive capabilities of the cancer cells. Conversely, LHX6 downregulation facilitates cell proliferation and tumorigenesis‐related processes, emphasizing its critical role in suppressing malignancy [523, 524]. As an important TF, LHX6 primarily mediates its tumor‐suppressive functions by negatively regulating the Wnt/β‐catenin signaling pathway. Through the silencing of CTNNB1, LHX6 reduces the pathway activity, leading to the suppression of four critical oncogenic downstream target genes. Therefore, it is inhibited the expression of c‐Myc and Cyclin D, key regulators of the G1 phase, thereby effectively impairing cell cycle progression. Subsequently, LHX6 diminishes tumor metastasis. Conversely, LHX6 downregulation activates the Wnt/β‐catenin pathway, driving tumorigenesis, metastasis, and resistance to chemotherapeutic agents such as erlotinib, an EGFR‐TKI, particularly in NSCLC [523, 524, 525]. Moreover, LHX6 promotes apoptosis by upregulating the p53 and p21, while simultaneously suppressing the expression of the Bcl‐2. These multifaceted mechanisms underscore LHX6’s pivotal role in inhibiting lung cancer progression and indicate its potential as a therapeutic target [523, 525, 526]. In addition, in vitro studies on NSCLC cell lines have been indicate that the upregulation of miR‐214 is detected as a negative regulator of LHX6 in lung cancer, directly reducing its mRNA level. This downregulation of LHX6 activates the Wnt/β‐catenin signaling pathway, enhancing tumor cell migration and contributing to resistance against erlotinib. Therefore, restoring LHX6 expression or inhibiting miR‐214 expression could potentially reverse erlotinib resistance and mitigate metastatic progression in NSCLC [525, 526]. Similarly, LHX9, ISL2, and LMX1A have also been implicated as potential tumor suppressors, though the precise mechanisms by which they exert their inhibitory effects on cancer progression remain to be fully elucidated [519, 521]. Recent evidence indicates that epigenetic silencing of LMX1A in lung cancer is caused by hypermethylation of its promoter, resulting in its downregulation in NSCLC cells. Additionally, LMX1A exhibits tumor‐suppressive properties in NSCLC and partly prevents NSCLC cell invasion by modulating EMT, angiogenesis, and ECM remodeling [527, 528].
SIX Homeobox Family's Role in Lung Cancer
4.3.1.1.11
Prior research has been demonstrated that aberrant expressions of the SIX genes can be implicated in tumor progression, tumorigenesis, and metastasis by promoting migration, angiogenesis, apoptosis, and cell proliferation [529, 530]. The sine oculis homeobox homologue 2 (SIX2), as a HD‐containing gene, is located at chromosome 2. Methylation of SIX2 has been detected in the CpG island spanning the promoter and its 3′ end in lung cancer. However, its precise role in lung cancer is still unclear and requires more extensive research [521]. There is evidence indicating that epigenetic silencing of SIX3 in NSCLC is caused by methylation of its promoter, resulting in its downregulation in LUAD. As a consequence, in LUAD tissue, methylation of SIX3 was downregulated compared with normal tissue adjacent to the tumor. There is evidence suggesting that SIX3 acts as a transcriptional repressor in NSCLC by regulating relevant oncogenes, thereby preventing the phenotype of NSCLC cells [531]. However, there is limited information available about SIX3’s role in tumorigenesis. Besides, in patients with LUAD and bronchioalveolar carcinomas, there was an observed significant correlation between the expression of SIX3 and improved patients’ OS rate and progression‐free survival. Consequently, SIX3 could serve as a valid prognostic biomarker for LUAD [531]. SIX6 gene was found to be hypermethylated in early‐stage NSCLC. In comparison with noncancerous lung tissues, SIX6 methylation is significantly higher in Stage I NSCLC. Furthermore, comethylation of SIX6 and SOX1 has been also observed in SCC and adenosquamous carcinoma samples from NSCLC patients; moreover, it is possible that their methylation contributed to the development of SCC. Its appears that comethylation of SIX6, BCL2, and retinoic acid receptor beta (RARB) might be caused by smoking. On the other hand, abnormal methylation of these three genes may be accurately used for diagnosing Stage I NSCLC due to their high‐sensitivity and specificity [49].
Distal‐Less Family's Role in Lung Cancer
4.3.1.1.12
DLX1 has been found to exhibit a complex expression pattern in lung cancer, specifically in LUAD. Research indicates that both methylation of the DLX1 gene and its upregulation are prominent features in lung cancer, particularly in LUAD [521, 532]. Despite these findings, DLX1 mutations are rare in LUAD (1.5%) and show no correlation with LUAD patient's OS. Studies have demonstrated that DLX1 mRNA levels are significantly elevated in LUAD tissues and cell lines compared with noncancerous controls. Notably, DLX1 overexpression and aberrant promoter methylation in two CpG islands are strongly associated with poor OS in LUAD patients, highlighting its role as an independent prognostic factor. In particular, DLX1 hypermethylation is believed to contribute to adverse survival outcomes in these patients. Additionally, Kaplan–Meier survival analyses reveal that elevated DLX1 expression is significantly correlated with reduced OS, PFI, and DSS in LUAD patients. Functionally, evidence suggests that DLX1 overexpression promotes proliferation and migration of LUAD cells. As a TF, DLX1 is involved in regulating multiple pathways, with potential links to TP53 activity and DNA replication; however, further research is needed to elucidate these associations. Furthermore, DLX1 has been implicated in immune evasion mechanisms in LUAD cells, underscoring its multifaceted role in lung cancer progression [532].
DLX4 plays a multifaceted role in the progression of lung cancer. Notably, studies have reported both increased expression and aberrant methylation of the DLX4 gene in lung cancer tissues [190, 521, 533]. Interestingly, methylation of the DLX4 in its CpG island has been reported in more than 80% of LUAD cases [534]. Furthermore, studies on NSCLC patient samples have highlighted the methylation status of the DLX4 promoter region, revealing that 49.5% of NSCLC cases exhibit DLX4 methylation. The prevalence of methylated DLX4 is particularly high in advanced stages, with 84.6% in Stage II and 93.1% in Stage III. Similarly, in vitro studies on metastatic lung cancer cell lines demonstrated a significant downregulation of DLX4 expression, while its re‐expression was associated with an improvement in the poor prognosis of lung cancer patients. Additionally, evidence suggests that methylated DLX4 could serve as a potential biomarker for poor prognosis, particularly in Stage I NSCLC following curative resection [99, 535]. However, the results of the new studies contradict those of previous studies, not only regarding the level of DLX4 expression but also its involvement in the upregulation associated with lung cancer progression and metastasis [99, 533, 535]. Recent findings underscore the oncogenic role of DLX4 in NSCLC, particularly in regulating cell proliferation and the cell cycle. In vitro studies on NSCLC cell lines demonstrate that silencing DLX4 significantly reduces tumor cell viability and induces G1/S phase cell cycle arrest. This cell cycle arrest is characterized by a higher proportion of cells in the G1 phase and a corresponding reduction in the S phase. This arrest results in a greater number of cells being blocked in the G1 phase, coupled with a decline in the proportion of cells progressing to the S phase. Functionally, DLX4 exerts its regulatory effects through the cyclin‐dependent kinase subunit 2 (CKS2)/Y‐box binding protein 1 (YB‐1) axis, a pathway critical for NSCLC progression. DLX4 positively regulates YB‐1, a multifunctional protein essential for tumor progression, particularly in NSCLC. Research has revealed a strong correlation between DLX4 and YB‐1 expression, with DLX4 silencing directly suppressing YB‐1 levels. Silencing YB‐1, in turn, inhibits CKS2 expression, leading to reduced cell proliferation and upregulation of critical tumor suppressors genes’ expression such as phosphatase and tensin homolog deleted on chromosome 10 (PTEN), p53, p21. By modulating the CKS2/YB‐1 axis, DLX4 drives tumor progression and highlights its central role in NSCLC growth [533].
DLX5 has gained considerable attention for its involvement in lung cancer. Studies on various lung cancer cell lines and NSCLC tissue samples have consistently reported that DLX5 is significantly overexpressed in cancerous tissues compared with noncancerous counterparts. Clinicopathologic analyses of NSCLC samples have further revealed that DLX5 upregulation is strongly correlated with increased cell proliferation, tumor growth, aggressive disease phenotypes, and reduced CSS, suggesting its utility as a potential biomarker for poor prognosis. Therefore, both in vivo and in vitro studies underscore the critical role of DLX5 as a growth factor in the development and progression of lung cancer. Functionally, DLX5 drives NSCLC progression by upregulating MYC transcription, a well‐established oncogene involved in promoting cell proliferation across various cancers. Silencing DLX5 expression in lung cancer cell lines significantly reduces cell proliferation, an effect attributed to the concurrent suppression of MYC expression. These findings highlight the oncogenic role of DLX5 in tumorigenesis and indicates its potential as a therapeutic target by regulating MYC‐dependent oncogenic pathways [536, 537, 538].
Paired‐Like Homeobox Family's Role in Lung Cancer
4.3.1.1.13
In early‐stage of NSCLC, PHOX2A has been found to be hypermethylated, with significantly higher methylation rates observed in Stage I NSCLC compared with noncancerous lung diseases. This suggests its potential as a biomarker for early detection [49, 50]. In addition to its epigenetic regulation, studies on lung cancer cell lines have revealed a distinct role for PHOX2A in cancer progression. Increased PHOX2A expression has been directly linked to enhanced invasiveness in lung cancer. In vitro experiments show that enforced PHOX2A expression significantly alters cell cycle dynamics and apoptotic regulation, leading to a higher proportion of cells in the S phase, increased invasive potential, and reduced apoptosis rates. These effects appear to be modulated by miR‐326, demonstrating a complex regulatory network involving PHOX2A in lung cancer progression [539]. In vitro studies on lung cancer cell lines have identified PHOX2A as a direct target of miR‐326, revealing a critical relationship between their expression levels and lung cancer progression. Mechanistically, miR‐326 binds to the 3′‐UTR of PHOX2A, resulting in reduced PHOX2A accumulation in lung cancer cell lines. Notably, miR‐326 levels are significantly downregulated in lung cancer; however, both in vivo and in vitro studies demonstrate that enforced miR‐326 expression suppresses PHOX2A accumulation, inhibits cell proliferation and migration, and promotes apoptosis. Furthermore, miR‐326 expression is negatively regulated by the lncRNAs HOTAIR, which suppresses its levels. In contrast, silencing HOTAIR, has been shown to elevate miR‐326 expression, further supporting its tumor‐suppressive role in lung cancer [539].
Orthodenticle Homeobox Family's Role in Lung Cancer
4.3.1.1.14
Numerous studies have identified OTX1 as an oncogene, demonstrating its involvement in promoting cell proliferation, migration, and tumor progression across various cancer types. Overexpression of OTX1 has been observed in cancers such as breast cancer, hepatocellular carcinoma, CRC, and lung cancer. While OTX1 overexpression is frequently observed in lung cancer, its downregulation has also been reported, indicating a complex regulatory role. Additionally, OTX1 hypermethylation at CGIs has been identified in 100% of SCC tumor samples, emphasizing its epigenetic significance in lung cancer [435, 534, 540, 541]. In NSCLC, OTX1 is markedly upregulated in tissues and cell lines, implicating its critical role in tumor progression. Elevated OTX1 expression is associated with poor OS in patients with NSCLC, suggesting its potential as a prognostic marker and therapeutic target. Mechanistically, in vitro studies on NSCLC cell lines reveal that OTX1 downregulation significantly impairs cell proliferation, migration, and invasion through multiple pathways. Downregulation of OTX1 induces G2/M phase arrest by reducing Cyclin B1 levels, which in turn suppresses the G2 to M phase transition and prevents cell growth. OTX1 suppression also decreases p‐ERK protein levels, indicating that its tumor‐promoting effects may rely on p‐ERK activation. Furthermore, OTX1 suppression impairs cell migration and invasion, likely by disrupting EMT through decreased expression of EMT‐related key markers such as N‐cadherin and vimentin. Finally, silencing OTX1 promotes apoptosis, as evidenced by enhanced levels cleaved PARP1 and active Caspase‐3, two key indicators of apoptotic activity. These results position OTX1 as a multifaceted regulator of NSCLC progression, with its diverse roles in cell proliferation, migration, invasion, and apoptosis [541].
Homeobox Genes Dysregulation in Other Four Cancers
4.3.1.2
Gene dysregulation of developmental regulators is a central theme in carcinogenesis, with homeobox genes emerging as pivotal modulators across diverse tumor types. Table 7 catalogues representative homeobox genes that are recurrently dysregulated in four major solid tumors breast, colorectal, prostate, and gastric cancers. Within each cancer type, the table distinguishes genes that are upregulated or downregulated and that have been functionally characterized as oncogenic drivers or tumor‐suppressive regulators. In addition, the table links these expression changes to the corresponding signaling pathways or axes and to the experimental evidence supporting their roles. Together, these data reveal both shared and tumor‑specific patterns of perturbation in developmental programs and highlight potential avenues for targeted therapy as well as candidate biomarkers for prognosis.
Clinical and Translational Implications of Homeobox Genes in Cancer Diseases
5
Given the broad involvement of homeobox genes across multiple cancer hallmarks and tumor types, there is growing interest in therapies that act through multiple, complementary strategies. Importantly, the oncogenic or tumor‐suppressive functions of homeobox gene dysregulation are not determined by altered expression alone but emerge from a broader regulatory network that includes DNA‐binding cofactors, intersecting signaling pathways, and multiple layers of ncRNA regulation such as miRNAs that directly target HOX transcripts [570, 571, 572, 573]. Pan‐cancer analyses further suggest that homeobox expression patterns are closely linked to features of the TME, which influence prognosis and may modulate responses to immunotherapy [17]. Moreover, in many malignancies, dysregulated oncogenic members of the homeobox family drive tumor initiation and progression while also contributing to intrinsic and acquired resistance to systemic therapy. For example, overexpression of HOXA5, HOXB7, and HOXB5 in estrogen receptor‐positive breast cancer promotes resistance to tamoxifen by activating related signaling networks [574, 575, 576]. Consequently, a wide range of therapeutic approaches has been proposed or developed to target homeobox complexes directly or to modulate this extended regulatory network.
Therapeutic strategies under investigation for cancers with dysregulated HOX genes span direct targeting of HOX complexes and modulation of their upstream and downstream networks. Notable approaches include disruption of HOX/PBX dimers using peptide inhibitors such as HXR9, which demonstrates antitumor activity in vivo across cancer types including PCa and NSCLC [573]. Epigenetic‐related strategies, including histone deacetylase (HDAC) inhibitors, have been computationally prioritized as compounds correlated with HOX downregulation and possessing antitumor properties [17]. Small‐molecule DNA ligands that disrupt HOXA9/DNA binding, such as DB1055 and DB818, have shown efficacy in suppressing proliferation and inducing cell death in HOXA9‐dependent AML models. RNA‐based approaches namely siRNA or shRNA‐mediated knockdown of oncogenic HOX genes also show promise; for example, targeted suppression of HOXD3 in CRC cell lines using siRNA and HOXD3‐specific shRNA delivered by lentiviral vectors led to marked reductions in HOXD3 transcripts in RKO cells, accompanied by impaired cell growth and increased apoptosis [577]. Homeobox genes are increasingly recognized as multilayered cancer biomarkers that provide diagnostically, prognostically, and therapeutically relevant information across biological levels. They can serve as diagnostic biomarkers by helping to distinguish tumor tissue from normal tissue or from benign disease. A prominent example is bladder cancer, where DNA methylation‐based urinary biomarkers improve risk stratification at diagnosis. Panels measuring methylation of genes such as HOXA9, POU4F2, ONECUT2, and PCDH17 in urine can differentiate bladder cancer from nonmalignant urological conditions with high predictive value, enabling many genuinely low‐risk patients to avoid invasive follow‐up examinations [578].
Discussion
6
Considering that homeobox genes are implicated in both malignant and noncancerous disorders, current evidence indicates that dysregulation of these genes is described far more extensively in cancers than in noncancer conditions. This imbalance reflects the central role of homeobox TFs, including both HOX clusters and non‐HOX families, in key cancer hallmarks such as proliferation, survival, EMT, invasion, and metastasis. Across numerous studies, HOX family members are frequently dysregulated either overexpressed with oncogenic activity or silenced with LOF of tumor‐suppressive effects across a wide range of tumors. In aggregate, dysregulated expression of homeobox genes across tumor types has repeatedly been linked to more aggressive clinicopathological features and poorer outcomes [3, 18, 187]. Systematic comparisons of five of the most common solid cancers illustrate how frequently homeobox alterations recur across distinct histological subtypes and molecular contexts. In lung cancer, one of the most prevalent solid tumors, large‐scale genomic and epigenomic analyses have identified extensive sets of aberrantly expressed and epigenetically deregulated genes; genome‐wide DNA methylation profiling shows that a broad spectrum of loci become hyper‐ or hypomethylated and that numerous genes exhibit altered expression across the major histological subtypes [3, 301].
Aberrant expression of homeobox genes plays a pivotal role in the pathogenesis of lung cancer through various mechanisms such as epigenetic modifications, and also regulatory interactions with miRNAs and lncRNAs [2, 190, 579]. Alterations in gene expression are integral to the onset and progression of lung cancer, with many such changes being driven by epigenetic mechanisms. Among these, abnormal DNA methylation has emerged as a critical factor in lung cancer development. For instance, hypermethylation of promoter regions in essential tumor suppressor genes often results in their transcriptional silencing, thereby facilitating tumor growth, invasion, and metastasis [190]. Any alteration in gene expression plays a critical role in the onset and progression of lung cancer. Numerous gene expression changes have been identified in this cancer, many of which are regulated by epigenetic modifications. Among these, abnormal DNA methylation has gained recognition as a critical factor in lung cancer development. For example, hypermethylation of promoter regions in essential tumor suppressor genes often leads to their transcriptional inactivation, thereby promoting tumor growth and metastasis. As outlined in our previous study, such epigenetic modifications in lung cancer frequently correlate with increased metastasis and malignancy [301]. Beyond methylation, the mis‐regulation of specific genes is closely tied to disruptions in crucial signaling pathways, such as the Wnt pathway, which is integral to cell proliferation and differentiation. Additionally, the homeobox genes superfamily, a well‐known group of TFs fundamental to developmental processes, warrants particular attention in this context. This superfamily comprises a broad array of genes with context‐specific expression patterns across various cancer types [580]. According to experiments conducted on lung cancer, HOX genes clusters exhibit different expression patterns across various types of cancer, especially lung cancer [1]. In this way, HOXA‐ and HOXB‐related genes from the 3′ end expression mostly observe in healthy adult lung tissues [381]. Moreover, the majority of homeobox genes are not expressed in adult tissues; therefore, their absence contributes to de novo methylation during malignancy progression [581]. As a consequence, aberrant expression of HOX gene clusters is a defining feature, with distinct gene families within the superfamily exhibiting unique expression profiles depending on the tumor type in lung cancer. This variability highlights the multifaceted roles of these genes in cancer biology, characterized by their remarkable complexity and duality. While some studies suggest an oncogenic role for certain HOX genes, others point toward their tumor‐suppressive properties [582]. Therefore, in human lung tissue, HOX genes have distinct patterns of expression [373]. In particular, depending on the type of lung cancer, these expression levels can vary, either upregulation or downregulation.
This indicates the complex and context‐dependent nature of HOX genes function in cancer. Understanding the intricate regulation of these genes could provide valuable insights into cancer mechanisms and pave the way for novel therapeutic approaches. Unraveling the complex interplay of HOX genes in cancer is also essential to develop more effective and targeted therapeutic strategies. Deciphering the precise role of individual HOX genes, and how they interact with other cellular pathways, will be crucial in leveraging their potential for cancer treatment. A comprehensive understanding of the HOX genes network in different solid tumor types may open up new avenues for developing more personalized and precise interventions [582]. The relationship between DNA methylation and homeobox gene expression is multifaceted and complicated, with significant implications for understanding tumor biology. Both hypermethylation and hypomethylation can influence the behavior of these critical genes, but in differing ways across various cancer types. In some instances, hypermethylation of homeobox genes has been associated with their transcriptional silencing and a potential tumor suppressor role. This epigenetic modification can lead to the downregulation of these genes, which may otherwise function to inhibit tumor development and progression. Conversely, hypomethylation of homeobox genes has been observed to result in their overexpression, potentially contributing to an oncogenic phenotype in certain cancers. This dysregulation of the normal expression patterns of these developmentally important genes can disrupt cellular homeostasis and promote malignant transformations [191, 583]. Aberrant DNA methylation patterns across different cancer types can alter HOX genes’ expression, which is a key factor in tumorigenesis and can serve as a biomarker for cancer prognosis and treatment [303, 407]. According to previous reports, several homeobox genes are abnormally methylated in the lung cancer cell lines [2]. Despite most of the CGIs in HOX gene clusters being highly methylated in lung cancer cells and primary lung tumors, some of the CGIs are still nonmethylated. Therefore, in the gene‐rich HOX cluster, both of these regions can occur adjacent to each other [2]. The studies evidence suggests that methylation of HOX genes is specifically tied to various types of cancer and may even be related to normal development and tissue‐specific differentiation [584]. Accordingly, HOX genes can provide a better understanding of some types of human diseases when considered in relation to cancers [82]. For instance, in 97 samples from NSCLC patients, HOXA2 (78%), HOXA10 (40%), and SHOX2 (39%) were found to be among the methylated genes [394]. Studies have previously reported that the HOXA and HOXD clusters substantially contain high levels of methylation, while the clusters HOXB and HOXC have a lower proportion of methylation in the lung SCC [585]. For instance, studies have reported hypermethylation of some HOX genes, such as HOXA3 and HOXD10, exhibit hypermethylation across a wide range of 16 cancer types. Additionally, HOXA9 and HOXB13 have been found to be hypermethylated in solid tumors, while HOXA genes are frequently hypermethylated in breast cancer [1, 191].
On the other hand, a comparison between normal and cancerous lung tissue revealed that several HOX genes from different clusters, such as HOXA1 [385], HOXA10 [428], HOXB7 [586], HOXB8 [69], and HOXC6 [587], HOXC13 [588] and HOXD9 [496], are highly expressed in lung cancer tissues [585]. Evidence suggests that the increased expression of certain HOX genes across various clusters is strongly associated with their oncogenic potential. Abnormal HOX genes’ expression may promote oncogenesis by activating antiapoptotic pathways, which contribute to the survival and proliferation of cancer cells. Consequently, it is postulated that HOX gene involvement in suppressing apoptosis plays a critical role in cancer progression, promoting malignancy through mechanisms such as cell proliferation, survival, migration, and invasion [150, 579]. Conversely, some HOX genes with downregulated expression are suggested to act as tumor suppressors, highlighting the dual roles of these genes in cancer biology. During HOX genes hypermethylation, the activity of tumor‐suppressor and/or apoptotic HOX genes is silenced, which contributes to tumorigenesis in multiple types of cancer [303]. Notable example is HOXA5, which has been identified as a tumor suppressor in NSCLC. HOXA5 demonstrates tumor‐suppressive roles by regulating cytoskeletal remodeling and inhibiting metastasis. Research indicates that the downregulation of HOXA5 expression, often due to promoter hypermethylation, is associated with NSCLC pathogenesis and poor prognosis. Conversely, its upregulation can inhibit cell proliferation by regulating the expression of p21 [399, 401, 402]. Conversely, other genes within the same cluster, such as HOXA10 and HOXA13, are frequently upregulated in lung cancer. HOXA10, particularly in LUAD, is implicated in promoting tumor progression and metastasis. As an oncogene, HOXA13 enhances tumorigenicity and metastasis through its regulation of the p53 and Wnt/β‐catenin pathways [351, 589, 590]. A similar duality in expression is observed in the HOXB cluster, where most genes are reported to be dysregulated in lung cancer, particularly in LUAD tumor tissue samples. These include HOXB2, HOXB3, HOXB4, HOXB6, HOXB7, HOXB8, HOXB9, and HOXB13 [303, 396, 438]. Among them, HOXB3, HOXB7, HOXB8, and HOXB9 are consistently upregulated in LUAD and are strongly associated with lung cancer pathogenesis. Specifically, the upregulation of HOXB9 and HOXB3 expression are correlated with poor cancer progression in patients with LUAD [69, 438, 443, 447]. In addition, several genes within the HOXC clusters, such as HOXC4, HOXC6, HOXC8, HOXC9, and HOXC10, are upregulated in lung cancer. Most of these upregulated genes predominantly exhibit oncogenic roles and are directly involved in tumorigenesis [379, 450, 452, 466]. For example, the oncogene HOXC8 has been studied for its critical role in promoting the lung tumor progression. Research demonstrates that HOXC8 enhances the proliferation and migration of lung cancer cells by upregulating TGFβ1, a key factor involved in tumor development and progression [379]. However, both downregulation and upregulation of genes’ expression in HOXD cluster, such as HOXD3 [483, 485], HOXD8 [436], and HOXD13 [474, 483], are reported. For instance, HOXD8 and HOXD9 exhibit elevated expression levels in NSCLC, suggesting their potential involvement in tumor development. Interestingly, their expression levels are associated with metastasis in NSCLC [491, 496]. In contrast, HOXD10 is downregulated in NSCLC, due to promoter methylation, indicating that it may have a distinct or opposing function, potentially [502, 522]. Consequently, compared with normal lung tumors, primary NSCLC exhibits dramatically higher transcription of most HOXC and HOXD cluster‐related genes [372, 399]. Furthermore, both of these clusters expressed in fetal lungs as well [381].
Notably, this review also highlights the dysregulation of gene expression in various homeobox families beyond the HOX clusters. Significant examples include genes within the IRX, SHOX, PITX, CDX, SIX, LHX, OTX, and DLX families. The LHX family, in particular, exhibits both upregulation and downregulation across different genes. For instance, LHX2 is hypermethylated, leading to its downregulation, whereas LHX3 is frequently upregulated [521]. It is important to emphasize that duality in expression patterns is a recurring theme across homeobox genes in lung cancer [3, 521]. Some studies report downregulation of specific genes in certain subtypes, while others observe upregulation in different contexts. Notable examples of this phenomenon include LHX2 [520], DLX4 [534], and HOXD13 [483], whose expression levels vary depending on the lung cancer subtype. This variability underscores the complex and context‐dependent roles of homeobox genes in lung cancer pathogenesis [3, 521]. In addition, the overexpression of LHX2 may function as an oncogenic driver, highlighting the complex role of LHX family genes, where individual members can either promote or inhibit tumorigenesis depending on their expression context. Given the altered expression patterns of LHX genes, such as LHX2 and LHX4, in cancerous tissues, these genes are being investigated as potential biomarkers for the diagnosis and prognosis of lung cancer. Their expression levels could offer valuable insights into tumor behavior and patient outcomes [90, 519]. As mentioned earlier, HOX genes are intricately linked with several oncogenic pathways in lung cancer. Dysregulation of homeobox gene expression is influenced by key signaling pathways, including the Wnt/β‐catenin, PI3K/AKT, and TGF‐β/SMAD3 pathways, all of which are known to be altered in various types of cancer, including lung cancer [377]. For instance, activation of the Wnt pathway has been associated with aberrant expression of specific HOX genes, contributing to tumorigenesis [301]. Similarly, upregulation of HOXB7 has been shown to promote activation of the TGF‐β/SMAD3 signaling pathway, directly driving tumor metastasis in LUAD patients [377]. These interconnected pathways underscore the multifaceted role of HOX genes in lung cancer and highlight their potential as targets for combinatorial therapeutic approaches.
LncRNAs and miRNAs are known to regulate the expression of HOX genes in lung cancer. Studies have revealed a complex interplay between lncRNAs, miRNAs, and HOX genes, demonstrating how they can influence homeobox gene regulatory networks directly or indirectly. Notably, lncRNAs and miRNAs are implicated in metastasis and drug resistance, underscoring their potential as novel therapeutic targets in lung cancer [329, 330, 591, 592]. Additionally, the interaction between lncRNAs and DNA methylation plays a crucial role in shaping the epigenetic landscape of lung cancer [326, 327]. Given their significant impact on lung cancer pathogenesis, further investigation into their specific roles in tumorigenesis is essential. In lung cancer, lncRNAs can function as either oncogenes or tumor suppressors, further complicating their roles in the disease. Several lncRNAs and also lncRNAs associated with HOX genes, such as HOXA‐AS2 and HOTAIR, are upregulated in lung cancer and are involved in cancer‐related processes. LncRNAs can also interact with their target genes by binding to related TFs, thereby repressing or promoting the transcription of these genes. Moreover, lncRNAs can act as ceRNAs, interacting with miRNAs. Consequently, lncRNAs regulate homeobox gene expression through multiple pathways, highlighting their critical role in lung cancer [329, 593]. Similarly, miRNAs can function as either oncogenic miRNAs (oncomiRs) or tumor suppressors, depending on their function and the specific target genes they regulate [594, 595, 596]. This duality is evident in lung cancer, where altered miRNA expression contributes to both tumor initiation and progression. For example, HOXA5 is a target gene of miR‐196a and miR‐891a‐5p in NSCLC, and both miRNAs have been identified as oncogenes in NSCLC [400, 593, 597]. Additionally, HOXA5, acting as a tumor suppressor, is also downregulated by HOTAIR, which is directly linked to increased cell migration and tumorigenesis in NSCLC. Interestingly, studies have shown that HOXA5, as an important TFs, can bind to the promoter of lncRNA LINC00312, suppressing NSCLC cell proliferation. However, LINC00312 levels are reported to decrease in the plasma of NSCLC patients [598]. Targeting lncRNAs and miRNAs in lung cancer has garnered significant attention as a potential therapeutic strategy. Notably, specific regulatory axes identified in lung cancer, such as the LINC00472/miR‐1275/HOXA2 axis, HCP5/miR‐17‐5p/HOXA7 axis, and LINC00466/miR‐144/HOXA10 axis, have been extensively studied due to the intricate regulatory networks of lncRNAs and miRNAs [395, 404, 426]. These discoveries underscore the potential of targeting these molecular pathways for the development of innovative treatment strategies.
Author Contributions
M.D. and S.R. conducted all literature searches relevant to the article's topic and contributed to drafting and writing the manuscript. Mo.D. and S.Y. assisted with data collection. M.A. and Sh.F. provided grammar and language editing of the final manuscript. S.R. and M.D. created all figures and finalized and edited the manuscript. S.R. is the corresponding author. All authors reviewed and approved the final version and confirm their agreement with its content and readiness for publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Ethics Statement
The authors have nothing to report.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1S. Bhatlekar and J. Z. Fields , “Boman BM. HOX Genes and Their Role in the Development of human Cancers,” Journal of Molecular Medicine 92 (2014): 811–823.24996520 10.1007/s 00109-014-1181-y · doi ↗ · pubmed ↗
- 2T. Rauch , Z. Wang , X. Zhang , et al., “Homeobox Gene Methylation in Lung Cancer Studied by Genome‐wide Analysis With a Microarray‐based Methylated Cp G Island Recovery Assay,” Proceedings of the National Academy of Sciences of the United States of America 104, no. 13 (2007): 5527–5532.17369352 10.1073/pnas.0701059104 PMC 1838508 · doi ↗ · pubmed ↗
- 3Y. Feng , T. Zhang , Y. Wang , et al., “Homeobox Genes in Cancers: From Carcinogenesis to Recent Therapeutic Intervention,” Frontiers in Oncology 11 (2021): 770428.34722321 10.3389/fonc.2021.770428 PMC 8551923 · doi ↗ · pubmed ↗
- 4M. P. Scott , “40 years of the Homeobox: Either It Is Wrong or It Is Quite Interesting,” Development (Cambridge, England) 151, no. 6 (2024): dev 202776.38493802 10.1242/dev.202776 · doi ↗ · pubmed ↗
- 5Y.‐F. Zhong and P. W. Holland , “The Dynamics of Vertebrate Homeobox Gene Evolution: Gain and Loss of Genes in Mouse and human Lineages,” BMC Evolutionary Biology 11 (2011): 1–13.21679462 10.1186/1471-2148-11-169PMC 3141429 · doi ↗ · pubmed ↗
- 6P. W. Holland , H. A. F. Booth , and E. A. Bruford , “Classification and Nomenclature of all human Homeobox Genes,” BMC Biology 5, no. 1 (2007): 47.17963489 10.1186/1741-7007-5-47PMC 2211742 · doi ↗ · pubmed ↗
- 7K. A. Hubert and D. M. Wellik , “Hox Genes in Development and Beyond,” Development (Cambridge, England) 150, no. 1 (2023): dev 192476.36645372 10.1242/dev.192476 PMC 10216783 · doi ↗ · pubmed ↗
- 8C. Mio , F. Baldan , and G. Damante , “NK 2 homeobox Gene Cluster: Functions and Roles in human Diseases,” Genes & Diseases 10, no. 5 (2023): 2038–2048.37492711 10.1016/j.gendis.2022.10.001PMC 10363584 · doi ↗ · pubmed ↗