Heart Failure: Epidemiology, Pathophysiology, and Management
Yujian Fan, Zhihua Yang, Qing Li, Meng Sun, Yumeng Pu, Ke Zhao, Yu Bao, Xianliang Wang, Jingyuan Mao, Zhiqiang Zhao

TL;DR
This review explores heart failure's epidemiology, calcium-related mechanisms, and management strategies to improve diagnosis and treatment.
Contribution
The paper introduces a mechanistic network linking calcium dynamics with inflammation and mitochondrial dysfunction in heart failure.
Findings
Calcium-handling differences vary across heart failure subtypes under physiological and pathological conditions.
Alterations in calcium-regulating proteins and pathways are integrated into a network model for mechanistic understanding.
Emerging therapies target specific mechanisms in heart failure through calcium homeostasis-related points and lines.
Abstract
Heart failure (HF) is one of the leading causes of hospitalization and mortality worldwide. Despite continuous updates to modern clinical guidelines regarding HF classification and management, mortality and rehospitalization rates remain persistently high. Enhancing the prevention and treatment of HF therefore represents a major challenge both now and in the future. In this review, we synthesize HF epidemiology and systematically mapped the calcium‑handling differences under physiological and pathological conditions, as well as the patterns of calcium‑homeostasis dysregulation across the major HF subtypes (heart failure with reduced ejection fraction; heart failure with preserved ejection fraction). Moreover, we summarize alterations in key calcium‐regulating proteins (points) and calcium homeostasis‐related pathways (lines), and further integrate these nodes into a network model that…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
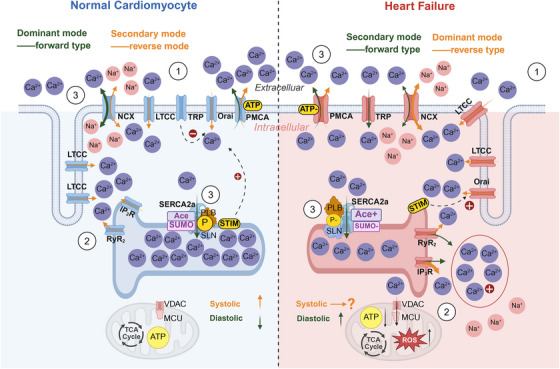 FIGURE 1
FIGURE 1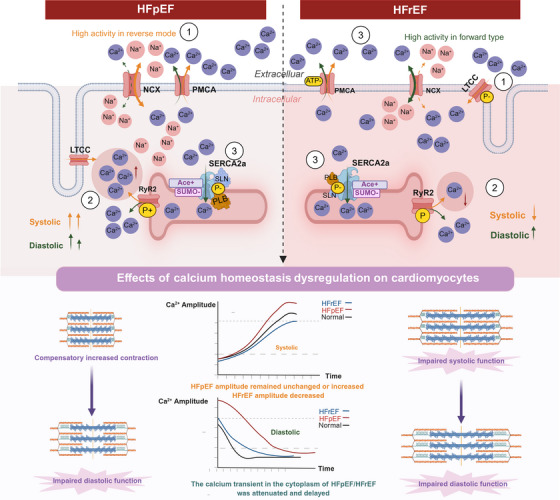 FIGURE 2
FIGURE 2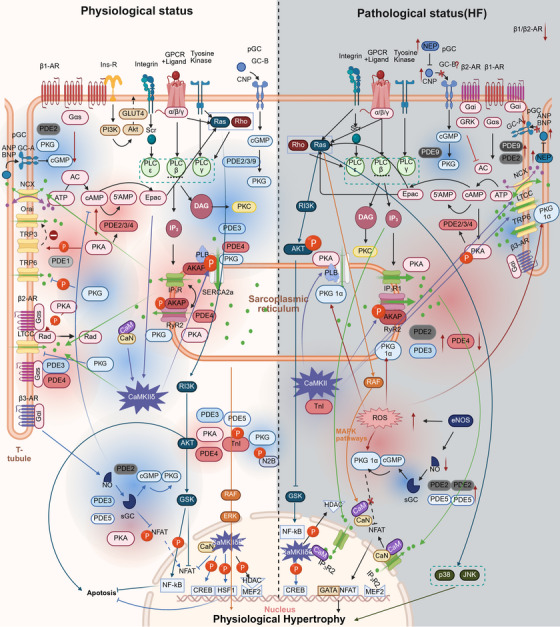 FIGURE 3
FIGURE 3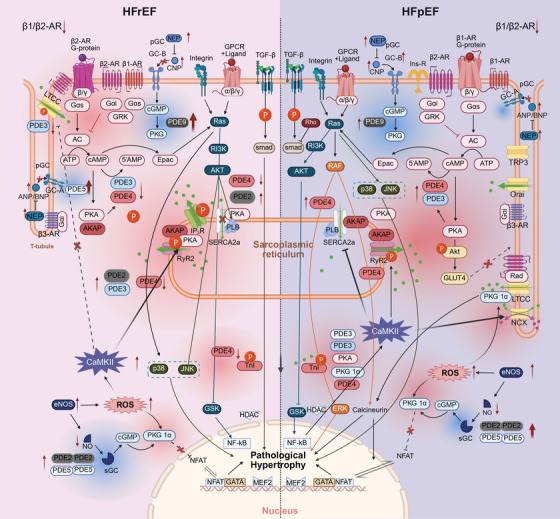 FIGURE 4
FIGURE 4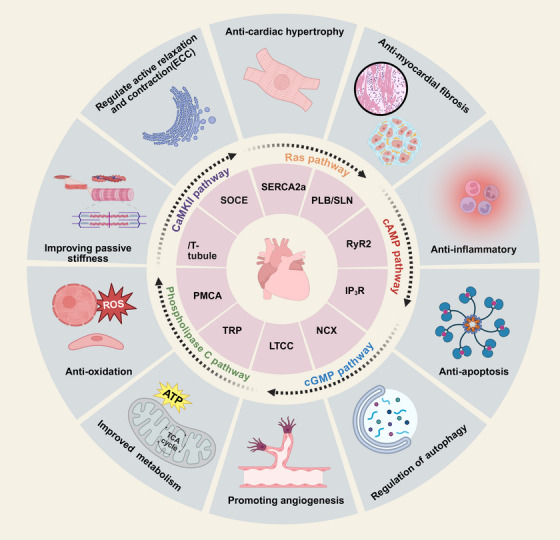 FIGURE 5
FIGURE 5| Medicine name | Mechanism | Research type | Animal | Proteins/pathways | HF type | References |
|---|---|---|---|---|---|---|
| Probenecid | Calcium homeostasis dysregulation, apoptosis, excitation–contraction coupling | Cell experiment | Primary cardiacmyocyte (C57) | TRP | HFrEF | [ |
| Danicamtiv | Calcium homeostasis dysregulation, mitochondrial function, passive stiffness | Cell experiment | Skinned muscle fibers and myofibrils | Titin | HFrEF | [ |
| Dantrolene | Calcium homeostasis dysregulation, metabolic disorder, oxidative stress | Cell experiment | Primary cardiacmyocyte (C57) | RyR2 | HFpEF | [ |
| Schaftoside | Calcium homeostasis dysregulation, autophagy, myocardial hypertrophy, inflammation | Cell experiment | AC16 cardiacmyocyte | CaMKII pathway | HFpEF | [ |
| Methyltransferase‐like protein 13 (Mettl13) | Calcium homeostasis dysregulation, oxidative stress, fibrosis | Animal experiment | C57BL/6 mice | SERCA2a | HFrEF | [ |
| PST3093 derivatives | Calcium homeostasis dysregulation, mitochondrial function | Animal experiment | SD rats | SERCA2a | HFrEF | [ |
| Sinapic acid | Calcium homeostasis dysregulation, inflammation, oxidative stress, myocardial fibrosis | Animal experiment | Wistar rats | Ras pathways | HFrEF | [ |
| Rhynchophylline | Calcium homeostasis dysregulation, mitochondrial function, apoptosis, oxidative stress | Animal experiment | C57BL/6 mice | RyR2, PLB, pPLB | HFrEF | [ |
| Dantrolene sodium | Calcium homeostasis dysregulation, oxidative stress | Animal experiment | Guinea pig | RyR2 | HFrEF | [ |
| Luteolin | Calcium homeostasis dysregulation, fibrosis, excitation–contraction coupling | Animal experiment | SD rats | SERCA2a, NCX, PLB, PI3k pathway | HFrEF | [ |
| Stachydrine hydrochloride | Calcium homeostasis dysregulation, myocardial hypertrophy, oxidative stress | Animal experiment | C57BL/6J mice | CaMKII pathway, LTCC, RyR2 | HFrEF | [ |
| CCG258208 | Calcium homeostasis dysregulation, mitochondrial function, metabolic imbalance | Animal experiment | C57Bl/6 mice | cAMP pathway | HFrEF | [ |
| Empagliflozin | Calcium homeostasis dysregulation, metabolic disorder | Animal experiment | C57Bl/6 mice | NHE | HFrEF | [ |
| Periplocin | Calcium homeostasis dysregulation, inflammation, oxidative stress, myocardial fibrosis | Animal experiment | SD rats | Ras pathways | HFpEF | [ |
| Nicotinamide | Calcium homeostasis dysregulation, metabolic imbalance, passive stiffness | Animal experiment | Leptin receptor‐deficient ZSF1 rats | SERCA2a, Titin | HFpEF | [ |
| Berberine | Calcium homeostasis dysregulation, mitochondrial function, autophagy | Animal experiment | C57BL/6J male mice | PLB, SERCA2a | HFpEF | [ |
| Triiodothyronine | Calcium homeostasis dysregulation, metabolic imbalance, fibrosis | Animal experiment | ZSF1 obese rats | RyR2 | HFpEF | [ |
| Acyl ghrelin | Calcium homeostasis dysregulation, metabolic imbalance, passive stiffness | Clinic trial | Human | Titin, cAMP pathway | HFrEF | [ |
| Adenosine A1‐receptor agonist neladenoson bialanate | Calcium homeostasis dysregulation, metabolic imbalance | Clinic trial | Human | SERCA2a, Titin | HFrEF | [ |
| AAV1/SERCA2a | Calcium homeostasis dysregulation | Clinic trial | Human | SERCA2a | HFrEF | [ |
| β3‐Adrenoceptor Agonist | Calcium homeostasis dysregulation | Clinic trial | Human | NCX | HFrEF | [ |
| Probenecid | Calcium homeostasis dysregulation, excitation–contraction coupling | Clinic trial | Human | TRPV | HFrEF | [ |
| Tranilast | Calcium homeostasis dysregulation, inflammation | Clinic trial | Human | TRPV | HFrEF | [ |
| CCBs | Calcium homeostasis dysregulation | Clinic trial | Human | LTCC | HFpEF | [ |
- —Jinmen Medical Talents
- —Scientific research projects in critical field of traditional Chinese medicine in Tianjin
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHeart Failure Treatment and Management · GDF15 and Related Biomarkers · Cardiac Fibrosis and Remodeling
Introduction
1
According to the 2023 ESC guidelines [1], heart failure (HF) is categorized by left ventricular ejection fraction into three subtypes: heart failure with reduced ejection fraction (HFrEF), heart failure with mildly reduced ejection fraction (HFmrEF), and heart failure with preserved ejection fraction (HFpEF). The guidelines recommend sodium–glucose cotransporter 2 inhibitors (SGLT2i) for patients across the HF spectrum, highlighting their efficacy in reducing the risk of HF hospitalization and cardiovascular mortality. However, a “one‐size‐fits‐all” therapeutic strategy remains inadequate given the complex and heterogeneous nature of HF. Advancing more precise management approaches and identifying superior therapeutic agents require a deeper understanding of the fundamental pathophysiological mechanisms underlying HF.
The onset and progression of HF are influenced by multiple factors and are closely associated with mechanisms such as calcium homeostasis dysregulation, inflammation, oxidative stress, energy metabolism disturbances, cardiac fibrosis, and apoptosis [2, 3]. Among these, calcium homeostasis dysregulation is increasingly recognized as a pivotal determinant of both systolic and diastolic dysfunction in HF [4]. The 11th edition of major cardiology textbooks has also recognized calcium dysregulation as a key contributor to HF pathogenesis [5]. However, current research on calcium homeostasis dysregulation in HF predominantly focuses on the HFrEF [6]. Mechanistically, HFrEF is characterized by abrupt alterations in specific calcium‐handling proteins within the calcium cycling machinery—such as impaired sarcoplasmic reticulum (SR) Ca^2^ ^+^ reuptake or abnormal Ca^2^ ^+^ influx through L‐type calcium channels (LTCCs). These abnormalities lead to altered cytosolic Ca^2^ ^+^ amplitude, shortened or markedly delayed calcium transients (CaT), and ultimately impaired systolic and diastolic performance [7]. In contrast, studies dedicated to HFpEF—where diastolic dysfunction is the predominant feature—remain relatively scarce. Existing mechanistic investigations in HFpEF have largely focused on phenotype‐specific contributors such as obesity and hypertension [8]. With the recent redefinition of HF in clinical guidelines, comparative analyses between HFrEF and HFpEF have only recently gained attention [9, 10], yet most remain centered on hemodynamic differences rather than mechanistic validation, particularly regarding cytosolic, endoplasmic reticulum, and mitochondrial calcium dynamics.
Recent studies [11] have experimentally examined calcium‐regulating proteins across HF subtypes, suggesting that divergent patterns of calcium homeostasis dysregulation may underlie the differences between HFrEF and HFpEF. Additional research has highlighted mitochondrial calcium dysregulation as a key mechanism specifically implicated in HFpEF [12]. The “calcium homeostasis–systolic/diastolic function” may represent the core “mechanistic–functional” distinction among HF subtypes and serves as a key differentiator between HFrEF and HFpEF. These insights underscore the need for etiology‐specific therapeutic strategies tailored to the distinct pathophysiological profiles of HFrEF and HFpEF.
This study uses calcium homeostasis as a central framework to explore HF pathogenesis, moving from individual calcium‐regulating proteins (“points”) to integrated calcium homeostasis‐related signaling pathways (“lines”). It examines the differential roles of calcium homeostasis dysregulation in HFpEF and HFrEF, aiming to fill critical gaps in the mechanistic understanding of HF and to inform more targeted clinical strategies. Restoring calcium homeostasis—through modulation of calcium‐regulating proteins or calcium‐related signaling pathways—has demonstrated efficacy in improving both systolic and diastolic function [13, 14]. By elucidating the pivotal role of calcium homeostasis dysregulation in HF development, this study offers new perspectives and potential therapeutic avenues for HF prevention and treatment.
Epidemiology
2
HF is a rapidly escalating global public health concern, affecting more than 40 million individuals worldwide [15]. In several European countries, its prevalence among adults is approximately 1–2% [16]. While the prevalence of HFrEF has shown a gradual decline, HFpEF and HFmrEF continue to rise annually and now account for nearly half of all HF cases [17]. In Asian regions such as China, the incidence of HF is 275 per 100,000 person‑years (287 per 100,000 in men and 261 per 100,000 in women), with approximately 3 million new cases occurring annually [18]. The prognosis of patients with HF is poor, with mortality rates of 20 and 53% at 1 and 5 years after diagnosis, respectively [19]. Since 2012, HF‑related mortality has steadily increased, with a particularly sharp rise observed between 2020 and 2021. Age‑adjusted mortality rates in 2021 exceeded those reported in 1999 [20]. Compared with HFrEF, HFpEF has a higher burden of comorbidities and an increased rate of noncardiovascular mortality [21]. A registry‐based cohort study from heart failure centers in China in 2024 reported that cardiovascular death was the leading cause of mortality among patients with HF, accounting for 71.5% of all‑cause deaths. Moreover, patients with HFrEF and HFmrEF exhibited substantially higher mortality rates compared with those with HFpEF [22]. In the past, therapies effective for HFrEF have failed to demonstrate long‑term benefit in HFpEF [23]. These trends underscore the urgent need to elucidate the underlying pathophysiology of HF.
Cardiac diastole consists of two key phases: rapid ventricular filling (linked to isovolumic relaxation) and then late filling (linked to atrial contraction) [24]. The chief mechanism by which cardiac myocyte relaxation occurs is through ATP‐driven uptake of calcium by the SR via SR Ca^2+^ ATPase 2a (SERCA2a) and the dissociation of calcium from myofilaments [25]. Direct quantification of Ca^2^ ^+^ flux among major Ca^2^ ^+^‑handling proteins in nonfailing human cardiomyocytes has shown that SERCA2a accounts for 77% of the decline in intracellular Ca^2^ ^+^ concentration ([Ca^2^ ^+^]i), whereas the Na^+^/Ca^2^ ^+^ exchanger (NCX) contributes the remaining 23% [26]. In HFrEF, these proportions shift to 64% for SERCA2a and 36% for NCX, indicating a 57% increase in NCX's relative contribution [25]. Comparable high‑resolution analyses in HFpEF are still lacking. Accumulating data have indicated that protracted changes in workload promote not only remodelling of ventricular geometry, but also changes in T‐tubule structure within cardiomyocytes [11, 27, 28]. During HFrEF, loss and disorganization of T‐tubules impairs Ca^2+^ homeostasis, and thereby cellular contraction. However, similarly detailed investigations in HFpEF remain scarce.
Given the current landscape of HF, there is an urgent need to clarify its pathophysiology. This study adopts calcium homeostasis as the entry point to explore the “critical points,” “functional pathways,” and “mechanistic networks” that contribute to HF progression.
Pathophysiology
3
The heart is regulated by multiple, interacting mechanisms, including neurotransmitter signaling, energy metabolism, and mitochondrial function, which exhibit extensive crosstalk. For example, β‐adrenergic signaling, a key target in HF therapy, can exert anti‐inflammatory effects, improve endothelial function, and modulate ion channels [29]. Under physiological conditions, cardiac glucose and lipid metabolism are well balanced and mitochondria sustain continuous ATP production. However, when exposed to chronic pressure overload, hemodynamic stress, and altered myocardial perfusion, metabolic remodeling precedes structural remodeling in the heart [30]. Cardiac metabolic derangements are mainly driven by three mechanisms: (i) mitochondrial structural and functional abnormalities, (ii) altered substrate availability and utilization, and (iii) intracellular Ca^2^ ^+^ overload. Mitochondrial dysfunction is manifested by impaired respiration, organelle shrinkage or swelling, and loss of mitochondrial membrane potential, ultimately leading to reduced ATP synthesis. As mitochondrial structure and function deteriorate, substrate utilization is reprogrammed, resulting in an imbalance between fatty acid and glucose metabolism, decreased glucose oxidation, increased glycolysis, and consequent lactate accumulation. In addition, SERCA2a deficiency ultimately contributes to intracellular Na^+^ and Ca^2^ ^+^ overload [31].
Here, we use Ca^2^ ^+^ homeostasis as an entry point to examine the pathophysiological mechanisms across different HF phenotypes.
Calcium Regulating Proteins in Cardiac Physiology
3.1
Calcium regulating proteins operate within a dynamic equilibrium and causal feedback loop. We describe their roles based on subcellular localization—extracellular space, cytosol, and SR—including components such as LTCCs and transverse tubule (T‐tube), NCX, plasma membrane calcium ATPase (PMCA), transient receptor potential (TRP), inositol‐trisphosphate receptor (IP_3_R), SERCA2a, phospholamban (PLN), sarcolipin (SLN), ryanodine receptor 2 (RyR_2_), and calcium release‐activated calcium channel (Orai), described across three phases: signal initiation, intracellular amplification, and calcium reuptake/recycling. Under normal physiological conditions, these calcium regulating proteins collectively maintain the systolic and diastolic function function of cardiomyocytes (Figure 1).
Mechanisms of calcium regulating proteins in physiological and pathological states (heart failure). All Ca2+ amounts in normal and HF were unified, where Ca2+ concentration in normal cardiomyocyte was as follows: SR > extracellular > cytoplasm, and Cart concentration in HF was as follows: cytoplasm > extracellular > SR. The flow of Ca2+ during systole/diastole is plotted, and the rising and falling trend of cytosolic Ca2+ during systole/diastole is indicated by arrows. Orange arrowheads indicate the direction of ion flux during systole, whereas green arrowheads denote the direction during diastole.
Plasma Membrane: Extracellular‐to‐Cytosolic Ca2
- Flux
3.1.1
The working mode of NCX on the plasma membrane of cardiomyocytes during systolic period is mainly reversed, with Na^+^ being expelled from the cells and Ca^2+^ entering the cytoplasm (transport ratio is 3:1) [32]. LTCCs distribute on T‐tubes and normally open to promote Ca^2+^ entry into cytoplasm [33]. When intracellular Ca^2+^ increases, the affinity and transport rate of PMCA for Ca^2+^ increase, promoting Ca^2+^ excretion to cells—the so‐called “calcium influx promotes calcium excretion,” a negative feedback mechanism that maintains intracellular calcium homeostasis [34]. TRP is often in a closed state. When the intracellular Ca^2+^ concentration decreases, it triggers a physiological response of intracellular TRP opening [35]. When Ca^2+^ concentration increases to a certain extent, the negative feedback mechanism inhibits TRP opening and restores calcium homeostasis [36]. Moreover, some uncommon calcium regulating protein also play a non‐negligible role in cardiomyocytes. Hyperpolarization‐activated cyclic nucleotide‐gated (HCN) channel is activated when the cell membrane hyperpolarizes [37]. There are four types of HCN channels—HCN1, HCN2, HCN3, and HCN4—which are predominantly expressed in cardiac and neuronal cells [38] and play a key role in regulating the rhythmic activity of cellular networks [39]. The binding of cyclic adenosine monophosphate (cAMP) molecules and the CNBD region at the C‐terminal of the HCN channel enhances the inward current during diastolic If, causing depolarization of the sinus node membrane potential close to the threshold of Ca^2+^ channel activation, triggering action potentials, thereby maintaining the rhythmic release of excitement [40]. Cardiomyocyte diastole is a different story. The working pattern of NCX on the plasma membrane is positive: Ca^2+^ is expelled from the cells and Na^+^ enters the cytoplasm. PMCA decreased its affinity and transport rate for Ca^2+^ and decreased its efflux of Ca^2+^.
Organelles: Cytosolic to Intracellular Organelles Ca2
- Transfer
3.1.2
Sarcoplasmic Reticulum. During the systolic phase of cardiomyocytes, the RyR_2_ and IP_3_R on SR are open, and Ca^2+^ is released into the cytoplasm in SR [41]. The myocardial T‐tube ensures that RyR_2_ and LTCCs on the plasma membrane are close to each other [42], and inositol 1,4,5‐triphosphate (IP3) initiates LTCCs influx thereby stimulating RyR_2_ to release more Ca^2+^ for optimal calcium‐induced calcium release (CICR) [43, 44]. In addition, IP_3_R activates the NCX reverse mode and increases the influx of Ca^2+^ into the cytoplasm, which is closely related to the “excitement” in the excitation–contraction coupling of the heart [45]. When IP_3_R and RyRs on SR interact, they simultaneously release Ca^2+^ into the cytoplasm. The stromal interaction molecule (STIM) on SR senses the depletion of Ca^2+^ in SR, thereby stimulating Orai to allow Ca^2+^ influx (the interaction between STIM and Orai occurs in a “point‐to‐point” manner) [46]. During diastole period, with the increase of Ca^2+^ in cytoplasm, calmodulin (CaM) was formed to activate TRPM4 and inhibit Ca^2+^ influx into Orai [47]. SERCA2a, when unaffected by PLN, SLN, or other modifications, remains in an open state under normal activity and expression, thereby facilitating the reuptake of cytoplasmic Ca^2+^ into the SR [48, 49].
Mitochondria. Related calcium regulating proteins on mitochondria also play a key role. For example, voltage‐dependent anion channel 1 is the main calcium ion transport channel on the outer membrane of mitochondria and participates in cellular metabolism by transporting ATP and other small metabolites, thereby regulating the TCA cycle and reactive oxygen generation [50]. The mitochondrial calcium uniporter (MCU) is located on the inner membrane of mitochondria [51]. It is influenced by mitochondrial calcium uptake 1 and mitochondrial calcium uptake 2 and can promote intracellular Ca^2+^ flow to mitochondria, thereby maintaining intracellular calcium homeostasis and oxygen free radical homeostasis [52].
Critical Points Linking Calcium Homeostasis Dysregulation to HF
3.2
There is currently no relevant research on HFmrEF calcium regulating proteins, and HFmrEF will not be included in the comparison range for the time being. (Figure 2 was constructed based on the pathological differences between HFrEF and HFpEF, and from a macroscopic perspective distinguishes the dynamic evolution of the normal state, HFrEF, and HFpEF.)
Pathological differences in calcium regulating proteins between HFrEF and HFpEF. At the molecular level, the forward and reverse modes of NCX are illustrated, together with the differential expression and posttranslational modifications of SERCA2a, RyR2, PMCA, and LTCC, as well as the distribution pattern of LTCC. At the functional level, CaT dynamics and sarcomeric states during systole and diastole are depicted. Orange arrowheads indicate the direction of ion flux during systole, whereas green arrowheads denote the direction during diastole.
Plasma Membrane: Extracellular‐to‐Cytosolic Ca2
- Flux
3.2.1
PMCA is an ATP‐driven Ca^2+^ pump capable of maintaining low Ca^2+^ concentrations at rest [53]. Four structurally similar PMCA subtypes have been identified in mammals (PMCA1–PMCA4), in which PMCA1 and PMCA4 are widely expressed without restriction [54]. PMCA has a CaM complex binding domain, which has an inhibitory effect on itself. When intracellular Ca^2+^ increases, more Ca–CaM complexes are formed, thus releasing the inhibition on PMCA and promoting Ca^2+^ efflux [55]. Several studies have shown that in both HFrEF and HFpEF, reduced expression or activity of PMCA impairs efficient Ca^2^ ^+^ extrusion, leading to delayed calcium clearance and contributing to cardiomyocyte systolic and diastolic dysfunction [56].
In HFrEF, the rate of Ca^2^ ^+^ extrusion via PMCA is reduced, whereas in HFpEF, the amount of Ca^2^ ^+^ expelled through PMCA is diminished [57]. In the future, specific experiments should be carried out to analyze the differences in PMCA expression and activity, so as to fill in the relevant mechanisms of calcium homeostasis in cardiomyocytes, which is helpful for further exploration in the field of HF.
NCX is a bidirectional ion transporter on the cardiomyocyte membrane [58]. Its main function is to use the potential energy of the sodium ion concentration gradient on both sides of the membrane established by sodium pump activity, and the direction of the current is consistent with the direction of sodium flow [59]. Sodium and calcium ions are exchanged on the cell membrane to maintain a low concentration of free calcium ions within the cell. The activity of NCX is regulated by many factors, including membrane potential, intracellular and extracellular calcium concentration, phosphorylation, and so on [60]. In HF, NCX expression and activity has always been a controversial issue. Despite differences in models and detection methods, the expression, activity, and predominant mode of NCX can yield completely opposite outcomes within the same disease context.
In HFrEF, Frisk suggested that there was no change in NCX activity [11], Rouhana showed a decrease in NCX activity [61], and Gupta experimental results showed an increase in NCX expression and phosphorylation [62]. Hu experiments showed that although NCX expression decreased, it was mainly in a positive high activity state, leading to sustained excretion of Ca^2+^, a decrease in intracellular Ca^2+^, and a contraction dysfunction [63].
In HFpEF, Frisk found increased NCX activity in ischemic and hypertensive types [11], and Rouhana found decreased NCX activity in deficient blood types [61]. Kamimura studies have shown that NCX is overexpressed and the negative mode activity increases (increased sodium ion concentration in myocardial cells reduces the forward type activity of NCX, enhances the negative type activity, and even directly converts to the reverse type) [64]. At present, NCX inhibitors are mostly applied to HFpEF [65]. In the future, the new trend of targeting drugs of different HF is to weaken the positive mode of NCX in HFrEF and the negative mode in HFpEF, so as to balance sodium and calcium homeostasis.
LTCC and T‐Tube: There are three voltage‐gated Ca^2+^ channels: Cav1, Cav2, and Cav3, of which Cav1 channels are also called LTCC. LTCC was first found in the heart and smooth muscle and includes four subtypes: Cav1.1, Cav1.2, Cav1.3, and Cav1.4 [66]. We know that these cardiovascular channels are almost entirely of the Cav1.2 subtype, and they are blocked by clinically used Ca^2+^ channel blockers such as nifedipine, amlodipine, verapamil, and diltiazem [67]. Mentioning LTCC has to mention the specific organelles on the myocardial cell membrane‐myocardial T‐tubes. Although T‐tubes are not proteins that regulate Ca^2+^, they are closely related to the coupling of cardiomyocyte excitation–contraction [68]. LTCC is rich in myocardial T‐tubes. Therefore, the density of T‐tubes is usually consistent with LTCC expression in cardiomyocytes and the intracellular calcium amplitude [69]. HF is typically characterized by T‐tube disorder, followed by impaired or redistribution of LTCC expression (mostly distributed on the ridge) [70, 71], eventually resulting in damage to CICR.
In HFrEF, reduced T‐tubule density and increased collagen deposition lead to downregulation of LTCC expression [11]. During systole, LTCC redistribution results in diminished Ca^2^ ^+^ influx from the extracellular space into the cytosol. This is accompanied by disruption of the spatial coupling between LTCCs and RyR_2_, forming isolated RyR_2_ domains [72]. Consequently, Ca^2^ ^+^ release from the SR is delayed, impairing CICR [73] and contributing to systolic dysfunction. However, other studies have shown that in HFrEF rats (induced by coronary artery ligation), LTCC protein expression remains unchanged, while LTCC phosphorylation is reduced. Since phosphorylation enhances LTCC open probability, open time, and conductance—thereby promoting Ca^2^ ^+^ influx—its reduction leads to decreased calcium entry, disrupts excitation–contraction coupling, and ultimately impairs systolic function [74].
In HFpEF, T‐tubule density is increased and the structures are dilated, while collagen deposition does not significantly infiltrate the T‐tubules [75]. Studies have shown that higher T‐tubule density and greater expansion are associated with impaired diastolic function. Among different HFpEF phenotypes, T‐tubule density follows a descending order: ischemic > hypertensive > diabetic [11]. In the hypertensive phenotype of HFpEF, LTCC expression is upregulated, leading to enhanced Ca^2^ ^+^ influx and excessive RyR_2_‐mediated Ca^2^ ^+^ release from the SR. This overactivation of CICR amplifies calcium amplitude, enhancing systolic function while concurrently impairing diastolic function [76].
TRP channels are a large family of nonselective cation channel proteins, with six isoforms families with similar sequences: TRPC, TRPM, TRPV, TRPA, TRPML, and TRPP (or PKD) [77]. In 1997, research found that capsaicin can activate the TRPV1 channel to allow Ca^2+^ to flow into the cell [78], and TRPV1 can also be inhibited from opening itself by intracellular Ca^2+^ binding with CaM.
In HFrEF, insufficient expression of TRPV1 may reduce myocardial contractility [79], whereas excessive TRPV1 expression can lead to mitochondrial calcium overload [80]. The increased expression and activity of TRPC channels activate the TLR‐mediated inflammatory signaling pathway, which facilitates Ca^2^ ^+^ leakage from the SR via RyR_2_ and IP_3_R, thereby impairing diastolic function [81, 82]. Low expression of TRPM channels contributes to cardiac hypertrophy and increased Ca^2^ ^+^ influx [47]. Despite their limited expression in cardiomyocytes, TRP channels have not been systematically investigated in the context of HFrEF versus HFpEF. Focused research on this channel may uncover novel pathophysiological mechanisms and address current gaps in HF characterization.
Orai. Store‐operated calcium entry (SOCE) is a ubiquitous mechanism for calcium signal generation and calcium homeostasis maintenance in animal cells [83]. SOCE is mediated by the STIM sensor on SR and the Orai on the plasma membrane [84]. In HF, excessive activation of SOCE or loss of SOCE results in immunodeficiency [85, 86].
In HFrEF, SOCE may be impaired. The reduced Ca^2^ ^+^ content in the SR fails to adequately activate STIM sensors, preventing the opening of Orai channels on the plasma membrane and thereby limiting Ca^2^ ^+^ influx into the cytosol [87].
In contrast, in HFpEF, SOCE may be excessively activated. The decreased SR Ca^2^ ^+^ content triggers STIM sensors on the SR to activate Orai channels on the plasma membrane, leading to enhanced Ca^2^ ^+^ influx into the cytosol. SOCE inhibitors, as immunosuppressants or anti‐inflammatory agents, can become new targets for drugs to treat HF [88].
Organelles: Cytosolic to Intracellular Organelles Ca2
- Transfer
3.2.2
Sarcoplasmic Reticulum
3.2.2.1
SERCA2a is a cardiac‐specific calcium pump located on the SR membrane. SERCA2a uses the energy from ATP hydrolysis to actively transport 75% of cytosolic Ca^2+^ into the SR during diastole, contributing to efficient cardiac relaxation and refilling of SR Ca^2+^ stores for subsequent contractions [89]. This process is inseparable from SERCA2a itself's activity, expression, posttranslational modification and regulation by transmembrane micropeptide [90]. Among them, ubiquitination, acetylation, and phosphorylation are the most important modification methods of SERCA2a. Considering the different modeling methods, most studies use the protein expression of SERCA2a/PLB and pPLB/PLB to reflect their functional roles [61]. A few studies used caffeine‐induced CaT to calculate the rate of Ca^2+^ reuptake and removal from cells to evaluate SERCA2a and NCX functions. Current studies have indicated that SERCA2a is a potential therapeutic target for both acute and chronic HF [91]. The activity of SERCA2a in HFrEF and HFpEF remains inconsistent, and no unified standard has been established to date.
Studies have shown that in HFrEF, both the expression and activity of SERCA2a are reduced, leading to impaired Ca^2^ ^+^ reuptake into the SR. Consequently, the rate of cytosolic Ca^2^ ^+^ clearance declines [11], CaT are attenuated, and myocardial contractility is diminished to a certain extent [90]. From a posttranslational modification perspective, decreased ubiquitination and phosphorylation, along with increased acetylation of SERCA2a, may contribute to its reduced activity [92]. Oxidation of SERCA2a at Cys498 leads to a decrease in its expression [93].
In HFpEF, the situation is more complex. In the ZSF‐1 rat model of HFpEF, which is based on diabetes and hypertension, SERCA2a expression is elevated, yet its activity is paradoxically decreased. Conversely, in HFpEF models induced by minor myocardial infarction, SERCA2a activity appears to be increased [94]. The direct role of SERCA2a dysfunction in HFpEF remains undetermined [25]. These experimental discrepancies likely arise from differences in disease modeling approaches, posttranslational modifications, and protein–protein interactions. Therefore, in addition to considering the subtype of HF, it is essential to account for the underlying pathogenic factors affecting SERCA2a when developing targeted pharmacological modulators.
PLB and SLN. exert inhibitory effects on SERCA2a, thereby reducing the capacity for Ca^2^ ^+^ reuptake into the SR and ultimately impairing cardiac systolic and diastolic function [95]. PLB is a calcium‐sensitive phosphoprotein; when phosphorylated (pPLB), its inhibitory effect on SERCA2a is attenuated, enhancing SERCA2a‐mediated Ca^2^ ^+^ uptake. In HFrEF, PLB expression is elevated while pPLB levels are decreased [76]. In HFpEF, PLB expression is also increased, yet the SERCA2a/PLB ratio is reduced [94]. In both HFrEF and HFpEF, the net effect is suppression of SERCA2a activity and impaired CaT dynamics. SLN is a low‐molecular‐weight protein homologous to PLB, sharing a similar transmembrane domain. Unlike PLB, SLN reduces Ca^2^ ^+^ accumulation within the SR without affecting ATP hydrolysis rate [96]. In HFrEF, SLN expression is downregulated, whereas SERCA2a expression is upregulated, leading to excessive SR Ca^2^ ^+^ loading and systolic dysfunction [97]. Whether SLN expression is elevated in HFpEF remains unclear due to a lack of definitive studies. Interestingly, SLN expression is significantly upregulated in diabetic HF models compared with nondiabetic counterparts [98].
*RyR_2_ *. RyRs are Ca^2^ ^+^ release channels located on the SR of cardiomyocytes. There are three subtypes of RyR_1_, RyR_2_, and RyR_3_. Among them, impaired RyR_2_ opening during systole or aberrant activation during diastole can disrupt SR Ca^2^ ^+^ homeostasis, thereby contributing to HF or cardiac arrhythmias [99]. In HFrEF, RyR_2_ expression during systole is reduced or functionally impaired, resulting in insufficient Ca^2^ ^+^ release from the SR into the cytosol. This limits actomyosin cross‐bridge formation and severely compromises systolic function [100]. In HFpEF, RyR_2_ exhibits excessive opening during systole, leading to markedly greater Ca^2^ ^+^ efflux from the SR compared with HFrEF, which is closely associated with the compensatory enhancement of cardiomyocyte contractility in HFpEF [101]. However, other studies have reported that RyR_2_ expression levels are comparable between HFrEF and HFpEF, yet both conditions display hyperphosphorylation of RyR_2_, with HFpEF showing a higher degree of phosphorylation [11]. This posttranslational modification may underlie the aberrant diastolic opening of RyR_2_, disrupted CICR, and impaired diastolic function.
IP_3_R is a membrane glycoprotein complex located in the endoplasmic reticulum that requires a second messenger to be activated by IP_3_ [102]. Among the IP_3_R subtypes, there are two types related to cardiomyocytes: IP_3_R2 and IP_3_R3. Atrial and ventricular myocytes in most animals mainly express IP_3_R2 and a small number express IP_3_R3 [103]. Their distribution in the atrium is much larger than that in the ventricle. The structure and function of IP_3_R are regulated by multiple factors such as phosphorylation, protein interactions, or negative calcium ion feedback. In HFrEF, RyRs expression is downregulated, while IP_3_R may be upregulated as a compensatory response, providing an alternative pathway for intracellular Ca^2+^ [100]. In HFpEF, phospholipase C (PLC) in cardiomyocytes is activated during diastolic phase, and IP_3_ stimulates IP_3_R to release Ca^2+^ stored in SR [104], thereby increasing intracellular Ca^2+^ concentration, leading to an increase in related diastolic tension.
Mitochondria
3.2.2.2
MCU is a highly selective Ca^2^ ^+^ channel located on the inner mitochondrial membrane, responsible for transporting cytosolic Ca^2^ ^+^ into the mitochondrial matrix [69]. In HFrEF, the expression level of MCU is compensated for increased, which may be to increase calcium uptake by mitochondria to maintain myocardial contractility and improve energy production [105].
Having identified calcium regulating proteins as critical points of calcium homeostasis disruption, we subsequently delineated the microdomain architecture, molecular homology classifications, and interrelationships among calcium homeostasis‐related signaling pathways (Figure 3).
Mechanisms of calcium homeostasis‐related signaling pathways in physiological and pathological states (heart failure). The diagram illustrates the interactions of the cAMP, cGMP, CaMKII, RAS, and PI3K pathways with calcium regulating proteins and calcium ions. Blue highlights denote cGMP microdomains (GC‐A and GC‐B), whereas red highlights denote cAMP microdomains. Under physiological conditions, cGMP microdomains predominate over cAMP microdomains; in HF, this relationship is reversed. The balance between the two influences calcium regulating proteins. In addition, the figure depicts how the CaMKII, RAS, and PI3K pathways differentially contribute to physiological versus pathological cardiac hypertrophy.
Calcium Homeostasis‐Related Signaling Pathways in Cardiac Physiology
3.3
cAMP Pathway
3.3.1
Neurohormones activate β‐adrenergic receptors on the plasma membrane to couple with heterologous triguanine nucleotide regulatory proteins (G proteins), causing βγ dimers to release active Gas and activating adenylcyclase (AC) to promote cAMP production [106]. The G protein‐coupled receptor kinases (GRK) family consists of seven members (GRK1–7), which can be classified into three subgroups based on gene structure and sequence homology: the visual or rhodopsin kinase subfamily (GRK1 and GRK7), the βAR kinase subfamily (GRK2 and GRK3), and the GRK4 subfamily (GRK4, GRK5, and GRK6) [107]. Among them, GRK2 and GRK5 are expressed in nearly all cardiac cell types [108, 109]. GRK2 phosphorylates activated βARs, which promotes the recruitment of β‑arrestins. This process uncouples the receptor from G proteins, leading to desensitization of βAR signaling [110, 111]. Similar to GRK2, overexpression of GRK5 leads to marked β‑adrenergic receptor desensitization [112]. Under physiological conditions, GRKs modulate the Ras signaling pathway, thereby influencing cardiac physiological hypertrophy [113].
This cAMP can be hydrolyzed by phosphodiesterases (PDEs). Among them, cAMP stimulated by β1‐AR is regulated by PDE2, PDE3, and PDE4, with a large microdomain. However, β2‐AR is regulated by PDE3 and PDE4, and the cAMP microdomain is restricted. cAMP and its downstream effector protein kinase A (PKA) are crucial biochemical messengers in regulating myocardial cell function. PKA can negatively feedback upstream AC and regulate stimulation of downstream PDEs. PKA is divided into multiple regions with the help of A‐kinase anchoring proteins [114]. Activation of the cAMP/PKA signaling pathway can directly phosphorylate several key proteins involved in excitation–contraction coupling, including the LTCC, PLB, RyR_2_, and troponin I (TnI) [115]. Short‐term activation of the cAMP pathway is beneficial to the heart. PKA phosphorylates guanosine 5′‐triphosphate (GTP)‐binding protein RAD, thereby enhancing Ca^2^ ^+^ influx—not through direct phosphorylation of LTCC. Activation of the cAMP pathway also facilitates systolic Ca^2^ ^+^ release by phosphorylating RyR_2_, accelerates Ca^2^ ^+^ reuptake into the SR via PLB phosphorylation and subsequent SERCA2a activation, and modulates myofilament sensitivity through phosphorylation of TnI. Additionally, short‐term activation of the cAMP pathway promotes HCN channel activity, contributing to the regulation of diastolic function.
Cyclic Guanosine Monophosphate Pathway
3.3.2
Cyclic guanosine monophosphate (cGMP) is the second messenger that regulates various physiological processes such as cardiac contraction, vascular tone, and cardiac remodeling. GTP is catalyzed by guanosine cyclase (GC) to synthesize cGMP. Upstream of cGMP are two types of GCs: (1) natriuretic peptides (NPs) activate particulate (membrane bound) GC (NP–GC, also known as pGC). For example, purine cyclase A (GC‐A, also known as NPR1 or NPRA) is activated by ANP and BNP, and purine cyclase B (GC‐B, also known as NPR2 or NPRB) is activated by CNP; (2) nitric oxide (NO) activates soluble (intracellular) GC (NO‐GC, also known as sGC) [116]. pGC‐A is located in the T tubule and pGC‐B is distributed throughout the sarcolma. The GC‐A microdomain has little effect on contractility, while the GC‐B microdomain has a positive inotropic effect. The activity of GC‐A microdomain is regulated by PDE2 and PDE9, while GC‐B microdomain is also influenced by PDE3. The cGMP microdomain produced by sGC is generated through β_3_‐AR and is regulated by PDE5 and PDE3. PDE9 primarily hydrolyzes cGMP derived from the pGC pathway, whereas PDE5 mainly hydrolyzes cGMP generated via the sGC pathway. Under physiological conditions, the abundance of PDE2 in cardiomyocytes is relatively low. Binding of cGMP to the regulatory domain of PDE2 stimulates cAMP degradation, thereby attenuating excessive cAMP responses [117].
Calcium/CaM‐Dependent Protein Kinase II Pathway
3.3.3
Calcium/CaM‐dependent protein kinase II (CaMKII) is a serine/threonine kinase comprising four distinct subtypes (α, β, γ, δ), with CaMKIIδ being the most prominent subtype in cardiomyocytes. CaMKIIδ is activated by Ca/CaM, reactive oxygen species (ROS), and exchange protein directly activated by cAMP. Activated CaMKIIδ produces a stimulating effect by phosphorylation of related calcium regulating proteins, such as increasing the inward current through LTCC and boosting the phosphorylation of PLB, which improves SR uptake of cytoplasmic Ca^2+^. In the nucleus, CaMKIIδB plays a protective role for the heart [118, 119]. CaMKIIδB mediates the phosphorylation of histone deacetylases (HDAC) and inhibits the transcription of the hypertrophy factor myocyte enhancer factor 2 (MEF‐2). Phosphorylation‐induced calcineurin (CaN), a regulator of the nuclear factors of activated T cells (NFAT) transcription factor involved in cardiac hypertrophy, also plays a role in transcription regulation [120]. CaMKIIδB increases phosphorylated heat shock factor 1 (HSF1), enhances intracellular heat shock protein 70 gene expression, and resists myocardial apoptosis [121]. Enhance MCU gene transcription by phosphorylated cAMP response element binding protein (CREB), facilitate the transfer of Ca^2+^ of SR to mitochondria, maintain intracellular calcium homeostasis, and prevent cardiac hypertrophy and diastolic dysfunction [122].
Ras Pathway
3.3.4
This classical pathway of Ras is called “pathway drug cocktail” and used to be the preferred target of cancer [123]. Its downstream PLCε, JNK, and GSK can affect HF fibrosis, inflammation, oxidative stress, mitochondrial function, and metabolic disorders, and interact with calcium homeostasis. Small G protein of Ras family (guanosine triphosphate binding) consists of enzymes that hydrolyze GDP and interact with various tyrosine kinase receptors (EGF, PDGF, etc.) and GPCR [124]. ERK pathway downstream of Ras is associated with physiological hypertrophy of heart induced by long‐term exercise. Phosphatidylinositol 3‐kinase (PI3K) pathway can inhibit NFAT nuclear translocation and phosphorylate NF‐kB, which plays a dual role in promoting survival and antiapoptosis of heart.
PI3K Pathway
3.3.5
PI3Ks protein family is involved in the regulation of multiple cellular functions such as cell survival, growth, metabolism and blood sugar homeostasis. Different PI3K isoforms have different effects.
PI3K γ stimulates cells to form PLC through G protein receptors [125]. Different isoforms of PLC can be activated by various upstream signals: PLCβ is activated by Gαq, PLCγ by receptor tyrosine kinases, and PLCε by Rho and Ras. PLC catalyzes the conversion of phosphatidylinositol 4,5‐bisphosphate (PIP2) into diacylglycerol (DAG) and IP_3_. DAG activates protein kinase C, while IP_3_ stimulates IP_3_R on SR. Notably, IP_3_R can colocalize with RyR_2_, allowing for mutual regulation and activation [126], which promotes calcium influx from the extracellular space (via LTCC and NCX), a process referred to as IP3‐induced calcium release.
PI3Kα phosphorylates PIP2 to produce PIP3, inhibiting sodium influx through the cytoplasmic Nav1.5 channel. Additionally, PI3Kα phosphorylates Akt, enhancing the phosphorylation of PLB, which increases SERCA2a activity and lowers intracellular Ca^2+^ levels. Inhibiting the expression and activity of IP_3_R reduces Ca^2+^ reaching mitochondria through MCU, reduces ROS, increases ATP, and further improves PMCA and SERCA2a activities. PI3Kβ inhibits phosphatase and tensin homolog, thereby reducing the dephosphorylation of PIP3 to PIP2.
Functional Lines Linking Calcium Homeostasis Dysregulation to HF
3.4
We next aimed to identify the specific stages at which calcium dysregulation emerges during HF progression and to clarify the “functional lines” that drive this imbalance. This analysis also highlights the mechanistic distinctions between HFrEF and HFpEF (Figure 4).
Pathological differences in calcium homeostasis‐related signaling pathways between HFrEF and HFpEF. The diagram depicts the differences in the cGMP pathway (GC‐A and GC‐B) between HFrEF and HFpEF, as well as the distinct interactions of the cAMP, CaMKII, Ras, and PI3K pathways with calcium regulating proteins.
cAMP Pathway
3.4.1
Remodeling of cAMP microdomains is currently a major focus in HF research, while sustained activation of cAMP can trigger HF [127]. In HF, both β1‐AR and β2‐AR are downregulated, with a reduced β1‐AR/β2‐AR ratio and redistribution of each receptor subtype. The microdomains between cAMP and PDE are diminished, and Ca^2^ ^+^ flux is altered. HFrEF and HFpEF exhibit distinct patterns of cAMP microdomain organization.
In HFrEF, cAMP metabolite concentrations are elevated [128], with neurohormonal activation serving as the primary driver. β1‐AR sensitivity is reduced, whereas β2‐AR ionotropic signaling is enhanced. Gαi coupling and recruitment of GRKs are increased. Elevated GRK2 levels simultaneously induce hypertrophic gene expression, impair insulin signaling, and promote myocardial fibrosis [108]. GRK5 contributes to impaired cardiac function and immune cell recruitment [113]. Due to T‐tubule defects, β2‐AR and LTCCs are redistributed to the crests or tops of the sarcolemma. Within β2‐AR‐associated cAMP microdomains, PDE3 and PDE4 activity is reduced, leading to decreased LTCC phosphorylation. In β1‐AR‐associated cAMP microdomains, PDE2 and PDE3 activity is increased while PDE4 activity is reduced, resulting in RyR_2_ hyperphosphorylation and PLN hypophosphorylation [129]. Consequently, systolic intracellular Ca^2^ ^+^ is reduced, and myocardial contractility declines.
In HFpEF, cAMP metabolite concentrations are lower [128]. Comorbidities drive cAMP signaling through inflammation, increased ROS, and reduced NO bioavailability. In HFpEF, β2‐AR interacts with the insulin receptor, thereby promoting Gαi coupling and GRK recruitment. However, some studies have reported that GRK2 and GRK5 show no significant increase compared with normal controls [130]. Within β2‐AR‐associated cAMP microdomains, PDE3 and PDE4 expression is upregulated, leading to PLN hypophosphorylation and reduced SERCA2a activity [131]. This increases Ca^2^ ^+^ transient kinetics or elevates diastolic cytosolic Ca^2^ ^+^ concentration, ultimately causing diastolic dysfunction. PKA‐mediated Akt phosphorylation regulates glucose uptake; however, sustained Akt activation and reduced GLUT4 translocation contribute to insulin resistance.
cGMP Pathway
3.4.2
In HF, although NEP and total NPs concentrations are increased, NPs are degraded at the cell surface by associated proteases, leading to a reduced ratio of mature (active) to immature (inactive) NPs and thereby diminished NP activation [132]. GC‐A activity is markedly reduced, whereas GC‐B activity remains unchanged or is increased [133]. Endothelial NO synthase generates pathological ROS, resulting in decreased NO and subsequent reduction of sGC activity. Expression of PDE2, PDE5, and PDE9 is upregulated, leading to enhanced hydrolysis of cGMP. PDE2 hydrolyzes both cGMP and cAMP, playing a critical role in crosstalk between the cGMP and cAMP pathways. At this stage, altered cGMP microdomain organization prevents cAMP degradation [134]. Downstream protein kinase G (PKG) is no longer activated but instead oxidized, while ROS directly reduce NO release from cardiovascular tissues.
In HFrEF, pGC‐associated cGMP microdomains are severely impaired. Expression of SERCA2a and pPLB is reduced, resulting in diminished SR Ca^2^ ^+^ uptake during diastole and elevated cytosolic Ca^2^ ^+^ [135]. This activates CaMKII, which alters LTCC distribution (restricted to crests), causing LTCC dysfunction and reduced systolic Ca^2^ ^+^ influx [136].
In HFpEF, pGC‐associated GC‐B microdomains are enhanced, whereas sGC‐associated cGMP microdomains are reduced [137]. Because sGC‐mediated NO activation is attenuated, inhibition of LTCC and TRPC6 is diminished, leading to increased systolic Ca^2^ ^+^ influx [138]. PLB phosphorylation is reduced, SR Ca^2^ ^+^ reuptake is impaired, and myofilament Ca^2^ ^+^ affinity is increased [139, 140], resulting in compensatory augmentation of contractile force in HFpEF [141]. However, Ca^2^ ^+^ activates CaN, which dephosphorylates NFAT, promoting its nuclear translocation and amplifying hypertrophic signaling [142]. This damages myofibrils and initiates a vicious cycle. Downstream PKG directly regulates diastolic function and is closely linked to HFpEF pathophysiology [143]. Nevertheless, isosorbide dinitrate, the only PKG modulator tested to date, worsens HFpEF‐related phenotypes, and other PKG‐targeted agents have not consistently improved outcomes in HF patients [144]. Future studies are warranted to further explore the cGMP signaling pathway.
CaMKII Pathway
3.4.3
In HF, nuclear CaMKIIδB expression is reduced, whereas CaMKIIδC expression is increased [121]. CaMKIIδB‐mediated phosphorylation of CREB and HSF1 is attenuated, leading to decreased MCU transcription. In contrast, CaMKIIδC enhances HDAC phosphorylation and nuclear translocation, diminishes repression of MEF‐2 expression, and promotes nuclear NFAT–GATA binding, thereby inducing cardiac hypertrophy [120]. Autophosphorylation of CaMKIIδC also modulates the cAMP‐binding protein CREB; phosphorylated CREB upregulates IP_3_R1 expression, facilitating IP_3_R1‐mediated Ca^2^ ^+^ release.
In HFrEF, CaMKII is hyperactivated, markedly increasing RyR_2_ phosphorylation to a greater extent than PKA‐mediated phosphorylation. Phosphorylation of RyR_2_ promotes systolic SR Ca^2^ ^+^ release and diastolic SR Ca^2^ ^+^ leak, activating inward NCX currents [118] and thereby triggering depolarization.
In HFpEF, CaMKII expression is reduced, accompanied by decreased Thr17 phosphorylation of PLB, which weakens SR Ca^2^ ^+^ reuptake [145]. However, RyR_2_ phosphorylation is excessively increased, resulting in an imbalance between elevated NCX expression and reduced SERCA2a expression [146].
Ras Pathway
3.4.4
In HF, chronic activation of Akt suppresses autophagy, while excessive activation of NF‐κB triggers inflammation and pro‐apoptotic signaling, both of which are key pathogenic factors [147]. Activation of the p38 and JNK pathways, as well as CaN downstream of Ras–RAF signaling, represent critical mediators of pathological hypertrophy. Clinically, RAS inhibitors are used more frequently in HFrEF than in HFpEF [148]; however, their application in basic experimental models is considerably more complex.
In HFrEF, the NF‐κB/p38/JNK pathway is activated [149]. JNK1 facilitates the interaction of Akt with PLB at Thr17, thereby inhibiting SERCA2a and preventing diastolic SR Ca^2^ ^+^ reuptake. Concurrently, the TGFβ/Smad pathway is activated, inducing inflammation and fibrosis, which further disrupt Ca^2^ ^+^ homeostasis.
In HFpEF, the Rho/ROCK pathway is upregulated, promoting actin–myosin contraction in cardiomyocytes. Rho/ROCK signaling suppresses Smad activity, thereby reducing TGFβ/Smad‐dependent signaling [150]. In addition, Ras/ERK signaling is upregulated, contributing to interstitial fibrosis. NF‐κB/p38/JNK signaling is also enhanced; increased NF‐κB activity stimulates CaMKII phosphorylation, which in turn promotes RyR_2_ phosphorylation and upregulation of NCX expression, leading to Ca^2^ ^+^ homeostatic imbalance.
PI3K Pathway
3.4.5
In HF, increased PI3Kγ, together with IP_3_ generated by PLC and Ca^2^ ^+^ released from RyR_2_, jointly regulate IP_3_R2 located on the nuclear envelope, thereby triggering nuclear Ca^2^ ^+^ signaling. This activates HDAC and NFAT shuttling, stimulating transcription factors that induce gene hypertrophy and ultimately drive cardiac remodeling [151]. Nuclear Ca^2^ ^+^ signaling can also activate CaMKII, which in turn promotes LTCC opening. By contrast, the activities of PI3Kα and PI3Kβ are relatively diminished, impairing their dual roles in suppressing autophagy and promoting mitophagy.
In HFrEF, RyR_2_ expression is reduced, whereas IP_3_R expression is compensatorily increased, with phosphorylation of both RyR_2_ and IP_3_R elevated in parallel [152].
In HFpEF, no fundamental experimental studies have yet been conducted on this pathway. Beyond Ca^2^ ^+^ homeostasis, this pathway is implicated in mitochondrial metabolism, apoptosis, inflammation, and other processes closely linked to the heterogeneity of HFpEF. The key protein IP_3_R within this pathway has also emerged as a promising target in the exploration of HFpEF phenotypes associated with atrial fibrillation, making it a recent focus of research.
This section focuses on analyzing the signaling pathways most frequently studied in HF over the past 5 years under conditions of calcium homeostasis dysregulation. Crosstalk between the cAMP and cGMP pathways is evident: under normal conditions, activation of the cAMP pathway is lower than that of cGMP, whereas in HF, cAMP pathway activation exceeds that of cGMP. Dysregulation of these two pathways, accompanied by inflammation and oxidative stress, contributes to calcium homeostasis dysregulation. The CaMKII and Ras pathways represent interconnected signaling cascades linking extracellular, cytoplasmic, and nuclear compartments of cardiomyocytes. Ca^2^ ^+^ imbalance drives the CaMKII pathway to induce secondary mechanisms such as fibrosis and apoptosis, while Ras pathway abnormalities simultaneously provoke calcium homeostasis dysregulation, mitochondrial dysfunction, inflammation, and impaired autophagy. PI3K, as a downstream branch of the Ras pathway, primarily regulates calcium homeostasis between the cytoplasm and intracellular organelles. It remains difficult to delineate the temporal sequence among these mechanisms or pathways; instead, calcium homeostasis dysregulation can only be described in terms of its specific localization within cardiomyocytes or its significance within a given pathway. Identifying critical points within calcium homeostasis, linking them to functional lines, and constructing mechanistic networks to drive effective clinical therapies remains a major challenge.
Mechanistic Network of HF
3.5
As previously discussed, dysregulation of calcium homeostasis can arise from posttranslational modifications of calcium regulating proteins—for example, acetylation or phosphorylation of SERCA2a, which may alter its expression or activity, or shifts in the forward and reverse modes of NCX that influence the direction of Ca^2^ ^+^ flux. Changes in calcium‑related signaling pathways are often driven by the activation or relocalization of specific components within these pathways. Moreover, disturbances in calcium homeostasis may also result from crosstalk with other mechanisms, whereby parallel pathways modulate calcium‑associated signaling or inflammatory mediators modify calcium regulating proteins. In this context, we discuss the bidirectional causal interplay between calcium homeostasis and other regulatory mechanisms. Nevertheless, the fundamental nature of these processes remains under active investigation. Calcium homeostasis dysregulation is closely associated with multiple mechanisms—including cardiac fibrosis, mitochondrial dysfunction, inflammation, oxidative stress, cardiomyocyte hypertrophy, and stiffness (Figure 5)—that collectively drive the onset and progression of HF.
Pathophysiological mechanism network in heart failure. Starting from calcium homeostasis, calcium‐regulating proteins, or calcium‐related pathways interact with other mechanisms to jointly drive the onset and progression of heart failure.
Cyclic Reinforcement
3.5.1
Calcium homeostasis dysregulation interacts with fibrosis, mitochondrial dysfunction, oxidative stress, and inflammation in reinforcing cycles. Each mechanism amplifies the others, creating a self‑perpetuating pathological loop that accelerates HF progression.
Dysfunction of calcium regulating proteins in SR or abnormalities in the cAMP/cGMP signaling pathways lead to calcium homeostasis imbalance [153], thereby disturbing mitochondrial calcium regulation [154]. In HFrEF, mitochondrial calcium overload alters membrane potential and elevates ROS levels, triggering mitophagy and oxidative stress [155, 156], which subsequently contribute to cardiomyocyte hypertrophy [157]. In HFpEF, reduced mitochondrial calcium impairs ATP production and induces metabolic disturbances [158], resulting in passive stiffness. These reactions promote apoptosis [159], which in turn activates Ras or PI3K signaling pathways, driving inflammation [160]. Inflammation further induces fibrosis, thereby affecting CaMKII signaling and once again disrupting calcium homeostasis, ultimately impairing excitation–contraction coupling and establishing a vicious cycle [161].
Dynamic and Bidirectional Interference
3.5.2
The relationship between calcium homeostasis dysregulation and other mechanisms is bidirectional and context dependent. Inflammation may arise as a consequence of calcium dysregulation yet also exacerbate it; oxidative stress both results from and worsens mitochondrial dysfunction. Such reciprocal coupling across pathways highlights the fluid and interconnected nature of the mechanistic web underlying HF.
In HFrEF, impaired calcium regulating proteins such as SERCA2a and RyR_2_ lead to cytosolic calcium overload. Excess calcium enters mitochondria, causing depolarization and ROS generation [162, 163]. The resulting oxidative stress further damages calcium cycling proteins [164], thereby reinforcing calcium homeostasis dysregulation in a bidirectional loop. In HFpEF, systemic inflammation and metabolic stress promote myocardial fibrosis and stiffness [165]. Increased stiffness impairs diastolic calcium reuptake, disturbing calcium homeostasis [166]. Calcium homeostasis dysregulation in turn activates inflammatory signaling pathways such as Ras pathway, which further drive fibrosis and stiffness [167, 168], forming a dynamic bidirectional interference.
Targeting the current pathophysiological mechanisms of HF provides opportunities for comprehensive patient management and the development of innovative therapeutic agents.
Management Strategies
4
At present, HF treatment still relies largely on conventional management strategies, including pharmacological and nonpharmacological interventions. The lack of individualized therapy has remained a persistent challenge in this field. In this section, we summarize guideline‐recommended management strategies for different HF phenotypes and, using calcium homeostasis as an illustrative example, review the current landscape of novel drug development, including both preclinical and clinical studies. To facilitate the future development of novel drugs directed at distinct mechanisms and molecular targets.
Current Standard Treatment: Holistic Comprehensive Management
4.1
Current management strategies for HF are individualized according to the type of HF and patient‐specific characteristics. Pharmacological therapy remains the cornerstone of HF management. For patients with HFrEF, guidelines recommend the use of diuretics, renin–angiotensin system inhibitors, β‑blockers, and mineralocorticoid receptor antagonists. For patients with HFimpEF, it is advised that guideline‐directed medical therapy (GDMT) be continued after improvement to prevent recurrence of HF and left ventricular dysfunction. For HFmrEF and HFpEF, diuretics and SGLT2i are recommended to alleviate symptoms and reduce the risk of hospitalization. Nonpharmacological interventions are integral throughout the course of HF management, including dietary regulation, exercise control, and device‐based therapies such as cardiac resynchronization therapy and implantable cardioverter‐defibrillators [169]. To prevent disease progression, dynamic monitoring of HF signs and biomarkers is required. Management of comorbidities such as hypertension, coronary artery disease, and atrial fibrillation is essential to reduce additional risk. Long‐term follow‐up and patient education are critical to improving adherence and optimizing outcomes.
Novel Horizons in Mechanism‑Oriented Treatment: Focusing on Calcium Homeostasis
4.2
Future development of pharmacological therapy is expected to follow a “critical point–functional line–mechanistic network” strategy to advance drug discovery for HF. Targeting calcium regulating proteins or calcium homeostasis‐related signaling pathways can alter ion flux direction and improve intracellular Na^+^–Ca^2^ ^+^ and acid–base balance in cardiomyocytes, thereby modulating excitation–contraction coupling and restoring both diastolic and systolic cardiac function.
To date, pharmacological agents targeting calcium regulating proteins or calcium homeostasis‐related signaling pathways in both HFrEF and HFpEF have been shown to effectively modulate pathological states [170]. Potential therapeutic targets for HFpEF include SERCA2a, NCX, PLB, and the Ras pathway [171]. Preclinical and clinical studies of calcium regulating proteins and calcium homeostasis‐related signaling pathways in HFrEF and HFpEF are summarized in Table 1, with classification according to molecular mechanisms.
Preclinical Experiments
4.2.1
Studies conducted in primary mouse cardiomyocytes and cardiomyocyte lines have identified several agents with potential therapeutic effects. For example, Probenecid and Danicamtiv target calcium regulating proteins, ameliorating the reduction of cytosolic calcium during systole in HFrEF, thereby restoring calcium homeostasis and modulating mitochondrial function [172, 173]. Schaftoside acts on calcium homeostasis‐related signaling pathways to improve delayed diastolic CaT in HFpEF, while also repairing mechanisms involving autophagy and inflammation [175]. Animal experiments have demonstrated that compounds such as Mettl13 and Luteolin can target calcium regulating proteins including SERCA2a, NCX, and PLB, enhancing SR Ca^2^ ^+^ reuptake during diastole, attenuating CaT in HFrEF, and regulating fibrosis as well as excitation–contraction coupling [176, 181]. The novel GRK2 inhibitor CCG258208, derived from paroxetine, improves left ventricular contractile function and limits adverse remodeling in HFrEF [183]. Berberine and Triiodothyronine, by targeting RyR_2_ and PLB, improve SR Ca^2^ ^+^ release during systole in HFpEF, augment CaT, and ameliorate fibrosis and mitochondrial dysfunction [187, 188]. Certain drugs regulate the activity or expression of calcium regulatory proteins by influencing their posttranslational modifications [93].
Clinic Trials
4.2.2
In clinical trials, the calcium channel blockers (CCBs) familiar to us act as calcium homeostasis modulators targeting LTCC. Evidence indicates that CCBs can restore disrupted calcium homeostasis, thereby improving clinical symptoms and prognosis in patients with HFpEF [195]. Acyl ghrelin and the adenosine A1‑receptor agonist neladenoson bialanate, on the other hand, target calcium regulating proteins such as SERCA2a and Titin, as well as calcium homeostasis‐related signaling pathways including cAMP, thereby influencing metabolism and cardiomyocyte stiffness in HFrEF [189, 190]. As a RyR_2_ inhibitor, M201‐A has been shown to enhance natriuresis and improve renal function in humans, suggesting its potential as a therapeutic target for HF [196]. Pharmacological agents that inhibit RyR_2_ phosphorylation may also hold potential for improving HFpEF. At present, clinical studies focusing on specific calcium‑related targets or pathways remain at an early stage.
Western pharmacotherapy is supported by a relatively complete modern theoretical framework and robust clinical evidence, whereas natural medicines exert therapeutic effects through multicomponent, multipathway, and multitarget mechanisms. Combination therapy has opened innovative avenues in pharmacokinetics. In the near future, advances in modern scientific technologies, building upon prior research, may enable precise targeting of calcium regulating proteins or calcium homeostasis‐related signaling pathways to restore calcium homeostasis, facilitate drug development, modulate the pathological networks of HF, and generate additional clinical and experimental evidence.
Unmet Needs and Future Directions
5
Beyond establishing personalized management strategies for HF, several critical yet often overlooked challenges persist. Current diagnostic pathways are relatively rigid, and the supporting research and technical infrastructure is still insufficient, underscoring an urgent need for further optimization and strengthening. Prospectively, increased financial investment, strengthened experimental capacity, improved interdepartmental coordination, and deeper international collaboration will be essential for establishing a more resilient HF diagnostic–therapeutic system and a comprehensive safety framework.
Current Problems and Challenges in HF
5.1
Reducing mortality, decreasing rehospitalization rates, and improving quality of life remain urgent clinical priorities. Achieving these goals requires not only comprehensive mechanistic exploration but also methodological innovation. Traditional single‑omics approaches face challenges of reproducibility and standardization, with substantial variability observed across laboratories, analytical platforms, and study populations [197]. As a result, validating omics‑derived biomarkers in independent patient cohorts and translating these findings into clinically actionable diagnostic tools remain difficult, limiting their clinical utility. The inherent complexity of biological systems further complicates efforts to accurately model cardiac physiology [198]. Current heart‑on‑chip platforms fall short in replicating the heart's highly intricate multicellular architecture and spatial organization [199]. In parallel, dissemination and implementation of HF management guidelines remain suboptimal, with marked disparities in diagnostic accuracy and therapeutic practices across countries, regions, and healthcare institutions [200].
Future Directions
5.2
Advances in emerging fields such as epigenetics, stem cell transplantation, and metabolomics may represent important regulatory factors influencing the onset of HF [201]. Systems biology approaches offer a holistic framework for elucidating how calcium‑regulating proteins shape the complex biological networks underlying HF and how these networks drive disease evolution. High‑precision 3D printing technologies, compared with traditional two‑dimensional cultures, demonstrate superior physiological relevance and enhanced biological functionality [202]. Pluripotent stem cell‑based biomimetic heart systems hold great potential for high‑throughput screening in drug development and toxicity assessment [203]. Clinically, large‑scale, multicenter trials are needed to generate high‑quality evidence. From a management perspective, it is not only essential to establish rigorous evidence‑based guidelines but also to strengthen oversight at the primary care level and to promote the dissemination of HF knowledge across schools, hospitals, and the general population [204].
Conclusion and Prospect
6
An expanding body of research underscores the pivotal role of calcium homeostasis in cardiovascular physiology and disease. Disruptions in calcium balance interact with multiple pathological processes, including mitochondrial dysfunction, dysregulated apoptosis and autophagy, and impaired energy metabolism [205, 206]. Notably, accumulating evidence indicates that such disturbances directly contribute to muscle dysfunction in cardiomyocytes [170]. Targeting specific calcium regulating proteins or calcium homeostasis‐related pathways may represent potential therapeutic strategies for HF.
Calcium antagonists that target LTCCs have been clinically validated as effective [207, 208]. Modulating SERCA2a expression at the genetic, protein, and posttranslational levels also represents a compelling approach to enhance SERCA2a‑based therapies for HF management [209]. Moreover, structural damage to T‑tubules further highlights calcium homeostasis as an independent pathogenic factor in both HFrEF and HFpEF [11]. Elucidating the mechanistic distinctions between these HF subtypes and developing therapeutic interventions aimed at correcting T‑tubule abnormalities may therefore offer new opportunities for subtype‑specific treatment.
Despite these advances, clinical studies directly targeting calcium homeostasis‐related signaling pathways for HF prevention and therapy remain lacking. Current research on calcium dysregulation in HFrEF versus HFpEF is fragmented, and methodological approaches are still evolving. Existing investigations have yet to bridge the gap between the “critical points” of individual calcium‑regulating proteins and the broader “functional lines” of calcium‑related signaling pathways. Furthermore, the inability to clearly define the initiation nodes of calcium homeostasis dysregulation in HF has hindered the transition from “functional lines” to “mechanistic networks” in deeper exploration.
These challenges underscore the need for interdisciplinary research strategies. Traditional ex vivo experiments and animal models alone are insufficient to fully capture the dynamic alterations in calcium‑regulating proteins across HF subtypes. Integrating multiomics technologies—including transcriptomics, proteomics, and metabolomics—with advanced imaging modalities may enable more direct visualization of the spatiotemporal features of calcium homeostasis dysregulation [210, 211]. In parallel, artificial intelligence and systems biology offer powerful tools for identifying key regulatory nodes and pathways within complex datasets, thereby accelerating target discovery and drug development [212]. Strengthening clinical translational research is equally essential, as the application of calcium‑related targets and therapeutics to patient stratification, prognostic assessment, and personalized treatment remains unresolved [213]. Future efforts should prioritize multicenter clinical trials to explore the predictive value of calcium homeostasis‐related biomarkers across different HF phenotypes and to assess the safety and efficacy of calcium‐targeted therapies in real‐world patient populations. Only through bidirectional integration of basic and clinical research can calcium homeostasis studies be effectively translated into HF prevention and treatment.
A comprehensive focus on the dynamic equilibrium of calcium homeostasis—together with its interplay with key pathological processes such as inflammation and metabolic dysfunction [214]—may facilitate the establishment of a more systematic pathological network model of HF. Clarifying the specific characteristics of different HF phenotypes, prioritizing calcium‐targeted drugs, and conducting classification‐based research with detailed therapeutic strategies hold great potential for supplementing the pathological mechanisms of HF, addressing the current clinical challenges of HFpEF, and accelerating novel drug development. Ultimately, these efforts will contribute to improved patient outcomes, enhanced cardiovascular risk prevention, and more effective management of heart disease.
Author Contributions
YF and ZY performed the literature search, selected relevant articles, interpreted data, and wrote the report. QL revised the manuscript. MS, YP, KZ, and YB performed the literature search. ZZ, XW, and JM designed and supervised this work. YF and ZY contributed equally to this work and shared the first authorship. All authors have read and approved the final submission.
Funding Information
This work was supported by Tianjin Municipal Health Commission's second batch of high‐level talent selection and training project in the health industry “Jinmen Medical Talents” (No: TJSJMYXYC‐D2‐052), the “Mechanism Research of Yangyin Shuxin Formula Inhibits Myocardial Calcium Overload via the PI3K/IP_3_R Pathway to Improve Diastolic Function in Heart Failure with Preserved Ejection Fraction” of Scientific research projects in critical field of traditional Chinese medicine in Tianjin (No: 2024003).
Ethics Statement
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1T. A. Mc Donagh , M. Metra , M. Adamo , et al., “2023 Focused Update of the 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure,” The European Heart Journal 44, no. 37 (2023): 3627–3639.37622666 10.1093/eurheartj/ehad 195 · doi ↗ · pubmed ↗
- 2J. Wen , L. Zhang , H. Liu , et al., “Salsolinol Attenuates Doxorubicin‐Induced Chronic Heart Failure in Rats and Improves Mitochondrial Function in H 9c 2 Cardiomyocytes,” Frontiers in pharmacology 10 (2019): 1135.31680945 10.3389/fphar.2019.01135 PMC 6797600 · doi ↗ · pubmed ↗
- 3Z. Liu , Y. Gan , Z. Shen , et al., “Role of Copper Homeostasis and Cuproptosis in Heart Failure Pathogenesis: Implications for Therapeutic Strategies,” Frontiers in pharmacology 15 (2024): 1527901.39850564 10.3389/fphar.2024.1527901 PMC 11754225 · doi ↗ · pubmed ↗
- 4Q. Guo , J. Wang , R. Sun , et al., “Comprehensive Construction of a Circular RNA‐Associated Competing Endogenous RNA Network Identified Novel Circular RN As in Hypertrophic Cardiomyopathy by Integrated Analysis,” Frontiers in Genetics 11 (2020): 764.32849787 10.3389/fgene.2020.00764 PMC 7399352 · doi ↗ · pubmed ↗
- 5D. P. Zipes , P. Libby , and R. O. Bonow , Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine (Elsevier/Saunders, 2019).
- 6D. Lei , Y. Liu , Y. Liu , et al., “The Gut Microbiota Metabolite Trimethylamine N‐oxide Promotes Cardiac Hypertrophy by Activating the Autophagic Degradation of SERCA 2a,” Communications Biology 8, no. 1 (2025): 596.40210720 10.1038/s 42003-025-08016-9PMC 11986001 · doi ↗ · pubmed ↗
- 7L. G. Dias , C. H. O. Reis , L. Dos Santos , et al., “Strength Training Improves Heart Function, Collagen and Strength in Rats With Heart Failure,” The Journal of Physiological Sciences 74, no. 1 (2024): 10.38365576 10.1186/s 12576-024-00899-3PMC 10873996 · doi ↗ · pubmed ↗
- 8C. Withaar , L. M. G. Meems , G. Markousis‐Mavrogenis , et al., “The Effects of Liraglutide and Dapagliflozin on Cardiac Function and Structure in a Multi‐hit Mouse Model of Heart Failure With Preserved Ejection Fraction,” Cardiovascular Research 117, no. 9 (2021): 2108–2124.32871009 10.1093/cvr/cvaa 256PMC 8318109 · doi ↗ · pubmed ↗