Chromatin Accessibility in Cancer: Biological Functions, Mechanisms, Therapeutic Potential, and Future Directions
Wentao Xia, Min Jiang, Yefei Huang, Kun Ding, Yansu Chen

TL;DR
This review explores how chromatin accessibility influences cancer development and treatment, highlighting new strategies and technologies for precision oncology.
Contribution
The paper systematically integrates chromatin accessibility with tumor biology and therapeutic strategies, which has been underexplored in prior reviews.
Findings
Chromatin accessibility is regulated by genetic, epigenetic, and environmental factors and plays a key role in cancer progression and therapy resistance.
Multiomics and AI integration offers novel perspectives for epigenetics-based precision tumor therapy.
Current limitations in translating chromatin accessibility research into clinical applications are identified, along with future directions.
Abstract
Cancer remains a major therapeutic challenge owing to its complex pathogenesis and the limitations of current treatments, such as poor specificity, toxicity, and multidrug resistance. Chromatin accessibility, which is dynamically regulated by genetic, epigenetic, and environmental factors, plays crucial roles in cancer initiation and progression. However, substantial obstacles persist in developing therapeutic strategies that target chromatin accessibility and translating them into clinical practice. This review comprehensively summarizes the biological functions and regulatory mechanisms of chromatin accessibility in tumors, encompassing tumorigenesis, progression, metabolic reprogramming, angiogenesis, stemness, tumor immune microenvironment, and therapy resistance. We integrate comparisons between human and murine models and detail key profiling technologies, including Assay for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
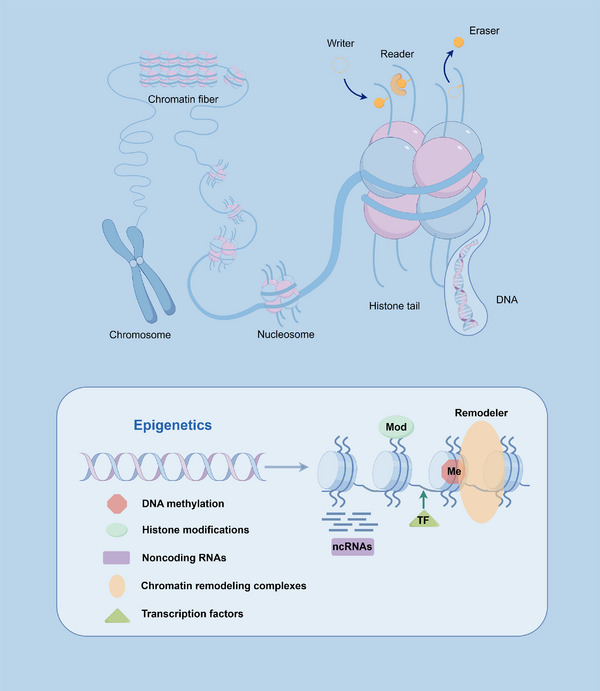 FIGURE 1
FIGURE 1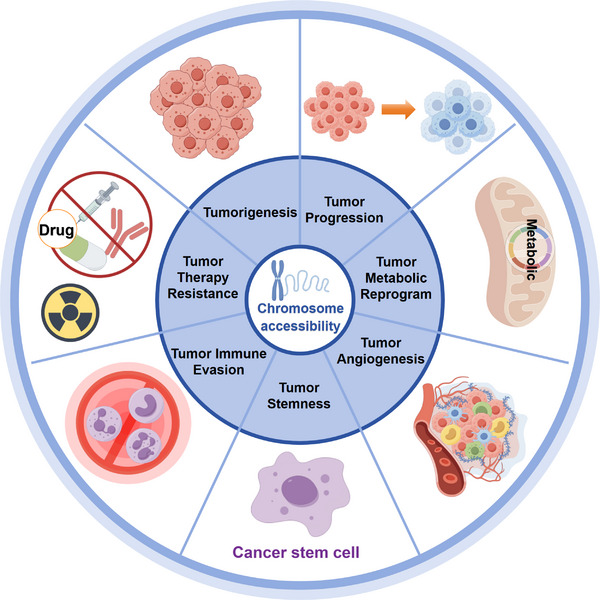 FIGURE 2
FIGURE 2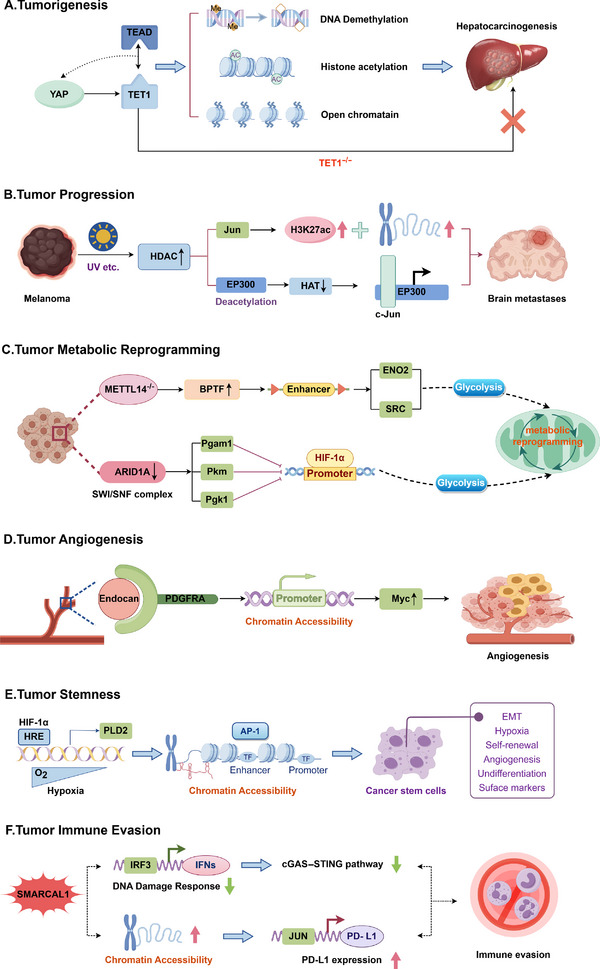 FIGURE 3
FIGURE 3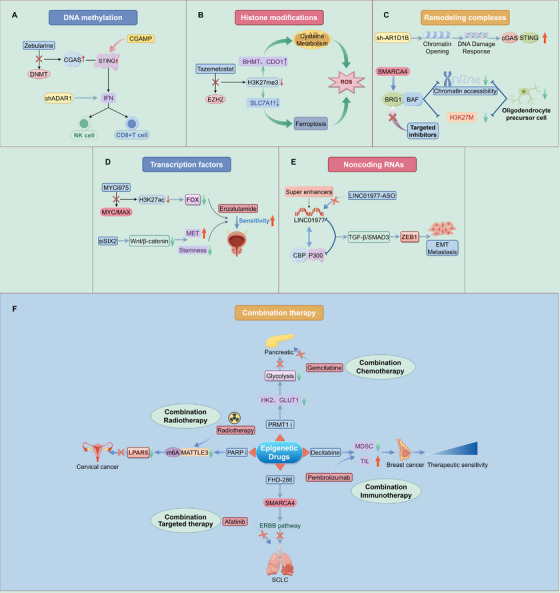 FIGURE 4
FIGURE 4 FIGURE 5
FIGURE 5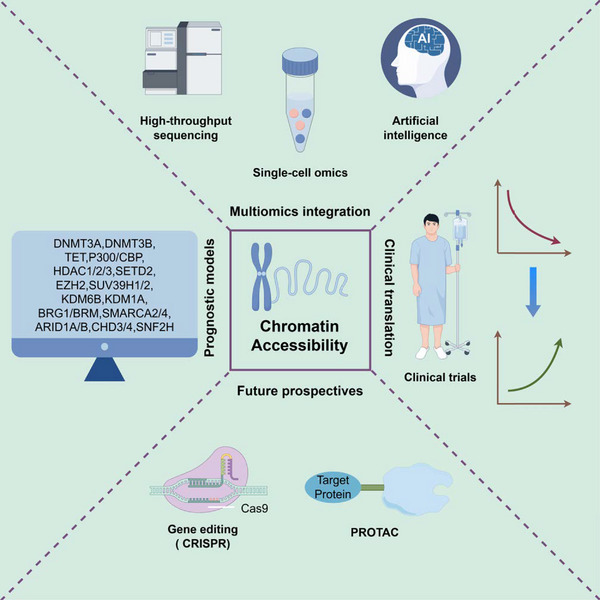 FIGURE 6
FIGURE 6| Target | Drugs | Mechanism | Function | References |
|---|---|---|---|---|
| DNMTs | Azacitidine | Nucleoside analogs that irreversibly bind to DNMT | Reversing DNA hypermethylation, inducing DNA damage, and inhibiting cancer progression | [ |
| DNMTs | Decitabine | Nucleoside analogs that bind irreversibly to DNMT | Reverses aberrant DNA hypermethylation to exert antitumor effects | [ |
| DNMTs | Zebularine | Nucleoside analogs, inhibits DNMT and cytidine deaminase | Sensitize cGAS–STING pathway to enhance antitumor immunity | [ |
| DNMTs | Guadecitabine | Nucleoside analog, inhibits DNMT | Increase HLA‐I accessibility and expression to inhibit prostate cancer | [ |
| DNMT3B | Nanaomycin A | Binds to the catalytic site of DNMT3B | Sensitize HCC cells to sorafenib | [ |
| DNMT1 | MG‐98 | Antisense oligodeoxyribonucleotide | Antitumor activity, well tolerated | [ |
| DNMT1 | GSK3685032 | Selective inhibitor of DNMT1, competes with the active site loop of DNMT1 | Induces DNA methylation deletion, transcriptional activation, and cancer cell growth inhibition | [ |
| HDACs | Vorinostat | Competitive binding to the catalytic site of HDAC | Induces apoptosis, impedes cell cycle progression, and inhibits cancer cell proliferation | [ |
| HDACs | Romidepsin | Interaction near the active site | Induces apoptosis and inhibits cancer progression | [ |
| HDACs | Belinostat | Prevents acetyl group removal | Induces cell cycle arrest and reduces tumor cell proliferation | [ |
| HDACs | Panobinostat | Inhibits aggregation of misfolded proteins and disrupts the cell cycle | Induces metabolic reprogramming and inhibits cancer progression | [ |
| HDACs | Trichostatin A | Competitively binds to the catalytic site of HDAC | Inhibit the growth of different cancer cells through cycle arrest and apoptosis | [ |
| HDAC8 | PCI‐34051 | Selective HDAC8 inhibitor | Enhances antitumor immunity and immune checkpoint blockade in hepatocellular carcinoma | [ |
| HDAC1/2 | MGCD0103 | Inhibits benzamide group‐dependent HDAC, induces hyperacetylation of histones | Induction of apoptosis with broad‐spectrum antitumor activity | [ |
| HDAC1 | CI‐994 | Class I‐specific HDACi, inhibits benzamide moiety‐dependent HDAC | Inhibits proliferation and induces apoptosis in vitro and in vivo | [ |
| HDAC1/2/3/10 | Tucidinostat | Novel benzamide‐based histone deacetylase inhibitor | Remodels tumor epigenome and activates antitumor immunity | [ |
| HDACs | LAQ824 | Novel pan‐histone deacetylase inhibitor | Inhibits tumor proliferation, inhibits epithelial–mesenchymal transition and induces apoptosis | [ |
| HMT | Tazemetostat | Specifically inhibits the methyltransferase activity of EZH2 | Reduces H3K27me3 levels and decreases HCC cell viability | [ |
| HMT | Valemetostat | EZH1/EZH2 dual‐target inhibitors | Reduce H3K27me3 level more thoroughly, overcome single target drug resistance | [ |
| HMT | GSK126 | Highly selective and potent EZH2 inhibitor, competes with S‐adenosyl‐methionine | Upregulates tumor suppressor genes to inhibit HCC | [ |
| HMT | Pinometostat | DOT1L inhibitor, competitively binds to S‐adenosyl‐methionine binding site | Blocked H3K79 methylation and inhibited MLL fusion protein‐driven leukemia cell proliferation | [ |
| HMT | UNC0642 | Inhibits G9a/GLP activity, reduces H3K9me2 labeling | Reduce the level of H3K9me2, restore oncogenic factors, and reduce the expression of oncogenic related proteins | [ |
| PRMT | Furamidine | Targets enzyme active domains | Inhibit pancreatic tumor growth and reverse chemotherapy resistance in pancreatic cancer | [ |
| PRMT | EPZ015666 | Highly selective PRMT5 inhibitor, competitive inhibitor of S‐adenosyl‐methionine | Antitumor activity through reprogramming of T‐cell‐mediated responses | [ |
| HDM | Phenelzine | Monoamine oxidase‐A inhibitor | Antidepressant, also found to reverse enzalutamide resistance in desmoplasia‐resistant prostate cancer | [ |
| HAT | C646 | Competitively binds p300/CBP catalytic domain and inhibits H3K27ac | Inhibits LINC00501 levels and inhibits gastric cancer growth in vitro and in vivo | [ |
| HAT | A‐485 | Highly selective p300/CBP catalytic domain inhibitor | Attenuates glycine/serine metabolism and inhibits proliferation of hepatocellular carcinoma cells | [ |
| BRD/BET | Pelabresib | Binds to the bromodomain of BRD4 and blocks its binding to acetylated histones | Potent cytotoxicity, limiting tumor cell proliferation and survival | [ |
| BRD/BET | Molibresib | Competitively binds to the bromo domain of BET proteins | Combination therapy with endocrine therapy may overcome endocrine resistance | [ |
| BRD/BET | JQ1 | Blocks the binding of BRD4 to acetylated histones by blocking its binding | Inhibits tumor growth by decreasing c‐Myc expression in endometrial cancer | [ |
| SMARCA2/4 | FHD‐286 | Binds to the ATPase domain of SMARCA2/4 and inhibits chromatin unwinding and gene transcription activation | Induces loss of neuroendocrine features in lung cancer and sensitizes SCLC tumors to afatinib | [ |
| SMARCA2/4 | AU‐15330 | PROTAC degrader of SMARCA2/4 that selectively degrades target proteins via the ubiquitin–proteasome system | Eliminates the structure of cis‐regulatory elements that bind to CHD6 and inhibits renal cancer growth | [ |
| SMARCA2/4 | PFI‐3 | Binds to the bromodomain of SMARCA2/4 and prevents its binding to acetylated histones | Targeting SWI/SNF sensitizes cancer cells to DNA damage | [ |
| SMARCA2 | A947 | Selective SMARCA2 protein hydrolysis targets chimeric molecules | Inhibited SMARCA4 mutant solid tumors | [ |
| SMARCA4 | JQ‐dS‐4 | Competitive binding to the bromo domain of BRD4 blocks its binding to acetylated histones | Inhibits progression of gliomas | [ |
| SMARCA2/4 | ADAADi | Inhibitor of the ATPase structural domain of ATP‐dependent chromatin remodeling proteins | Ability to block migration and invasiveness of cancer cells and promote apoptosis of cancer cells | [ |
| CHD4 | ED2–AD101 | Dual‐targeted inhibitor of CHD4/SMARCA5 | Sensitized ovarian cancer cells to cisplatin | [ |
| BPTF | C620‐0696 | Inhibitor of the BPTF bromodomain | Inhibits NSCLC progression by inhibiting c‐Myc transcription | [ |
| MYC | MYCi975 | Binds directly to MYC and disrupts MYC/MAX dimerization | Sensitize drug‐resistant prostate cancer cells to enzalutamide | [ |
| ONECUT2 | CSRM‐617 | Suppresses lineage plasticity reprogramming induced by enzalutamide | Blocking or delaying the emergence of desmoplasia‐resistant prostate cancer | [ |
| Epigenetic drug | Combination treatment | Clinical trial ID | Cancer type(s) | Status | Phase of clinical trial | |
|---|---|---|---|---|---|---|
| Combination chemotherapy | Azacitidine or decitabine | Cytarabine, doxorubicin, etoposide, etc. | AML | Active, not recruiting | II | |
| Decitabine | Cytarabine | AML | Unknown | II | ||
| Azacytidine | Fludarabine, cytarabine | ALL, AML | Completed | I | ||
| Decitabine | Tetrahydrouridine | Pancreatic cancer | Completed | I | ||
| Chidamide | Epirubicin, cyclophosphamide, docetaxel | Breast cancer | Unknown | II | ||
| Decitabine, vorinostat | Fludarabine, cytarabine | AML | Completed | I | ||
| Decitabine | Idarubicin, cytarabine | AML, MDS | Terminated | II | ||
|
Decitabine, panobinostat | Temozolomide | Metastatic melanoma | Terminated | I/II | ||
| Azacitidine | Homoharringtonie, cytarabine | AML | Unknown | II | ||
| CC‐486 | Abraxane | Metastatic melanoma | Withdrawn | II | ||
| 5‐Azacitidine | Carboplatin, paclitaxel | NSCLC | Completed | Not Applicable | ||
| RRx‐001 | Cisplatin, etoposide | NSCLC, ovarian cancer | Completed | II | ||
| Decitabine | Cytarabine, etoposide, mitoxantrone hydrochloride | AML, myelodysplastic syndromes | Completed | I/II | ||
| RRx‐001 | Gemcitabine, cisplatin | Advanced cholangiocarcinoma | Terminated | II | ||
| Decitabine | Daunorubicin, cytarabine | AML | Completed | III | ||
| Chidamide | CMOP regimen | Peripheral T‐cell lymphoma | Unknown | Not Applicable | ||
| Decitabine | Temozolomide | Metastatic melanoma | Completed | I/II | ||
| Combination immunotherapy |
Chidamide, azacitidine | Sintilimab | ENKTL | Unknown | II | |
| Decitabine | Nivolumab | NSCLC | Completed | II | ||
| Chidamide | Anti‐PD‐1 antibody | NK/T cell lymphoma | Recruiting | II | ||
| Guadecitabine | Pembrolizumab | Advanced lung cancer | Active, not recruiting | I | ||
| Guadecitabine | Atezolizumab | Urothelial carcinoma | Active, not recruiting | II | ||
| Tetrahydrouridine–decitabine | Nivolumab | NSCLC | Completed | II | ||
| EDO‐S101 | Nivolumab | Advanced melanoma | Unknown | I | ||
| SGI‐110 | Ipilimumab | Metastatic melanoma | Unknown | I | ||
| Azacitidine | Pembrolizumab | Colorectal cancer, NSCLC | Terminated | I/II | ||
| Guadecitabine | Atezolizumab | Ovarian, Fallopian tube, primary peritoneal cancer | Completed | I/II | ||
| CC‐486 | Pembrolizumab | NSCLC | Active, not recruiting | II | ||
| Decitabine | Pembrolizumab | NSCLC, esophageal carcinomas | Terminated | I/II | ||
| Decitabine | Nivolumab | Mucosal melanoma | Active, not recruiting | I/II | ||
| Combination targeted therapy | Decitabine | Eltrombopag | AML | Terminated | II | |
| Decitabine | Anlotinib | Digestive system tumors | Unknown | I/II | ||
| Combination radiotherapy | Decitabine | Radiotherapy | AML | Completed | II | |
| Vorinostat | Radiotherapy | NSCLC | Completed | I | ||
| Vorinostat | Radiotherapy | Brain metastases | Completed | I | ||
| Panobinostat | Radiotherapy | Prostate, esophageal, head and neck | Completed | I | ||
| Combination endocrine therapy |
Decitabine, LBH589 | Tamoxifen | Breast cancer | Terminated | I/II | |
| CC‐486 | Fulvestrant | Metastatic breast cancer | Terminated | II | ||
| Decitabine | Enzalutamide | Metastatic castration resistant prostate cancer | Withdrawn | I/II | ||
| Multiple combined therapy | Azacytidine | Carboplatin, paclitaxel, durvalumab, etc. | NSCLC | Not yet recruiting | I/II | |
| Azacitidine | Avelumab, utomilumab, gemcitabine, oxaliplatin | DLBCL | Terminated | III | ||
| Decitabine | Adebrelimab, paclitaxel, gemcitabine | Metastatic pancreatic cancer | Recruiting | I/II | ||
| Chidamide | Rituximab, gemcitabine plus oxaliplatin | DLBCL | Completed | II | ||
| Azacytidine | Carboplatin, paclitaxel, durvalumab | NSCLC | Not yet recruiting | I/II | ||
| Vorinostat |
Pembrolizumab, tamoxifen | Breast cancer | Terminated | II | ||
| CC‐486 | nab‐Paclitaxel IV, duravalumab | NSCLC | Completed | II | ||
| Decitabine | Radiotherapy, pembrolizumab | Solid tumors, lymphoma | Active, not recruiting | I | ||
| Vorinostat | Radiotherapy, temozolomide | Glioblastoma multiforme | Completed | I/II | ||
| Tazemetostat | Radiotherapy, docetaxel | Sinonasal carcinoma | Not yet recruiting | II | ||
| Vorinostat | Radiotherapy, temsirolimus | Diffuse intrinsic pontine glioma | Completed | I | ||
| Vorinostat | Radiotherapy, cisplatin | Head and neck squamous cell carcinoma | Withdrawn | II | ||
| Belinostat | Radiotherapy, temozolomide | GBM | Unknown | II | ||
| Tazemetostat | Radiotherapy, docetaxel, 5‐FU | Sinonasal carcinoma | Not yet recruiting | II | ||
|
Azacitidine, romidepsin |
nab‐Paclitaxel, gemcitabine, durvalumab | PDAC | Unknown | I/II | ||
| Chidamide | Regorafenib, iparomlimab, tuvonralimab | Advanced colorectal cancer | Recruiting | II |
| Therapeutic target/strategy | Cancer type/process | Key findings in different models | Consensus and discrepancies | Potential implications/translational insights |
|---|---|---|---|---|
| Targeting DNA methylation | ||||
| DNA methyltransferase inhibitors (DNMTis) | Myelodysplastic syndrome/acute myeloid leukemia | Human clinical trials: Azacitidine and decitabine have been approved for the treatment of MDS and AML, reactivating silenced tumor suppressor genes through hypomethylation [ | Consensus: DNMTis exert antitumor effects through demethylation in both human and mouse models. | Human‐derived models such as PDOs can better predict clinical responses and drug resistance, thereby guiding patient stratification. |
| Mouse model: In an AML mouse model, DNMT inhibitors were demonstrated to induce tumor differentiation and prolong survival [ | Differences: Mouse models often demonstrate more pronounced efficacy to DNMT inhibitors than human patients, suggesting that the tumor microenvironment and heterogeneity in human tumors are more complex. | |||
| Patient‐derived organoids (PDOs): Leukemia PDOs are employed to assess sensitivity to DNMT inhibitors and to identify mechanisms of resistance [ | ||||
| Targeting histone modifications | ||||
| BET bromodomain inhibitor | Lymphoma/multiple myeloma/colon cancer | Human clinical trials: Preliminary efficacy has been demonstrated in multiple myeloma and lymphoma, though single‐agent activity remains limited and resistance develops readily [ | Consensus: BET proteins are key transcriptional coactivators, and their inhibition suppresses tumor growth across multiple models. | The value of employing advanced human models, such as 3D models, for testing drug permeability and combination strategies prior to clinical trials has been emphasized. |
| Mouse model: In lymphoma patient‐derived xenograft (PDX) models, BET inhibitors effectively downregulate the expression of key oncogenes such as MYC [ | Discrepancies: The toxicity and drug resistance observed in human clinical trials prove difficult to fully replicate in mouse models. | |||
| 3D bioprinted models: For evaluating the permeability and efficacy of epigenetic drugs within simulated tumor spatial structures [ | ||||
| Targeting chromatin remodeling complexes | ||||
| SWI/SNF complex (e.g., ARID1A deficiency) | Breast cancer/ovarian cancer/colorectal cancer | Human studies: ARID1A mutations occur in certain breast cancers, leading to altered chromatin accessibility and potentially conferring therapeutic vulnerability [ | Consensus: Deletion of SWI/SNF complex subunits creates novel therapeutic dependencies in humans and mice. | Provides a theoretical foundation for precision therapies based on synthetic lethality. PDOs can be employed to validate the efficacy of treatments targeting mutations in specific chromatin remodeling complexes. |
| Mouse model: Synthetic lethality with EZH2 inhibitors was validated in an ARID1A‐deficient ovarian cancer mouse model [ | Differences: The intensity and specificity of synthetic lethal effects may vary depending on genetic background and cellular environment, necessitating validation in human‐derived models. | |||
| Colorectal cancer PDOs: Research has confirmed that chromatin remodeling complexes are key drivers of colorectal cancer progression and represent potential therapeutic targets [ | ||||
| Targeting transcription factors | ||||
| AP‐1 transcription factor family | Colorectal cancer | Human organoid research: Utilizing colorectal cancer CiPDOs models, AP‐1 has been identified as pivotal in maintaining tumor oncofetal state plasticity. Inhibiting AP‐1 diminishes this plasticity whilst enhancing chemotherapy sensitivity [ | Consensus: Transcription factors such as AP‐1 play a central role in driving tumor plasticity and malignant progression. | CiPDOs provide a unique platform for investigating human‐specific transcription factor targets associated with cellular state plasticity. |
| Mouse models: Frequently employed to validate the in vivo function of transcription factors in tumorigenesis and tumor progression. | Differences: The CiPDOs model has for the first time stably captured a human CRC‐specific “fetal‐like state” in vitro, revealing the unique and pivotal role of AP‐1 within it. This particular cellular state proves difficult to maintain stably over extended periods and study effectively in mouse models. | |||
| NF‐κB pathway | Triple‐negative breast cancer | Human cell lines/mouse models: Research indicates that the NF‐κB pathway mediates chemotherapy resistance in triple‐negative breast cancer (TNBC). Inhibiting this pathway downregulates cellular activity and enhances chemotherapy sensitivity [ | Consensus: NF‐κB is a key pathway in promoting survival and inflammatory responses, and its activation is associated with therapeutic resistance. | Combination therapies or local tumor delivery strategies may represent more viable approaches for targeting such key pathways. |
| Differences: The pathway's critical role in normal immunity limits the therapeutic window for systemic inhibition, suggesting the need for tumor‐specific targeting strategies. | ||||
| Targeting noncoding RNAs | ||||
| miRNA | Triple‐negative breast cancer | Human cell lines/mouse models: Radiation prevents tumor progression by inhibiting the miR‐93‐5p/EphA4/NF‐κB pathway in triple‐negative breast cancer [ | Consensus: Noncoding RNAs are key regulators of chromatin states and gene expression. | This reveals novel opportunities for overcoming resistance by combining targeted epigenetic modulators with standard therapies such as radiotherapy. |
| Differences: The expression and function of noncoding RNAs are typically highly cell‐type and species‐specific. | ||||
| Combination chemotherapy | ||||
| PRMT1 inhibitor + gemcitabine | Pancreatic cancer | Human cell lines/mouse models: Pharmacological inhibition of PRMT1 in combination with gemcitabine has a synergistic effect on pancreatic tumor growth in vitro and in vivo [ | Consensus: Drugs targeting chromatin accessibility can reshape tumor cell states, thereby sensitizing them to conventional chemotherapy. | Support the combination of epigenetic therapies with standard chemotherapy to overcome drug resistance and enhance therapeutic efficacy. |
| Discrepancy: The extent of synergistic effects may vary due to tumor heterogeneity and requires validation in models more closely resembling human tumors. | ||||
| Combination immunotherapy | ||||
| EZH2 inhibitor + immune checkpoint inhibitor (ICI) | Multiple solid tumors | Human clinical trials: EZH2 inhibition enhances therapeutic efficacy by directly acting on CAR‐T cells, thereby improving immunotherapy outcomes in patients with B‐cell lymphoma [ | Consensus: Altering chromatin states can modulate the tumor immune microenvironment, thereby enhancing the efficacy of immunotherapy. | This underscores the urgent need to develop complex 3D models incorporating immune components—such as organoid‐immune cell coculture systems—to more accurately predict the efficacy of combined immunotherapies. |
| Human cell lines/mouse models: EZH2 inhibitors have been demonstrated to enhance CD8+ T cell infiltration and function, producing synergistic antitumor effects when combined with anti‐PD‐1 antibodies [ | Discrepancies: The mouse immune system differs from that of humans, and current human in vitro models require refinement in immunological coculture approaches. | |||
| Limitations of 3D bioprinting/PDOs: Current PDOs and bioprinted models typically lack a complete immune microenvironment, restricting their use in simulating interactions with immunotherapy [ | ||||
| Combination targeted therapy | ||||
| Targeted drug delivery | Hepatic metastatic colorectal cancer | 3D bioprinted models: An advanced 3D bioprinted liver metastasis colorectal cancer (CRC) model demonstrates that oncolytic viruses carrying 5‐fluorouracil prodrugs can specifically target and penetrate CRC tumor regions, achieving targeted chemotherapy effects equivalent to higher doses of systemic administration [ | Consensus: Virus‐mediated targeted delivery enhances the tumor specificity of epigenetic drugs or chemotherapeutic agents. | Provides robust proof‐of‐concept for virus‐based targeted therapeutic strategies and demonstrates the unique advantages of 3D bioprinted models in evaluating complex therapeutic modalities such as viral delivery |
| Distinction: For the first time, 3D bioprinted models provide a visual demonstration of the spatial specificity of this targeted delivery and local activation within human‐derived tissue—an achievement difficult to replicate in conventional mouse models. | ||||
| Combination radiotherapy | ||||
| BET inhibitor/NF‐κB pathway inhibitor + radiotherapy | Breast cancer | Human cell lines/mouse models: Research indicates that targeting the NF‐κB–MIR155HG axis or BET proteins can reverse radiotherapy resistance in breast cancer stem cells, enhancing radiotherapy efficacy in both in vitro and in vivo models [ | Consensus: Epigenetic regulators serve as key mediators of radiotherapy resistance. | Demonstrated the clinical potential of drugs targeting chromatin accessibility as radiotherapy sensitizers |
| Discrepancy: In vivo studies of radiotherapy response remain highly dependent on models that fully replicate the tumor microenvironment and immune system. | ||||
- —Qinglan Project of Jiangsu Province of China10.13039/501100013088
- —Graduate Research and Innovation Projects of Jiangsu Province10.13039/501100012154
- —Natural Science Research of Jiangsu Higher Education Institutions of China10.13039/501100010023
- —Natural Science Foundation of Jiangsu Province10.13039/501100004608
- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChromatin Remodeling and Cancer · Protein Degradation and Inhibitors · Genomics and Chromatin Dynamics
Introduction
1
Cancer continues to pose a significant therapeutic challenge, primarily attributed to its intricate pathogenesis and the inherent limitations of current treatment modalities—including poor specificity, systemic toxicity, and the development of multidrug resistance [1, 2, 3]. Beyond genetic mutations, acquired epigenetic abnormalities have emerged as key drivers of oncogenic gene expression programs and the hallmarks of tumor biology [4, 5, 6, 7]. The term “epigenetics,” first coined by Conrad Waddington, broadly describes heritable regulatory mechanisms governing gene activity without altering the DNA sequence, encompassing DNA methylation, histone modifications, chromatin remodeling, and noncoding RNA (ncRNA) regulation [4, 5]. As a central pillar of epigenetic regulation, chromatin accessibility—defined as the degree of physical openness of chromatin, which determines the binding capacity of transcription factors, regulatory proteins, and other molecules—plays a pivotal role in defining cell identity and function [8]. Its dynamic regulation integrates genetic, epigenetic, and environmental cues, making it a pivotal interface in cancer development and progression.
The study of chromatin accessibility has a long‐standing history, tracing back to mid‐20th century observations of nuclease‐sensitive genomic regions, which first hinted at structurally open chromatin associated with active transcription. Milestone discoveries have progressively unveiled the multifaceted mechanisms governing this accessibility. These include the establishment of histone acetylation as a marker linked to increased chromatin openness and gene activation [9], the elucidation of DNA methylation in promoter regions as a mechanism for silencing gene expression by reducing accessibility (notably, intragenic DNA methylation is typically associated with gene activation) [10], and the characterization of how three‐dimensional (3D) chromatin architecture (e.g., loops and topological‐associated domains [TADs]) modulates the proximity of regulatory elements to influence accessibility and gene expression [11]. Chromatin accessibility is thus coordinately regulated by an integrated network involving DNA methylation, histone modifications, chromatin remodeling complexes (e.g., switch/sucrose nonfermentable [SWI/SNF]), transcription factors, ncRNAs, and higher‐order chromatin folding [9, 10, 11]. These findings underscore the crucial role of chromatin accessibility in epigenetic regulation, which in turn dictates gene expression patterns in cells. Advances in profiling technologies, particularly Assay for Transposase Accessible Chromatin with high‐throughput sequencing (ATAC‐seq) [12], now enable genome‐wide mapping of these dynamics, revealing widespread alterations in tumors that influence oncogenesis, metastasis, metabolic reprogramming, and therapy resistance [13, 14, 15, 16, 17, 18, 19].
Despite the growing recognition of its importance, previous reviews have often focused either on the general landscapes and regulatory mechanisms of chromatin accessibility [20] or its broad pathological roles across human diseases [21]. A systematic and comprehensive integration of chromatin accessibility with the hallmarks of cancer biology and, crucially, with translational therapeutic strategies (spanning from mechanistic insights to clinical trial evidence), remains underexplored. This critical knowledge gap provides the rationale for the present review.
This review aims to synthesize the latest research to provide a comprehensive overview of how chromatin accessibility shapes tumor biology and can be leveraged for cancer treatment. We will specifically explore: (1) the key epigenetic mechanisms governing chromatin accessibility during tumorigenesis and progression; (2) its functional impact across various tumor processes including metabolism, angiogenesis, stemness, immunity, and therapy resistance; and (3) current and emerging therapeutic strategies, compiling representative epigenetic drugs and their associated clinical trials when used in combination with other modalities. Furthermore, we will examine the utility of chromatin accessibility as a diagnostic and prognostic biomarker.
To address these objectives, the review is structured as follows. We first outline the biological basis of chromatin accessibility, covering its structural foundations, key regulatory mechanisms, and core profiling technologies. We then detail its multifaceted roles in tumors, discussing its contributions to tumorigenesis, progression, and the tumor microenvironment. Subsequently, we compile and analyze treatment strategies and clinical trials involving chromatin accessibility, reviewing its use as a biomarker and a therapeutic target in various combination regimens. Finally, from a multiomics and interdisciplinary perspective, we discuss current challenges in clinical translation and propose future directions for epigenetics‐driven precision oncology. This logical progression from fundamental concepts to biological functions, and ultimately to therapeutic applications, is designed to offer a cohesive and insightful resource for both researchers and clinicians in the field.
Biological Basis of Chromatin Accessibility
2
This section lays the foundational framework for understanding chromatin accessibility in the context of cancer. We begin by delineating the fundamental concepts of chromatin architecture and the principle of accessibility. We then dissect the sophisticated, multilayered regulatory network governing its dynamics. Finally, to bridge mechanistic understanding with empirical research, we detail the pivotal experimental and computational technologies that enable the mapping and interpretation of chromatin accessibility landscapes.
Chromatin Structure and Accessibility
2.1
The Basic Structure of Chromatin
2.1.1
Chromatin is a stable yet highly dynamic nucleoprotein complex composed of DNA, histones, nonhistone proteins, and small amounts of RNA, serving as the primary carrier of genetic material in eukaryotic cells [22]. The fundamental structural unit of chromatin is the nucleosome, formed by 147 base pairs of DNA wrapped around a histone octamer. This octamer consists of an H3–H4 histone tetramer and two H2A–H2B dimers. Chromatin plays a crucial role in genome compaction while dynamically regulating various nuclear processes, with nucleosomes constituting the core regulatory machinery [23, 24]. We will subsequently elaborate on the multifaceted regulatory functions of nucleosomes in these biological processes.
Characterization of Open and Closed Regions of Chromatin
2.1.2
Chromatin accessibility—a critical property defined by the permissibility of physical interactions between macromolecular complexes and chromatin DNA—fundamentally reflects the openness of chromatin architecture. Within the nucleus, chromatin exists in a spectrum of accessibility states: (1) highly accessible (“open chromatin”) characterized by nucleosome‐depleted regions, (2) moderately accessible (“permissive chromatin”) with dynamic nucleosome repositioning, and (3) low‐accessibility/repressive (“closed chromatin”) marked by stable nucleosome occupancy [8]. Open chromatin regions exhibit decompacted structures with exposed DNA, predominantly localizing to gene promoters, enhancers, and cis‐regulatory elements to facilitate transcriptional activation. In contrast, closed chromatin adopts condensed configurations with limited DNA exposure, typically occupying transcriptionally silent loci and heterochromatic regions. Under specific circumstances, open chromatin region can also promote gene repression (not only promote gene activation), for example, opening of insulator or repressor regions can promote nearby gene's silencing. The opposite scenario also holds true [25]. This structural dichotomy between accessible and inaccessible chromatin provides critical mechanistic insights into the relationship between chromatin topology and epigenetic regulation, which will be systematically explored in subsequent sections.
Relationship Between Chromatin Accessibility and Gene Expression
2.1.3
Chromatin accessibility is primarily influenced by the spatial distribution and occupancy patterns of nucleosomes and other DNA‐binding factors [26]. This accessibility determines the availability of DNA for interactions with transcription factors, regulatory proteins, and RNA polymerase, thus directly regulating transcriptional activity. Notably, accessible chromatin regions constitute merely 2–3% of the genome, with over 90% of these regions remaining unoccupied by transcription factors under basal conditions [27]. Transcription factors are a class of proteins that can bind to specific sequences of DNA. In regions of open chromatin, where DNA is more exposed, transcription factors can readily access and bind to key regulatory sequences, such as promoters and enhancers. For pioneer transcription factors, they can access the nucleosome occupied regions directly, thereby initiating or enhancing gene transcription [8]. Conversely, in closed chromatin regions, limited DNA exposure hinders transcription factor binding, effectively repressing gene transcription [28]. Thus, it is easy to see that the relationship between dynamic changes in chromatin accessibility and transcription factor binding and gene transcriptional activity as one of the central mechanisms of gene expression regulation.
Mechanisms Regulating Chromatin Accessibility
2.2
Chromatin accessibility is dynamically regulated through an integrated network of epigenetic mechanisms, including DNA methylation, histone modifications, ncRNAs, chromatin remodeling complexes, transcription factors, and chromatin 3D structure. In this section, we systematically delineate the principal mechanisms governing chromatin accessibility and their coordination in transcriptional control (Figure 1).
Basic chromatin structure and regulatory mechanisms. The basic structure of chromatin and the mechanisms by which it is subject to multiple epigenetic regulations, including DNA methylation, histone modifications, noncoding RNAs, chromatin remodeling complexes, and transcription factors.
DNA Methylation
2.2.1
DNA methylation is a crucial epigenetic modification mechanism that directly regulates gene expression by adding methyl groups to the DNA molecule. This process typically occurs initially on cytosine–phosphate–guanine (CpG) islands within the promoter regions of genes. This modification can silence genes or increase mutation probability by deaminating 5‐methylcytosine (5mC) to 5mU, which later will be repaired as T. DNA methylation is primarily catalyzed by three enzymes, DNMT1, DNMT3A, and DNMT3B. DNA methyltransferase (DNMT) mediates the addition of a methyl group to the fifth carbon of the cytosine base to form 5mC. DNMT3A and DNMT3B are the enzymes responsible for ab initio DNA methylation, while DNMT1 is the enzyme necessary to maintain DNA methylation during DNA replication [29, 30]. Methylation at the 5‐carbon (5mC) of cytosine ring in CpG dinucleotides was the first identified form of epigenetic modification and remains the most extensively studied chromatin modification [31, 32, 33]. DNA methylation plays a crucial role in epigenetic regulation and is involved in a variety of nuclear processes such as gene expression, DNA repair, and recombination [34, 35].
DNA methylation affects chromatin accessibility through multiple mechanisms. First, DNA methylation alters the structural properties of DNA, including geometric conformation, mechanical stability, and physicochemical properties [36, 37, 38], and it should be noted that the magnitude of the effects induced by CpG methylation is highly dependent on the local DNA sequence environment around the methylation site [39]. Second, numerous studies have demonstrated that DNA methylation‐induced changes in DNA geometry and mechanical properties offer the prospect of gaining insight into how DNA methylation regulates nucleosome structure and dynamics [37, 40, 41].
Emerging evidence highlights the context‐dependent role of DNA methylation in regulating nucleosome positioning and transcription. However, findings remain inconsistent across different investigations [42]. A subset of investigations demonstrates that unmethylated CpG‐rich promoter regions exhibit nucleosome depletion, whereas DNA hypermethylation correlates with increased nucleosome occupancy [43, 44, 45]. Conversely, alternative studies report an inverse association, where CpG methylation destabilizes nucleosome–DNA interactions and promotes nucleosome eviction [38, 46, 47]. A possible explanation for these contradictory results may lie in the existence of nucleosomes located in different genomic regions, which may respond differently to DNA methylation. DNA methylation of promoter regions and CpG islands usually leads to closing of open chromatin regions and gene silencing. But DNA methylation of gene body regions usually connects to gene activation [43, 48]. Finally, DNA methylation is also involved in the regulation of the 3D structure of chromatin, and most studies have shown that DNA methylation can be recognized by reader proteins, leading to gene silencing and chromatin compression [10, 38]. In conclusion, DNA methylation can significantly affect the physicochemical properties of DNA in a sequence‐dependent manner, inducing changes in nucleosome stability and DNA packaging that ultimately modulate DNA accessibility.
Extensive research has unequivocally established DNA methylation as a pivotal epigenetic hallmark of carcinogenesis, characterized by two complementary aberrations: locus‐specific hypermethylation at CpG islands within tumor suppressor gene (TSG) promoters and genome‐wide hypomethylation promoting chromosomal instability (CIN) [1, 49]. In cancer cells, the predominant alteration is the hypermethylation of TSGs, leading to their inactivation and promoting tumor growth. In contrast, activation of prometastatic genes induced by DNA hypomethylation promotes tumor invasion and metastasis [50]. Hypermethylation‐mediated silencing of TSGs is a dominant mechanism in cancer. For instance, Sun et al. found that hypermethylation disrupts TP53 binding to the ZNF334 promoter, suppressing its transcription and accelerating hepatocellular carcinoma (HCC) progression [51]. Similarly, Yellow et al. identified hypermethylation by DNMT1 as a driver of triple‐negative breast cancer (TNBC). This process suppresses estrogen receptor (ER) expression, induces epithelial–mesenchymal transition to enable metastasis, and enhances autophagy and cancer stem cell (CSC) proliferation in TNBC [52].
Global DNA hypomethylation is a hallmark of CIN in aggressive cancers [53]. Endo et al. linked genome‐wide hypomethylation to poor prognosis and occult metastasis in pancreatic cancer, suggesting its utility as a predictive biomarker [54]. In small cell lung cancer (SCLC), Na et al. found that DNMT3A downregulation—mediated by KMT2C loss—induces hypomethylation of prometastatic genes, facilitating tumor dissemination [55]. Guo et al. further showed that DNA hypomethylation silences antitumor immune genes in early prostate cancer (PC) while retaining proproliferative drivers, enabling immune evasion during metastasis [56].
DNA methylation intersects with key cancer pathways. Han et al. found that PHF14‐mediated hypermethylation of SMAD7 activated TGF‐β signaling to drive lung adenocarcinoma (LUAD) metastasis [57]. In colorectal cancer (CRC) with KRAS mutations, Huang et al. found that SLC25A22‐mediated glutamine catabolism reduced DNA demethylation, which enhanced Wnt/β‐catenin signaling and promoted tumorigenesis and cancer stemness [58]. Collectively, these findings underscore DNA methylation's indispensable role in shaping tumor biology through chromatin remodeling and pathway dysregulation.
Histone Modifications
2.2.2
Histones are core proteins essential for regulating processes such as DNA packaging, chromatin acquisition, gene expression, and DNA repair [59]. Posttranslational modifications (PTMs) of histones—including acetylation, methylation, phosphorylation, SUMOylation, and ubiquitination—dynamically modulate chromatin structure and function. These modifications occur predominantly on the N‐terminal tails of histones and influence nucleosome stability, chromatin accessibility, and recruitment of effector proteins [60]. Histone modifications are key epigenetic mechanisms that regulate chromatin accessibility and gene expression, dynamically regulating chromatin accessibility by altering chromatin structure and recruiting effector proteins. Transcriptional activation or repression of genes is affected by aberrant histone modifications, which also affect many processes, including DNA replication and recombination, thereby impairing cellular homeostasis and controlling tumor formation [61, 62]. Histone tails and cores undergo a variety of PTMs, including acetylation, phosphorylation, methylation, SUMO acetylation, and ubiquitination. These PTMs can establish different chromatin environments that regulate a variety of nuclear processes such as gene expression, replication, repair, and regulation of genome structure [63]. In this section, we will briefly describe how several important and well‐studied PTMs (acetylation, methylation, and phosphorylation) affect chromatin accessibility in open and closed chromatin conformations [59, 64].
Histone Acetylation
2.2.2.1
Histone acetylation, a hallmark epigenetic modification, is catalyzed by histone acetyltransferases (HATs), which transfer acetyl groups to lysine residues on histone N‐terminal tails [65]. Chromatin dynamics can be determined by nucleosome stability, which can be directly affected by histone core structural domain acetylation [66]. Histone acetylation significantly improves chromatin accessibility by neutralizing histone positive charge and attenuating its interaction with negatively charged DNA, leading to loosening of chromatin structure [67]. For example, Kouzarides et al. found that elevated acetylation enhances DNA accessibility, enabling transcriptional machinery to engage with target genes [62]. Similar claims were validated in a later study by Mathias Wenes, who found that mitochondrial pyruvate carrier (MPC) inhibition‐induced metabolic flexibility promotes acetyl coenzyme‐A production from glutamine and fatty acid oxidation, thereby enhancing histone acetylation and chromatin accessibility on promemory genes [68]. Scholars such as Sheu et al. found that cilium 5‐HTR6 stimulation activates the nonclassical Gαq/11–RhoA pathway, which regulates nuclear actin and increases histone acetylation, thereby increasing chromatin accessibility [69]. Histone acetylation dysregulation is increasingly implicated in tumorigenesis. He et al. highlighted acetyl‐CoA's role as a metabolic bridge, connecting lipid metabolism to histone acetylation to fuel cancer growth, proliferation, and metastasis [70]. Miziak et al. emphasized that aberrant acetylation patterns alter chromatin architecture and gene expression, serving as potential biomarkers for cancer progression and prognosis [71]. Thus, these studies underscore histone acetylation's dual role as a regulator of chromatin structure and a driver of oncogenic processes.
Histone Methylation
2.2.2.2
Histone methylation represents a fundamental epigenetic modification primarily occurring on lysine (K) and arginine (R) residues of histones H3 and H4. This dynamic process is mediated by histone methyltransferases (HMTs) and reversed by demethylases (KDMs). Unlike acetylation, methylation does not alter histone charge but regulates chromatin state by recruiting specific effector proteins [72]. Histone methylation of lysine 4 (H3K4me3), the most extensively studied modification, marks transcriptionally active chromatin regions [73]. Methylation of cytosine in CpG dinucleotides, histone lysine and arginine residues has been suggested by Li et al. to be a chromatin modification that plays a key role in regulating genome integrity, replication, and accessibility [74]. Posttranslational methylation of histone lysine or arginine residues plays an important role in gene regulation and other physiological processes. Aberrant histone methylation patterns resulting from genetic alterations (mutations, translocations, or gene overexpression) are strongly implicated in disease pathogenesis, particularly cancer [75]. Enhancer of Zeste Homolog 2 (EZH2) overexpression correlates with poor overall survival (OS) in lung cancer patients [76]. Other recent studies have found that abnormal histone methylation levels contribute to skin tumorigenesis and summarized the efficacy of several epigenetic inhibitors targeting histone methylation‐modifying enzymes in skin cancer, suggesting that histone methylation‐modifying enzymes could serve as a new class of targets for skin cancer therapy [77].
Histone Phosphorylation
2.2.2.3
Histone phosphorylation is a dynamic PTM regulated by the coordinated activity of protein kinases (e.g., Aurora B, MSK1/2) and phosphatases (e.g., PP1/PP2A). Predominantly occurring on serine/threonine residues of histones H3 and H2A, this modification modulates chromatin structure and function during critical cellular processes such as transcription and mitosis [78]. It has been shown that phosphorylation of histone H3 is unique because it binds to open chromatin during gene activation on the one hand and marks highly condensed chromatin during mitosis on the other [79]. In tumorigenesis, phosphorylation of serine 10 on histone H3 (H3S10ph) is emerging as an important player in cancer development and dissemination as it promotes malignant transformation of cells and is involved in essential cellular functions [80]. Large‐scale proteogenomic analyses have further identified pan‐cancer patterns linking phosphorylation to dysregulated DNA repair and acetylation to altered metabolic–immune crosstalk, revealing distinct tumor subpopulations with shared epigenetic vulnerabilities [81].
Histone modifications are critical regulators of chromatin structure, gene expression, and tumorigenesis. In addition to the above three histone modifications, there are a variety of less prevalent and atypical PTMs, such as ubiquitination, lactylation, succinylation, citrullination, ADP‐ribosylation, 5‐hydroxytryptophanization, and serotoninization. For example, posttranslational histone modifications are one of the mechanisms used by cellular processes to remodel chromatin and gain access to potential DNA templates [82], and deletion of the deubiquitinating enzyme BAP1 promotes H2AK119ub modification, remodeling the accessibility of chromatin and thus disrupting transcriptome patterns in human liver‐like organs [83]. Merkuri et al. demonstrated that glycolysis induced histone lactylation of neural crest‐associated genes, thereby increasing their chromatin accessibility, combining metabolic state of embryonic cells with chromatin organization and gene regulatory network activation [84]. Xu et al. found that histone lactylation plays a crucial role in cancer progression and that anaerobic metabolism promotes breast cancer survival through histone‐3 lysine‐18 lactylation mediating the PPARD axis [85]. Upon study, Wang et al. found that Zeb1 in epithelial‐like cells transcriptionally regulate the expression of several key glycolytic enzymes, thereby predisposing tumor cells to utilize glycolysis for energy metabolism. In the process, lactate accumulation‐mediated histone lactylation enhances chromatin accessibility and cellular plasticity, including induction of neurogenic gene expression, thereby promoting neuroendocrine PC (NEPC) development [86]. Jing et al. found that lysine succinylation (Ksucc) is a newly identified histone PTM and that this succinylation affects nucleosome dynamics and is important in regulating DNA accessibility and chromatin dynamics [87]. Kamo et al. found that the citrullination at R53 in H1.2 resulted in the reduced electrostatic interaction with DNA and the reduced binding affinity to nucleosomes [88]. Smith et al. demonstrated that histone poly(ADP‐ribosylation) factor 1‐dependent histone ADP‐ribosylation triggers chromatin relaxation to facilitate the recruitment of repair factors at DNA damage sites [89]. Similarly, Martinez‐Zamudio et al. proposed that PARP‐1 enzyme activity promotes gene transcription by increasing promoter accessibility through histone ADP‐ribosylation [90]. Recently, it has also been found that histone 5‐hydroxytryptophanylation, serotoninylation may also be a potential marker of chromatin activity [91, 92]. In conclusion, PTM is an integral part of tumor cell adaptation and response to intracellular and environmental changes, and more in‐depth studies are needed on the PTM control processes that lead to cancer development and progression.
Regulation of histone proteins affects gene expression through multiple mechanisms including exchange with histone variants. Beyond PTMs, the selective incorporation of histone variants represents another critical mechanism governing chromatin structure and accessibility. Histone variants (e.g., H3.3, H2A2, H2BE) differ in amino acid sequence from their canonical counterparts and are often incorporated into chromatin in a DNA replication‐independent manner, conferring unique biophysical properties to the nucleosome. These variants directly remodel the chromatin accessibility landscape by altering nucleosome stability and histone–DNA interactions [93, 94]. In cancer, the dysregulated expression of histone variants is a frequent event. They drive gene expression programs linked to malignant phenotypes by promoting or restricting specific chromatin states. For instance, in glioblastoma (GBM), histone variant macroH2A2 shapes chromatin accessibility at enhancer elements to antagonize transcriptional programs of self‐renewal [95]. In addition, Filipescu et al. report that macroH2A deficiency in cancer‐associated fibroblasts leads to altered chromatin looping and elevated inflammatory gene expression, thereby affecting immune cell function and limiting the antitumor response in melanoma [96]. Consequently, acting as “oncohistones,” histone variants play a deterministic role in tumorigenesis and progression by establishing unique patterns of chromatin openness.
Noncoding RNAs
2.2.3
ncRNAs, broadly defined as RNA transcripts not translated into functional proteins, have emerged as pivotal regulators of chromatin dynamics and gene expression. Advances in transcriptome‐wide analyses have expanded the catalog of ncRNAs, revealing their diverse roles in epigenetic modulation [97]. ncRNAs can be broadly categorized into small (<200 nucleotides) and long (>200 nt) ncRNAs. Small ncRNAs include endo‐siRNAs, microRNAs, piwi‐interacting RNAs, small nuclear RNAs, small nucleolar RNAs, and tRNA‐derived fragments [98, 99]. Among these, siRNAs and miRNAs are known to have regulatory roles in epigenetics [100]. SiRNAs are derived from double‐stranded RNA precursors that are cleaved by DICER to form 19 to 24 base RNAs, and studies have shown that siRNAs act through the RNA interference pathway to silence gene expression through DNA methylation and histone modification [101]. MiRNAs are short ncRNAs (∼21 nt) that act as posttranscriptional regulators with a large number of targets [102]. Shui et al. used ATAC‐seq to analyze the chromatin accessibility landscape of colon tissues expressing K‐RasWT and K‐RasG12D, and the data showed that overactivation of K‐Ras induced a significant increase in K‐Ras expression. Ras overactivation induces full de‐repression of miRNA targets that is dependent on miRNA expression levels [103].
It is well known that many lncRNAs play regulatory roles in cell growth, development, and disease processes. Lu et al. identified a long‐chain ncRNA called lncRNA Muscle Regeneration Enhancer Factor, which interacts with Smarca5 to promote chromatin accessibility when muscular satellite cells are activated and begin to differentiate, thus facilitating the p300/CBP/H3K27ac genomic binding [104]. Terroba et al. found an increase in overall chromatin accessibility upon overexpression of lncRNA metastasis‐associated LUAD transcript 1 (MALAT1), suggesting that individual lncRNAs can drive LUAD metastasis through reprogramming of the tumor microenvironment [105]. Deng et al. identified a mechanism of chromatin accessibility and gene transcriptional regulation jointly mediated by RNA m6A formation and DNA demethylation, highlighting the importance of the interplay between RNA m6A and DNA modifications in physiological and pathogenic processes [106]. Similarly, Li et al. revealed through their study that the CFL1–METTL3–seRNA m6A–YTHDC2/MLL1 axis plays a role in the epigenetic regulation of local chromatin status and gene expression [107]. Confirmed by a growing body of evidence, we can assume that regulatory ncRNAs play an important role in epigenetic control.
Beyond the sequences of ncRNAs themselves, their posttranscriptional chemical modifications—core components of epitranscriptomics—represent an additional critical layer of gene expression regulation. Key RNA modifications (e.g., m6A, m5C, m3C) dynamically and reversibly regulate RNA fate, including stability, splicing, nuclear export, and translation efficiency, through dedicated “writer,” “eraser,” and “reader” proteins [108]. Notably, profound crosstalk exists between this RNA‐level regulation and chromatin structure. For instance, Li et al. demonstrate the cofilin family protein CFL1 as a METTL3 cofactor that helps super‐enhancer (SE) RNA m6A methylation formation. Then SE RNA m6A promotes local chromatin accessibility and oncogene transcription in pancreatic ductal adenocarcinoma (PDAC) [109]. Furthermore, Lee et al. report that METTL8 links mt‐tRNA m3C modification to the HIF1α/RTK/Akt axis to sustain GBM stemness and tumorigenicity [110]. Thus, RNA modifications, along with their potential interactions with chromatin regulators, fine‐tune chromatin accessibility and gene expression programs.
Chromatin Remodeling Complexes
2.2.4
Chromatin remodeling complexes are a class of ATP‐hydrolysis‐dependent molecular machines that dynamically regulate chromatin accessibility by altering nucleosome position and composition [111, 112]. Chromatin remodelers were originally discovered and validated in yeast, and eukaryotic cells contain four chromatin remodeling complexes that are classified based on similarities and differences in ATPase subunits, including SWI/SNF, Imitation Switch (ISWI), Chromodomain Helicase DNA binding domain (CHD), and inositol requiring 80 (INO80) [113].
SWI/SNF Complexes
2.2.4.1
The SWI/SNF complex is the most studied chromatin remodeling complex in mammals and has been found to be involved in a wide variety of life activities [114]. The SWI/SNF complex can be divided into three major modules—ATPase, actin‐related protein (ARP), and somatic module [115]. The SWI/SNF chromatin remodeling complex (SCRC) is a subfamily of ATP‐dependent chromatin remodeling proteins that play a wide range of roles in the regulation of gene expression by modifying chromatin structure [116]. The SCRC acts by displacing nucleosomes near important regulatory sites, which promotes the binding of transcription factors, thereby facilitating the expression of genes and gene activation in unicellular and multicellular eukaryotes [117, 118]. Genomic abnormalities of the SCRC subunit occur in approximately 20% of cancers [119, 120]. The first clue linking the SWI/SNF complex to cancer appeared in the late 1990s, when mutations in the gene encoding the SMARCB1 subunit were identified in rhabdomyosarcoma [121]. Additional studies have supported this idea, and inactivating mutations in AT‐rich interaction domain 1A (ARID1A) are prevalent in a wide range of cancers, including up to 62% of clear cell carcinomas of the ovary [122], endometrioid carcinomas [123], and gastrointestinal tumors such as gastric, colorectal, and pancreatic cancers [124]. The mechanisms by which mutations in each individual subunit promote tumorigenesis and the function of mutant SWI/SNF complexes in cancer is currently an active area of research, and in addition to this, a number of studies have elucidated the pathways regulated by the SWI/SNF complexes, and how subunit mutations that disrupt the expression programs of these genes can promote cancer [125]. Concepcion et al. found that SMARCA4/BRG1 encodes one of the two mutually exclusive ATPases present in the mammalian SCRC, which is frequently mutated in lung cancer and drives cancer progression, leading to an increased incidence of development and metastasis of highly complex undifferentiated malignancies [126]. Therefore, we can assume that aberrant expression and mutation of the SWI/SNF complex is prevalent in cancer and that it plays a broad role in regulating gene expression by modifying chromatin accessibility and structure.
ISWI Complexes
2.2.4.2
It has been shown that Smarca5 (also known as Snf2h), an important enzyme in the SWI/SNF family with remodeling activity, alters gene expression by promoting chromatin accessibility, and a transposase‐accessible chromatin analysis of zebrafish and newborn fetuses has shown that Smarca5 is responsible for maintaining chromatin accessibility to promoters of hematopoiesis‐related genes in fetal HSPCs. Smarca5 interacts with nucleolin to promote chromatin remodeling, which in turn promotes genomic binding of transcription factors to regulate expression of hematopoietic regulators such as bcl11ab [127]. The ISWI family is an important component of ATP‐dependent chromatin remodeling complexes and consists of two ATPases, SNF2L (SMARCA1) or SNF2H (SMARCA5), which alternatively bind to complex‐specific auxiliary subunits [128]. Deletion of SNF2H in mammalian cells results in genome‐wide changes in nucleosome organization, accompanied by an increase in nucleosome repeat length and a decrease in the binding of specific transcription factors (e.g., CCCTC‐binding factor [CTCF]), which coincides with reduced chromatin accessibility [129, 130]. Unlike the related SCRC, the ISWI complex fails to evict nucleosomes, but instead regulates nucleosome sliding to maintain appropriately spaced nucleosome arrays, which dynamically affects chromatin accessibility [131, 132]. Iurlaro et al. investigated this and found that the core subunit of the nucleosome remodeling factor nucleosome remodeling factor (NURF), bromodomain PHD finger transcription factor (BPTF) leads to a strong reduction in chromatin accessibility and SNF2H ATPase localization around CTCF sites. The study further revealed a mechanistic link between NURF‐mediated chromatin remodeling and the structural function of CTCF. [133]. High‐throughput sequencing and a growing number of basic and clinical studies have identified altered function or composition of ISWI‐containing complexes as critical for tumorigenesis and progression. Genetic abnormalities are major determinants of the levels of certain ISWI subunits in specific types of cancer and contribute to the tumor phenotype [134]. For example, Buganim et al. identified BPTF as a gene involved in transcriptional regulation and chromatin remodeling and showed that BPTF may play a procarcinogenic role in tumors carrying chromosomal aberrations in 17q [135]. Itamochi et al. obtained tumor from 55 Japanese women diagnosed with ovarian clear cell carcinoma (OCCC). Tissue samples and matched blood samples were obtained from 55 Japanese women diagnosed with OCCC, and whole‐genome sequencing using the Illumina HiSeq platform revealed that mutations in the ISWI ATPase SNF2L (SMARCA1) were associated with OCCC [136]. In addition, some ISWI subunits were strongly associated with patient prognosis. In HER2+ breast tumors, high levels of BAZ1A are associated with deleterious recurrence‐free survival (RFS) and very poor OS [137]. Pietrzak et al. found that TIP5 expression and high PTEN‐del expression in prostate tumors were strongly associated with reduced prostate‐specific antigen RFS [138]. Dai et al. found that BPTF was highly expressed in non‐small cell lung cancer (NSCLC) tumor tissues, and BPTF cooperated with p50 NF‐κB to promote COX‐2 expression and tumor cell growth in lung cancer and was positively associated with advanced clinical stage, more lymph nodes, and distant metastasis [139]. This series of studies on ISWI has also demonstrated that it influences tumorigenesis and development through its involvement in transcriptional regulation and chromatin remodeling.
CHD Complexes
2.2.4.3
CHD proteins are ATP‐dependent chromatin modifiers involved in the structural organization of chromatin and act as gatekeepers for genome access [140]. The hallmark feature of CHD remodelers is the presence of a bichromatin structural domain in its N‐terminal region and SNF2‐like ATPase/helicase core, which mediates binding to chromatin by directly binding to the histone H3 tails to mediate binding to chromatin through direct binding to methylated lysines [141]. The CHD family is a family of chromatin regulators that are frequently lost or inactivated in a variety of human cancers, and CHD proteins affect chromatin compression and thus access to DNA by cellular mechanisms. The CHD family consists of nine members, CHD1–9, with CHD1 serving as the founding member of the CHD family, was originally discovered to be a DNA‐binding protein. It has been shown that yeast Chd1 is primarily responsible for chromatin assembly, and that the nucleosome remodeling deacetylase (NuRD) complex remodeling agent helps to repress gene binding to chromatin, regulating gene transcription, genome stability, and developmental signaling [142]. A study found that exposure to cigarette smoke was associated with hypermethylation of the CHD1 promoter [143]. Factors that interact with components of the transcriptional machinery and histone modifiers converge upstream of CHD1 to regulate its expression. For example, the Pol II‐associated factor hPAF2/PD2 mediates MLL‐mediated deposition of H3K4me2/3 covalent modifications characteristic of transcriptionally active genes and promotes CHD1 expression in pancreatic cancer cells [144]. In addition, CHD enzymes act downstream of key signaling pathways that are disrupted during tumorigenesis, and the chromatin remodeling activity of CHD enzymes appears to be critical for translating information from ligand‐mediated signaling pathways into the transcriptional machinery [145, 146, 147]. It is easy to see that CHD controls fundamental processes, including transcription, proliferation, and DNA damage repair, by controlling access to DNA by the cellular machinery.
INO80 Complexes
2.2.4.4
The INO80 complex is an evolutionarily conserved ATP‐dependent chromatin remodeling complex, and like SWI/SNF, INO80 can be divided into ATPase, ARP, and body modules. INO80 is the major ATPase subunit of the INO80 complex and has a wide range of effects on a variety of cellular processes, including transcriptional regulation, DNA replication and repair, telomere maintenance, and chromosome segregation [148, 149]. In yeast, INO80 has also been implicated in the removal and degradation of ubiquitinated RNAPII from chromatin [150]. Gowans demonstrated that the INO80 complex mediates metabolic signaling in chromatin, remodeling coordinated metabolic homeostasis and cell division [151]. Chakraborty and Magnuson found that INO80 promotes repression of sex‐linked gene expression during spermatogenesis in mice by regulating chromatin accessibility [152]. In cancer, the INO80 chromatin remodeling complex plays an important role in many tumors. INO80 is important for the maintenance of genomic stability, and inactivation or depletion of INO80 results in aneuploidy and chromosomal structural abnormalities [153, 154]. Since there is a causal relationship between genomic instability and tumorigenesis [155], these findings suggest that INO80 may act as a tumor suppressor [156]. Less consistent with this conventional account, however, Lee et al. found that INO80 haploinsufficiency inhibits colon cancer tumorigenesis by increasing apoptosis through activation of replication stress‐induced ATR–Chk1 signaling [156]. Belk et al. demonstrated, by in vivo clustered regularly interspaced short palindromic repeats (CRISPR) screenings in mouse and human tumor models, that the relationship between INO80 and the perturbation of the BAF chromatin remodeling complex improved T cell persistence in tumors [157]. In addition, Prendergast et al. showed that the ATP‐dependent chromatin remodeling INO80 complex promotes R‐loop resolution, and counteracting the R‐loop promotes cancer cell proliferation and avoids DNA damage‐induced death [158]. These results suggest that the INO80 complex plays a critical role in transcriptional regulation, DNA damage repair, and stem cell maintenance by affecting chromatin accessibility.
Transcription Factors
2.2.5
Transcription factors are widely recognized for their ability to bind to conserved motifs (unmethylated DNA sequences) within promoters to modulate transcription [159]. There is a bidirectional regulatory relationship between transcription factors and chromatin accessibility, and this interaction constitutes a central aspect of gene expression regulation.
This is first reflected in the active regulation of chromatin accessibility by transcription factors. Transcription factors are key regulators of cellular processes, and the activity of transcription factors can be modulated either by regulating the abundance of their active forms (including transcriptional, translational, and posttranslational regulation) or by regulating the accessibility of their binding sites (including epigenetic processes and cell‐type‐specific chromatin states). Once bound, transcription factors can open chromatin for or prevent binding of other factors and activate or repress transcription of genes [160]. Pioneer transcription factors play an important role in regulating chromatin accessibility, thereby defining the epigenetic landscape of the cell. This is most evident for the so‐called pioneer TF class, whose definition is based on their ability to bind to closed chromatin [160, 161]. Brennan et al., by studying TF binding data and chromatin accessibility data in early Drosophila embryos, showed that chromatin accessibility during genome activation follows complex sequence rules and is mediated by both the pioneer TF and the transcriptional activators driven in distinct steps [162]. There is also growing evidence that nonpioneer transcription factors can regulate chromatin, and Benveniste et al. has shown through a large‐scale computational study that histone modifications can be predicted very accurately from transcription factor binding profiles, outlining known interactions between transcription factors and chromatin‐modifying enzymes [163]. A study also showed by single‐cell transcriptional and chromatin accessibility analyses that chromatin accessibility correlates with cell type‐specific transcription factor activity and chromatin interaction networks [164].
Second is the regulation of transcription factors by chromatin accessibility. Enhancers are a class of cis‐regulatory elements that are key drivers of cell‐type‐specific gene expression, and because enhancers require TF binding, they rely heavily on chromatin accessibility to trigger transcriptional activity. Chromatin accessibility is therefore an important regulator of enhancer function [165]. Klemm et al.’s observation that ∼94% of all ENCODE TF ChIP‐seq peaks belong to accessible chromatin supports this view, and that the organization of accessible chromatin throughout the genome reflects a network of permissive physical interactions in which enhancers, promoters, insulators, and chromatin‐binding factors synergistically regulate gene expression through this network [8]. Wang et al. determined that chromatin accessibility determines the potential of bile acid‐dependent transcription factors to regulate antimicrobial peptides (AMPs) at the pretranscriptional level, shaping the regional heterogeneity of AMPs between the small and large intestine [166].
As for the role of transcription factors in cancer by regulating chromatin accessibility, some studies have verified this. Liu et al. generated high‐quality single‐cell chromatin accessibility profiles of epithelial cells from 29 CRC patients and found that subtype‐specific transcription factors bind to different sets of target genes and contribute to the similarity and diversity of chromatin accessibility and RNA expression among patients. In addition, the CpG island methylator phenotype was identified and the chromatin status of the CIMP‐high subtype was characterized and TF regulators identified [167]. Chen et al. found that CD8+ T cells from individuals with cancer or chronic viral infections express high levels of Nr4a transcription factors and show enrichment of Nr4a binding motifs in accessible chromatin regions. Tumor‐infiltrating lymphocytes targeted for Nr4a1/2/3 triple knockdown display robust effector functions: reduced expression of the inhibitory receptor, increased cytokine production, and strong enrichment of accessible chromatin for motifs involved in effector function transcription factors [168]. Helminen et al. have identified, by genome‐wide techniques, several key features of the glucocorticoid receptor (GR) action on PC cells, and that in enzalutamide (ENZ)‐exposed PC cells, GR substitution for the androgen receptor (AR) occurs almost exclusively at accessible chromatin loci displaying occupancy of the pioneer factor Forkhead box A1 (FOXA1). Silencing of FOXA1 enhances the chromatin‐binding and transcriptional activity of GR, identifying chromatin accessibility and FOXA1‐mediated repression as important regulators of GR action in PC, pointing to novel pathways to counter steroid receptor‐mediated antiandrogen resistance [169]. These studies reveal molecular pathways by which transcription factors play key roles in cancer by remodeling chromatin accessibility.
Chromatin 3D Structure
2.2.6
3D chromatin architecture refers to the form of spatial organization of genomic DNA in the nucleus formed by multiple levels of folding, including TADs, chromatin loops, and active (A) or repressive (B) compartments [170]. Recent studies have emphasized the importance of the 3D structure of chromatin in regulating various cellular processes, especially transcription. This is achieved through a dynamic chromatin structure that controls spatial accessibility and thus dynamically regulates gene expression. Quiroga et al. have argued that the 3D structure of chromatin plays an important role in gene regulation and cellular identity by regulating contacts between motifs and gene promoters through reattachment [171]. Kirkland et al. showed that LamC deletion may reduce chromatin accessibility of cardiomyocytes by decreasing their expression of transcription factors as well as cytoskeletal regulators [172]. A study proposes that T cell activation is associated with disruption of long‐range chromatin interactions as well as partitioning of TADs and remodeling of their TAD boundaries. Newly formed/enhanced TAD boundaries are associated with higher nucleosome occupancy and lower accessibility [173]. Chromatin loops typically connect enhancers and promoters and play a crucial role in the regulation of gene transcription [174]. Also due to dynamic long‐range preferential interactions, chromosomes segregate into two forms of mutually exclusive chromatin: compartments A and B. Compartment A corresponds to active transcription and open chromatin regions, whereas compartment B is compressed and enriched with repressive chromatin features [175, 176]. These studies serve as a demonstration of the role of the 3D structure of chromatin in influencing chromatin accessibility, which in turn determines the activation or repression of genes.
The 3D structure of chromatin plays a key role in development, gene regulation, and cellular identity. Alterations in this structure can have profound effects on cellular phenotypes and have been linked to a variety of diseases, including many types of cancer. Alterations in 3D chromatin structure through a variety of different mechanisms have been found to be associated with the development of various cancers. Mutations in adhesins, one of the most common mutations in cancer, have been shown to lead to dysregulation of DNA cycling within chromosomes, thereby affecting genome organization and gene expression [177]. Disruption of the 3D structure of chromatin in cancer often leads to activation of proto‐oncogenes or silencing of oncogenes. For example, some studies have found that chromatin is disrupted in some cancers as a result of genomic rearrangements or structural variants of this genomic structure, thereby affecting the regulatory landscape of cancer cells [178]. Sui et al. found that the MLL–AF9 fusion disrupts the 3D chromatin landscape and may contribute to dramatic transcriptome remodeling in MLLr acute myeloid leukemia (AML) by comprehensively analyzing 3D genomic structure, chromatin accessibility, and gene expression in samples of MLL–AF9 AML, a genetically edited aggressive with AML [179]. Luo et al. showed that HOTTIP‐mediated R‐loop formation directly enhances CTCF chromatin boundary activity and TAD integrity to drive oncogene transcription and leukemia progression [180]. Lai et al. found that knock‐in of the C‐terminus of hematopoietic‐specific nucleophosmin 1 reshaped the TAD topology, resulting in disruption of cell cycle regulation as well as aberrant chromatin accessibility and aberrant homologous gene expression, leading to blocked myeloid differentiation [181]. Recent in vitro investigations of PC metastasis have shown that the metastatic potential and aggressiveness of PC are also closely linked to chromatin compartmentalization and dynamic genomic alterations. During cancer progression, there is extensive genomic compartmentalization, which leads to significant changes in the nuclear chromatin activation environment, resulting in increased mixing and interactions in the A compartment [182]. In addition, in metastatic pancreatic cancer cells, there is an increase in the number of chromatin loops, along with the appearance of cell‐specific chromatin loops. LIPC is a gene that promotes pancreatic cancer metastasis and is associated with tumor cell migration and invasion. LIPC expression is regulated by enhancer 3 and enhancer 4, while tissue‐specific chromatin loops form progressively during distant metastasis of pancreatic cancer, enhancing LIPC expression [183]. These studies suggest that aberrant changes in the 3D structure of chromatin can drive tumorigenesis by remodeling chromatin accessibility and gene regulatory networks.
Technologies for Profiling Chromatin Accessibility and 3D Genome Architecture
2.3
The rapid advancement of chromatin accessibility research is closely linked to breakthroughs in high‐throughput sequencing technologies. From early population‐level (bulk) analyses to the current single‐cell and spatial multiomics profiling, technological evolution has not only mapped the dynamic chromatin landscape in cancer but also profoundly revealed the epigenetic basis of tumor heterogeneity, plasticity, and therapy resistance. This section will systematically review classical and cutting‐edge technologies for analyzing chromatin accessibility and 3D conformation, with a focus on their innovative applications and potential for clinical translation in cancer research.
ATAC‐seq and DNase‐seq
2.3.1
Genome‐wide chromatin accessibility profiling was initially driven by DNase‐seq, which uses DNase I digestion followed by high‐throughput sequencing to map DNase I hypersensitive sites at base‐pair resolution and thereby identify active regulatory elements across the genome. DNase‐seq has been instrumental in the ENCODE project and in defining regulatory landscapes in diverse human cell types, including multiple cancer models [184, 185].
The development of ATAC‐seq, which relies on a hyperactive Tn5 transposase to simultaneously cut DNA and insert sequencing adapters, has substantially simplified chromatin accessibility profiling and reduced input requirements to as few as hundreds of cells. Compared with DNase‐seq, ATAC‐seq requires less material and handling time, provides comparable resolution for regulatory elements, and additionally preserves nucleosomal “laddering” patterns that inform nucleosome positioning and transcription‐factor occupancy [186, 187].
In cancer research, bulk ATAC‐seq and DNase‐seq have been widely used to map tumor‐specific regulatory elements and infer transcriptional regulatory networks. Large‐scale efforts have generated chromatin accessibility atlases of primary human cancers, linking tumor‐type‐specific open chromatin to oncogenic drivers and noncoding risk variants [188, 189]. Recent work is extending these assays to clinically relevant specimens: for example, optimized ATAC‐seq protocols for formalin‐fixed paraffin‐embedded (FFPE) samples and spatial FFPE–ATAC‐seq now enable in situ accessibility profiling in archived tumor tissues with preserved tissue architecture, opening a path toward retrospective clinical studies and routine translational applications [190, 191].
Single‐Cell Chromatin Accessibility and Multiomics
2.3.2
Bulk assays average signals across heterogeneous cell populations and therefore obscure rare cell states and evolutionary trajectories that are central to cancer biology. Single‐cell ATAC‐seq (scATAC‐seq) overcomes this limitation by resolving chromatin accessibility profiles at single‐cell resolution, enabling the reconstruction of regulatory cell states, lineage relationships, and subclonal architectures within tumors and their microenvironment [192]. Recent pan‐cancer studies have generated large‐scale scATAC‐seq atlases from primary or archival tumor samples, revealing tumor‐type‐specific regulatory programs, immune‐cell state transitions, and the impact of copy‐number alterations on chromatin landscapes [193, 194]. Beyond accessibility alone, single‐cell multiomics technologies jointly measure chromatin accessibility, transcriptomes, and in some cases additional layers such as DNA methylation or protein abundance in the same cell. Early coassays such as sci‐CAR and SHARE‐seq established that simultaneous profiling of ATAC and RNA can directly link distal regulatory elements to their target genes and predict “chromatin potential” of future transcriptional states [195, 196]. Newer platforms such as Parallel‐seq and related methods have further increased throughput and robustness, facilitating the application of single‐cell multiomics to clinically annotated tumor cohorts and therapy‐response studies [197].
In cancer immunology, single‐cell multiomics is beginning to resolve how chromatin accessibility and transcriptional programs jointly shape T‐cell exhaustion, myeloid cell reprogramming, and neoantigen‐driven immune responses, thereby directly informing epigenetic‐based combination immunotherapies and minimal residual disease monitoring [198, 199]. From a mechanistic standpoint, these approaches provide a unique opportunity to causally link chromatin accessibility dynamics to changes in cell state plasticity, stemness, and therapy resistance.
3D Chromatin‐Conformation Assays
2.3.3
Chromatin accessibility is tightly coupled to higher‐order 3D genome architecture. Chromosome‐conformation capture‐based methods, particularly Hi‐C and in situ Hi‐C, quantify physical contacts between distal genomic loci, enabling the discovery of TADs, chromatin loops, and long‐range enhancer–promoter interactions [200, 201]. In cancer, 3D genome reorganization can disrupt TAD boundaries, create oncogenic enhancer hijacking events, and rewire regulatory hubs, thereby reshaping chromatin accessibility and gene expression programs that drive tumorigenesis and progression [202, 203].
Newer 3D genome assays, including Micro‐C, HiChIP, and PLAC‐seq, increase resolution or enrich for specific protein‐anchored chromatin interactions, allowing finer mapping of regulatory loops around oncogenes and tumor suppressors [204, 205, 206]. When combined with ATAC‐seq and histone‐mark ChIP‐seq, these methods delineate “3D regulatory hubs” in which clusters of accessible enhancers physically converge on key transcription factors and lineage‐defining genes, providing a structural explanation for SE‐driven transcriptional addiction and for context‐specific vulnerabilities that may be exploited therapeutically [207, 208].
Importantly, 3D chromatin‐conformation assays are beginning to be applied to patient‐derived organoids (PDOs), ex vivo coculture models, and longitudinal biopsy samples, offering a route to study how therapy reshapes genome topology and how emergent 3D configurations correlate with minimal residual disease or acquired resistance [209, 210, 211]. Integrating these 3D maps with the chromatin accessibility and multiomics datasets discussed above will be critical for moving from correlative associations toward mechanistic models of genome folding in cancer.
Collectively, these complementary technologies—bulk ATAC‐seq and DNase‐seq, single‐cell multiomics, and 3D chromatin‐conformation assays—provide a multiscale view of chromatin accessibility in cancer, ranging from nucleotide‐level regulatory elements to cell‐type‐specific programs and higher‐order genome topology. Their joint application in clinically annotated cohorts will be essential for identifying robust accessibility‐based biomarkers, for disentangling tumor‐intrinsic versus microenvironment‐driven regulatory changes.
Role of Chromatin Accessibility in Tumors
3
We explore the multidimensional role of chromatin accessibility in tumors, including tumorigenesis, progression, metabolic reprogramming, angiogenesis, tumor stemness, immunity and microenvironment, and tumor therapy resistance (Figures 2 and 3).
The role of chromatin accessibility in tumors. The role of chromatin accessibility in tumors, including: tumorigenesis, tumor progression, tumor metabolic reprogramming, tumor angiogenesis, tumor stemness, tumor immune evasion and microenvironment, and tumor therapy resistance.
Schematic representation of the mechanisms underlying the role of chromatin accessibility in tumors. (A) YAP–TEAD complex interacts with TET1 to demethylate DNA at YAP target genes in the liver. Loss of TET1 reverses YAP‐induced chromatin and transcriptional changes and suppresses YAP‐induced hepatomegaly and tumorigenesis. (B) Stress‐induced (e.g., UV irradiation) HDAC8 activity regulates an invasive melanoma cell state, deacetylates EP300, inhibiting its enzymatic activity, switching its association from MITF sites to Jun sites and increases H3K27 acetylation and chromatin accessibility at Jun‐target gene transcriptional sites, thereby increasing the development of melanoma brain metastases. (C) BPTF constituted super‐enhancers that activate downstream targets like enolase 2 and SRC proto‐oncogene nonreceptor tyrosine kinase, leading to glycolytic reprogramming of METTL14−/− cells; ARID1A loss increased chromatin accessibility and enhanced HIF‐1α binding to the promoter regions of Pgam1, Pkm, and Pgk1, similarly promotes glycolytic reprogramming. (D) Endocan, a proteoglycan secreted by endothelial cells, can directly bind to the PDGFRA receptor and activate its activity, thereby enhancing the chromatin accessibility of the Myc promoter and upregulating Myc expression, which in turn confers enhanced proliferation, migration, and angiogenesis capabilities to GBM cells. (E) HIF‐1α activates PLD2 transcription through hypoxia response elements, increased accessibility of AP‐1 transcription factor binding sites induces cancer stem cells formation. (F) The DNA translocase SMARCAL1 favors tumor immune evasion by two distinct mechanisms: it suppresses the cGAS–STING pathway by limiting endogenous DNA damage and induces PD‐L1 expression by modulating chromatin accessibility at a PD‐L1 regulatory element.
Tumorigenesis
3.1
Tumorigenesis is a complex, gradual process primarily driven by the activation of oncogenes and the inactivation of TSGs [212]. In tumor cells, alterations in chromatin accessibility play a crucial role in tumorigenesis and progression.
Chromatin accessibility regulates proto‐oncogenes and oncogenes by modulating promoter and enhancer activity [213]. For example, protein arginine methyltransferase (PRMT) 1 promotes PDAC by increasing chromatin accessibility at glycolytic gene promoters and enhancers [214]. Similarly, SE RNA m6A modification enhances chromatin accessibility, driving PDAC‐associated gene transcription, ultimately driving PDAC development [107]. SET domain containing 2 (SETD2), a frequently mutated chromatin modifier in cancer, facilitates KRAS‐driven lung tumorigenesis by increasing enhancer accessibility and upregulating polycomb repressive complex 2 (PRC2) and KRAS signaling genes [215].
Epigenetic mechanisms—including DNA methylation, histone modifications, transcription factors, and ncRNAs—further influence tumorigenesis by altering chromatin structure. The YAP–TEAD complex upregulates TET1, which promotes DNA demethylation, H3K27ac modification, and chromatin opening at YAP target genes, ultimately accelerating liver tumorigenesis [216]. In addition, recent studies have found that lactate‐induced lactic acidification further affects lactate production, recycling, and utilization, which in turn promotes tumorigenesis [217]. Lactate, through histone lactylation, modifies chromatin structure to regulate gene expression, linking metabolic reprogramming to tumor progression [218]. In melanoma, PHB2 recruits MLL2 to the CANT2 promoter, enhancing H3K4me3 and activating this oncogenic lncRNA while suppressing the tumor suppressor CCBE1 [219].
In addition, the role of chromatin remodeling complexes in tumorigenesis cannot be ignored. SMARCA5, an ISWI complex ATPase, maintains aberrant chromatin accessibility in leukemia by recruiting DDX5 and SP1 to activate AKR1B1, which reprograms fructose metabolism and worsens patient outcomes [220]. CARM1 promotes the development of breast cancer by methylating the NuRD complex to upregulate cell cycle genes [221]. Similarly, Wei et al. found that a JmjC family protein, JARID2, is highly expressed in several types of cancers including breast cancer, and JARID2 promotes breast tumorigenesis by interacting with the NuRD complex, especially playing a key role in the adipocyte‐derived leptin response [222].
The impact of chromatin accessibility extends beyond these examples. Studies have highlighted its role in the progression of various cancers, including CRC [223], gastric cancer [224], PC [225], and ovarian cancer (OC) [226], underscoring the multifaceted contribution of chromatin remodeling in tumorigenesis.
Tumor Progression
3.2
Chromatin accessibility, as a core hub of epigenetic regulation, plays a key role in tumor proliferation, invasion, and metastasis, and recent studies have revealed that it can contribute to the molecular panorama of tumor malignant evolution through multidimensional mechanisms.
Dysregulated chromatin accessibility fuels tumor proliferation by activating oncogenic pathways. For instance, HMGA1 drives myeloproliferative tumor progression by enhancing chromatin accessibility at the GATA2 locus, recruiting activating histone marks to stimulate proproliferative gene networks [227]. Similarly, BRD8 sustains GBM proliferation via the EP400 complex, which deposits the histone variant H2AZ at p53 target loci to repress cell cycle arrest signals [228]. In neuroblastoma, activator protein‐1 (AP‐1) remodels chromatin to expose IRF2BP2 binding sites, activating ALK signaling through SEs to drive proliferation [229]. Microtubule‐associated serine/threonine kinase‐like (MASTL) has a key function in mitotic regulation, and it has been shown that IL‐6 and TNF‐α stimulation induces trimethylation of H3K4Me3 at the MASTL promoter to promote chromatin accessibility, thereby promoting HCC proliferation [230].
Chromatin accessibility dysregulation facilitates tumor invasion by rewiring oncogenic signaling pathways. For example, in early‐stage LUAD, SE activation at the LINC01977 locus drives malignancy by hijacking the TGF‐β/SMAD3 pathway, linking chromatin remodeling to invasive progression [231]. Similarly, in melanoma, imprinted site regulator BORIS, also known as CCCTC binding factor, which mediates altered chromatin accessibility promotes a proinvasive transcriptional signature [232]. In PDAC, chromatin accessibility profiling identified KRAS‐activated FOSL2 as a critical mediator of invasion. FOSL2 upregulates CCL28 to promote tumor cell migration and invasion, correlating with poor patient prognosis [233]. Zheng et al. showed that HBV X protein (HBx) is the most frequently integrated viral gene sequence after HBV infection. HBx increases chromatin accessibility and ETV4 expression to regulate dishevelled‐2 and promote HCC cell migration and invasion, and high expression of ETV4 is correlated with poor prognosis of HCC patients [234].
Dynamic chromatin accessibility changes are central to epigenetic reprogramming during metastasis, enabling transcriptional plasticity that drives tumor dissemination. For example, histone deacetylase (HDAC) 8 upregulation in melanoma increases H3K27ac levels and enhances c‐Jun binding site accessibility, promoting invasive phenotypes and brain metastasis [235]. Similarly, the lncRNA MALAT1 fuels LUAD metastasis by amplifying chromatin accessibility at inflammatory loci, upregulating CCL2 to remodel the tumor microenvironment [105]. Inactivation of tumor suppressors further reshapes metastatic potential. LKB1 loss in LUAD alters chromatin landscapes to accelerate metastatic progression [236]. In HCC, the transposon‐derived lncMER52A—dependent on chromatin accessibility for its expression—promotes metastasis by interacting with p120‐catenin [237]. Epigenetic regulators like Cat eye syndrome chromosome region, candidate 2 (CECR2) also play key roles: in breast cancer, CECR2 increases chromatin accessibility to activate NF‐κB signaling, fostering immunosuppressive microenvironments and metastasis [238]. SETD2 mutations exemplify another mechanism, where loss of H3K36me3 activates enhancers that drive prometastatic transcription, creating dependencies on histone chaperone complexes [239]. These mechanisms extend across cancers, including colorectal [240], osteosarcoma [241], pancreatic [242], bladder [243], and prostate malignancies [244]. These results suggest that dynamic alterations in chromatin accessibility in tumor cells can drive aberrant tumor proliferation, invasion, and metastasis by remodeling the transcriptional program and regulating the activation of oncogenes and metastasis‐related pathways. Future studies need to combine multiomics data to resolve the driving mechanisms of accessibility changes in specific tumor types and develop precise therapeutic strategies targeting open regions of chromatin.
Regulation of Metabolic Reprogramming
3.3
Metabolic reprogramming—a hallmark of cancer—enables tumor cells to adapt to nutrient demands through enhanced glycolysis (the Warburg effect), dysregulated glutamine metabolism, and increased lipid synthesis [245, 246]. Emerging research highlights chromatin accessibility as a central regulator of these metabolic shifts.
Tumor microenvironment metabolites reciprocally remodel chromatin landscapes. Lactate, for instance, induces histone lactylation, altering chromatin spatial configuration to regulate DNA accessibility and gene expression. Spatial lactate gradients link metabolic activity to epigenetic rewiring, positioning lactate as a dual driver of tumor energetics and malignant progression [218]. Similarly, in NEPC, Zeb1 promotes metabolic plasticity by modulating glycolytic enzymes and reshaping chromatin accessibility to facilitate lineage plasticity [86].
Meanwhile, key transcription factors regulate metabolic enzyme expression through chromatin accessibility. For example, in CRC, the Lyn/RUVBL1 complex enhances FOXA1 chromatin accessibility, activating arachidonic acid (AA) metabolism to accelerate liver metastasis [247]. In renal cell carcinoma (RCC), METTL14 loss stabilizes BPTF mRNA, amplifying glycolysis‐associated SEs and fostering prometastatic epigenetic memory [248].
Chromatin remodeling complexes also directly regulate metabolic enzyme expression. For example, ARID1A deficiency disrupts SWI/SNF complex function, opening chromatin regions to activate glycolytic genes and sensitizing lung tumors to glutaminase inhibitors—a “synthetic lethal” vulnerability with therapeutic potential [249].
Collectively, chromatin accessibility dynamics orchestrate metabolic reprogramming by modulating enzymes, signaling pathways, and epigenetic crosstalk. These insights underscore the need to target chromatin–metabolism interplay for novel cancer therapies.
Tumor Angiogenesis
3.4
Tumor angiogenesis, a process by which tumor cells acquire oxygen and nutrients by inducing neovascularization, is critical for tumor growth, invasion and metastasis [250]. A growing body of evidence identifies chromatin accessibility as a critical regulator of this process via diverse epigenetic mechanisms.
Chromatin remodeling by transcriptional complexes activates proangiogenic gene networks. For example, in breast cancer, SIN3A‐associated protein, 30 kDa (SAP30), a subunit of the SIN3 corepressor complex, paradoxically enhances chromatin accessibility at promoters of genes linked to angiogenesis, lymphangiogenesis, and metastasis, driving tumor progression [251]. Similarly, in HCC, nuclear actin dynamics increase chromatin accessibility to coordinate extracellular matrix remodeling and angiogenesis‐related gene expression, promoting metastasis [252]. Resistance to antiangiogenic therapies is closely associated with dynamic changes in chromatin accessibility. For instance, amilotinib‐resistant tumor cells exhibit increased accessibility at angiogenesis‐associated genomic regions, with TFAP2A driving resistance via proangiogenic transcriptional programs [253]. In GBM, endothelial‐secreted Endocan activates PDGFRA signaling, enhancing MYC promoter accessibility to stabilize tumor‐promoting phenotypes and underscoring vascular–tumor crosstalk in disease progression [254]. In addition, angiogenesis is epigenetically linked to metabolic reprogramming. For example, Li et al. found that EZH2‐mediated H3K27me3 deposition suppresses transsulfuration pathway genes, but inhibition of EZH2 (e.g., Tazemetostat) restores chromatin accessibility, reactivating cysteine–methionine metabolism and lipid homeostasis while suppressing angiogenesis [255].
These results suggest that altered chromatin accessibility plays an important role in tumor angiogenesis by modulating the expression of proangiogenic factors and endothelial cell function. Future studies should further resolve the heterogeneity of angiogenesis‐associated chromatin opening patterns in different tumor types and develop epigenetic drugs targeting specific accessibility regions to optimize strategies for antiangiogenic therapy.
Tumor Stemness
3.5
Tumor stemness refers to the key characteristics of tumor cells with self‐renewal, multidirectional differentiation, and tumorigenic ability, which are closely related to tumorigenesis, metastasis, and recurrence [256]. Recent studies have shown that dynamic remodeling of chromatin accessibility plays a central role in maintaining the self‐renewal and differentiation of CSCs by regulating the spatiotemporal expression patterns of stemness‐related genes.
Metabolic–epigenetic crosstalk modulates CSC properties. In leukemia, nuclear hexokinase 2 (HK2) enhances chromatin accessibility at DNA repair and stemness loci, reinforcing leukemic stem cell identity [257]. Furthermore, hypoxia drives CSC's characteristics through epigenetic reprogramming. For example, Hypoxia further reprograms CSCs via HIF‐1α, which activates AP‐1 binding sites and increases PLD2 transcription to drive chemoresistance in OC [258]. Similarly, abnormalities in the SWI/SNF chromatin complex affect stemness homeostasis, and ARID1A is a key subunit of the SCRC, and Wang et al. found that ARID1A deficiency increased the expression of hepatic stem/progenitor cell markers and enhanced cellular self‐renewal, as well as remodeled chromatin accessibility of genes associated with liver function. Thus, ARID1A deficiency may increase the number of stem/progenitor‐like cells by dysregulating the expression of these genes associated with cell stemness, differentiation, and liver function, leading to cancer development [259]. Similarly, MUC1‐C promotes prostate CSC self‐renewal by activating the PBAF complex to enhance chromatin accessibility, linking redox homeostasis to stemness [260]. Targeting MUC1‐C inhibits NEPC CSC tumorigenicity and therapy resistance [261].
ncRNAs and histone variants fine‐tune chromatin landscapes. In HCC, CircHULC drives CSC growth by inducing chromatin reprogramming and genomic instability [262]. Nikolic et al. revealed that the histone variant macroH2A2 shapes chromatin accessibility at enhancer elements to antagonize the epigenetic program of stemness in GBM [263].
Based on the above studies, it is easy to see that chromatin accessibility alterations maintain the self‐renewal and differentiation potential of CSCs by regulating stemness‐associated transcription factors and epigenetic modifications. Future studies should further resolve the heterogeneity of chromatin opening patterns in different tumor types and develop precise therapeutic strategies targeting the specific accessibility regions of CSCs to overcome tumor resistance and recurrence.
Tumor Immunity and Microenvironment
3.6
Tumor immune evasion and the immunosuppressive tumor microenvironment are critical barriers to effective cancer therapy [264]. Chromatin accessibility plays a pivotal role in this process by regulating immune checkpoint molecules and immune cell functionality.
Chromatin remodeling drives immune evasion by upregulating immunosuppressive signals [265]. For example, SMARCAL1, a DNA translocase, collaborates with the transcription factor JUN to maintain chromatin accessibility at the PD‐L1 promoter, enhancing its expression and enabling tumor cells to escape immune detection [266]. Similarly, the transcription factor FLI1 amplifies IDO1 expression by coordinating CBP and STAT1 activity, promoting IFN‐γ‐induced kynurenine production and suppressing T cell responses [267].
Tumor‐infiltrating regulatory T cells exhibit distinct chromatin accessibility patterns compared with peripheral blood Tregs, with enriched AP‐1 motifs in enhancers linked to immune suppression and leukocyte differentiation [268]. Combining DNMT and EZH2 inhibitors reduces promoter methylation, increases chromatin accessibility, and reactivates interferon‐stimulated genes, restoring antitumor immunity in HCC [269]. In NSCLC, IL‐4 suppresses TAP2 expression via chromatin remodeling, impairing antigen presentation and promoting immune evasion [270].
Chromatin accessibility regulates extracellular matrix (ECM) remodeling and metabolic pathways to shape TIME. In GBM, RUNX1 interacts with NPM1 to maintain chromatin openness at ECM‐associated genes, fostering an immunosuppressive niche [271]. WDR6 promotes HCC progression by enhancing TNF‐α expression through chromatin remodeling, while MPC activity preserves cytotoxic T cell function by maintaining histone acetylation and promemory gene accessibility [68, 272].
In addition, Loss of tumor suppressors (NF1, TSC1, TGF‐β RII) alters chromatin landscapes, activating the IL6‐JAK3‐STAT3/6 pathway and recruiting immunosuppressive LAG3+ T cells [273]. In nasopharyngeal carcinoma, EBV infection reduces CTCF levels, driving CD74‐mediated T cell exhaustion and epigenetic reprogramming [274]. Moreover, SETD2 inactivation impairs H3K36me3 deposition, reducing NR2F1 transcription and activating STAT1 to enhance PD‐1 expression, thereby reshaping TIME [275].
These findings suggest that altered chromatin accessibility plays a critical role in tumor immune evasion and immune microenvironment remodeling through multilevel regulation of immune‐related gene expression. The integrated multiomics analysis and the establishment of organoid models will help to deeply understand the regulatory mechanism of chromatin accessibility in the dynamic changes of TIME and provide new ideas for the development of novel immunotherapy strategies.
Tumor Therapy Resistance
3.7
Tumor therapeutic resistance is a major challenge in clinical cancer treatment, including various forms of chemotherapy resistance, targeted therapy resistance, endocrine therapy resistance, immunotherapy resistance and radiotherapy resistance. Recent studies have found that chromatin accessibility plays a central role in multiple drug resistance mechanisms by dynamically regulating key pathways such as drug metabolism, target expression, DNA damage repair, and immune microenvironment [18, 218, 276].
Chemotherapy Resistance
3.7.1
Chemoresistance is a major challenge in tumor therapy. Y‐box binding protein 1 (YBX1) stabilizes CHD3 mRNA via m5C modification recognition, enhancing chromatin accessibility to promote homologous recombination and platinum resistance in OC [277]. Similarly, the histone demethylase KDM4 increases promoter accessibility of senescence‐associated genes, fostering chemoresistance in PC, while KDM4 inhibition reverses this phenotype [278]. In leukemia, nuclear HK2 elevates chromatin accessibility at DNA repair and stemness loci, reducing double‐strand breaks and conferring chemoresistance in leukemia stem cells [257]. CBX2 drives CRC chemoresistance by maintaining chromatin accessibility via the RUNX1–CBX2–MAP4K1–pERK axis, with CBX2 knockdown inducing epigenetic reprogramming and sensitizing tumors to therapy [223]. In squamous cell carcinoma, DLGAP1–AS2 enhances H3K27ac‐marked chromatin accessibility at the FAM3D locus, activating YAP signaling through phosphatidic acid synthesis to promote chemoresistance [279]. Hypoxia induces chemoresistance in OC by stabilizing HIF‐1α, which increases AP‐1 binding site accessibility and activates PLD2 transcription. PLD2 overexpression mimics hypoxia to enhance cisplatin and carboplatin resistance [258]. Overall, dynamic changes in chromatin accessibility can dynamically orchestrate the stress response and survival adaptation of cancer cells to chemotherapeutic agents by modulating DNA repair capacity, remodeling epigenetic status, and activating key signaling pathways, thereby driving the onset and development of chemoresistance in a wide range of solid and hematologic tumors.
Targeted Therapy Resistance
3.7.2
Recent studies have reported the role of epigenetic changes, particularly chromatin accessibility, in resistance to targeted tumor therapies. In EGFR‐mutant lung cancer, the SCRC promotes osimertinib resistance by altering chromatin landscapes, facilitating survival despite tyrosine kinase inhibition [280]. Similarly, SMARCA4 inactivation in small‐cell lung cancer (SCLC) increases chromatin accessibility at neuroendocrine (NE) transcription factor loci, driving ERBB pathway activation and sensitizing tumors to afatinib [253]. In anrotinib‐resistant lung cancer, chromatin accessibility profiling revealed enrichment of angiogenesis‐related pathways. TFAP2A accelerates resistance by activating proangiogenic programs, highlighting the interplay between chromatin remodeling and vascular signaling [281]. In addition, in OC, CYP1B1, a member of the cytochrome P450 family of enzymes, promotes PARP inhibitor resistance through histone H1.4 interactions and increased chromatin accessibility [282].
In line with the above, the aberrant expression and adaptive signaling activation of targeted therapy‐related genes are coordinated through dynamic mechanisms such as remodeling chromatin structure, modulating transcription factor activity, enhancing DNA repair capacity, and mediating tumor cell state plasticity, thereby driving the onset and evolution of targeted therapy resistance in a wide range of lung, myeloma, ovarian, and NE tumors.
Endocrine Therapy Resistance
3.7.3
Endocrine therapy resistance is also a common problem in tumor therapy, and the role of chromatin accessibility in its mechanism is gradually being revealed. In a study on endocrine therapy resistance in breast cancer, YAP was found to bind to TEAD to increase local chromatin accessibility to stimulate transcription of nearby genes, and transcriptional repression of ER by YAP was demonstrated revealing the Hippo pathway as a therapeutic target in ER+ breast cancer [283]. Blawski et al. indicated that the pioneer factor FOXA1‐driven another chromatin‐accessible state in invasive lobular carcinoma (ILC) of the breast, FOXA1 regulates its own expression in a feed‐forward mechanism by binding to the unique FOXA1 enhancer site in ILC. This results in the FOXA1–ER axis promoting transcription of genes associated with tumor progression and tamoxifen resistance [284]. Chen et al. showed that AR‐indifference is a mechanism of resistance to hormone therapy in PC, in which one cut homeobox 2 (ONECUT2) regulates gene expression through promoter binding, enhanced genome‐wide chromatin accessibility, and SE reprogramming, which activates resistance with multiple drivers associated with adenocarcinoma, stem cell‐like, and NE variants [285]. The use of AR inhibitors in PC increases the plasticity of the cell line, leading to resistance to AR‐targeted therapies. In a similar study to Chen, Leppanen et al. examined the chromatin landscape of AR‐positive PC cells after exposure to the AR inhibitor ENZ, identified a novel regulator of cellular plasticity, the homology‐frame transcription factor Sine oculis homeobox homolog 2 (SIX2), whose motifs are enriched in accessible chromatin regions after treatment, and suggested that depletion of IX2 might be a possible strategy for overcoming the androgen‐resistant cellular plasticity of PC mechanism [286].
Overall, it appears that aberrant activation of hormone receptor‐dependent and nondependent pathways is coordinated through dynamic mechanisms such as activation of transcription factor complexes, remodeling of pioneer factor‐mediated chromatin states, driving SE reprogramming, and inhibition of key receptor signaling, which can affect endocrine therapy resistance.
Immunotherapy Resistance
3.7.4
Immunotherapy resistance is driven by chromatin accessibility changes that suppress antigen presentation, immune checkpoint activity, and antitumor immune responses. In multiple myeloma (MM), gamma‐secretase inhibitors induce antigen shedding by altering chromatin accessibility, reducing surface antigen expression and enabling immune evasion [287]. Similarly, acute lymphoblastic leukemia cells resistant to immunotherapy exhibit reduced CD19 and CD22 promoter accessibility, downregulating antigen expression while increasing dependence on BTK signaling [288]. Conversely, HDAC inhibitors like CXD101 restore antitumor immunity in checkpoint blockade‐resistant models by enhancing chromatin accessibility and H3K27 hyperacetylation at IFNγ‐responsive genes, synergizing with immune checkpoint inhibitors (ICIs) to activate STAT1‐driven immunity [289]. In NSCLC, IL‐4 signaling in NSCLC reduces TAP2 promoter accessibility, impairing antigen processing and promoting reversible immune evasion [270].
These results suggest that reduced accessibility of drug resistance‐related gene promoters leads to downregulation of their expression, and chromatin accessibility dynamically coordinates tumor cell regulation of immune checkpoints, silencing of antigenic expression, and adaptive remodeling of immune responses, thereby driving the onset of immunotherapeutic drug resistance and adaptive immune escape in a variety of malignancies, including MM, acute lymphoblastic leukemia, and NSCLC.
Radiotherapy Resistance
3.7.5
Chromatin accessibility alterations significantly influence radiotherapy resistance through diverse mechanisms, as evidenced by emerging studies. For example, Yang et al. that exosomal DEK binds directly to chromatin, increasing genome‐wide accessibility. This process triggers the quiescent state of breast cancer CSCs, thereby reducing their sensitivity to chemo‐ and radiotherapy [290]. Similarly, Dawn et al. identified a clinical correlation between the cancer/testis antigen GAGE and radiotherapy resistance in cervical cancer. Mechanistically, GAGE mediates radioresistance by modulating chromatin accessibility, suggesting its role as a biomarker for treatment failure [291]. In contrast, Maja et al. found a higher ratio of alpha radiation‐induced DNA damage in the invasive breast cancer cell line MDA‐MB‐231 cells, which may be explained by the basal heterochromatin marker higher levels and suggests that dense chromatin is associated with poor tumor prognosis and resistance to photon radiotherapy [292].
These findings suggest that altered chromatin accessibility plays a key role in tumor therapy resistance through multidimensional regulation of gene expression programs. An in‐depth understanding of the regulatory mechanisms of chromatin accessibility in drug resistance will provide an important theoretical basis for the development of novel anticancer strategies.
Chromatin Accessibility in Tumor Treatment Strategies and Clinical Trials
4
In recent years, with the development of high‐throughput sequencing technologies, such as ATAC‐seq and DNase‐seq, the role of chromatin accessibility in tumor therapy has been continuously explored. This section will focus on the following points.
Chromatin Accessibility as a Tumor Marker
4.1
Diagnostic Markers
4.1.1
Chromatin accessibility dynamics reflect epigenetic reprogramming events early in tumorigenesis. Abnormalities in open chromatin regions emerge earlier than those detected by traditional mutation analysis and can be noninvasively identified via liquid biopsies, such as circulating free DNA (cfDNA) [293, 294].
A recent multicohort study found that coverage of cfDNA in promoter regions can be used as an indicator of chromatin status and reflect gene expression levels in living cells. A comprehensive survey of plasma cfDNA from 546 individuals concluded that inferred chromatin accessibility alterations derived from cfDNA profiles have the potential to be used not only for cancer screening, but also for diagnostic and preoperative evaluation [295]. A simplified cfDNA methylation assay targeting OTOP2 and KCNA3 enables accurate diagnosis of esophageal cancer, addressing the unmet need for reliable blood‐based biomarkers [296]. In Taklifi et al.’s review, it was also shown that chromatin accessibility differences are reflected in the fragmentation patterns of free DNA, and a new pipeline that integrates chromatin accessibility status into the design of liquid biopsy‐targeted sequencing panels is described for identifying labeled regions of free DNA for cancer detection, as well as cancer‐specific markers that have potential use in liquid biopsy testing [297].
In addition, the role of differential expression of chromatin accessibility in distinguishing tumors from paraneoplastic or normal tissues, as well as in the classification of cancer subtypes, has been confirmed by a number of studies. Liu et al. comprehensively investigated the genome‐wide DNAme landscapes of breast cancer and 10 other cancers and their neighboring normal and healthy breast tissues, obtained eight CpGs with large differences in chromatin accessibility status, and constructed a logistic regression model, by which it was possible to differentiate between breast cancers and normal samples as well as other cancers with high sensitivity and specificity [298]. Similarly, Peter et al. analyzed whole‐genome sequencing data from more than 1000 cfDNA samples from cancer patients and healthy controls using a self‐developed bioinformatics pipeline that inferred accessibility of transcription factor binding sites from cfDNA fragmentation patterns and showed that inferring transcription factor binding from cfDNA can help predict and early detect tumor subtypes [299]. In a similar vein, Doebley et al. developed Griffin, a framework for analyzing cfDNA nucleosome conservation and accessibility, which employs a GC correction procedure tailored to variable cfDNA fragment sizes, allowing accurate analysis of chromatin accessibility of cfDNA for cancer subtype prediction and potentially direct personalization of therapies to improve patient prognosis [300].
Chromatin accessibility markers exhibit high tissue specificity and early detection potential. Integrating these with multiomics data could establish an “epigenetic liquid biopsy” platform, revolutionizing early cancer screening, diagnosis, and subtype stratification.
Prognostic Markers
4.1.2
Chromatin accessibility alterations are closely associated with tumor progression, metastasis, and clinical outcomes, offering significant potential as prognostic biomarkers.
Dynamic chromatin accessibility changes serve as real‐time indicators of tumor aggressiveness. In clear cell renal carcinoma, BAP1 mutations reduce chromatin accessibility, while PBRM1 mutations increase it, with copper–cyanophorin promoting tumor–stromal interactions to drive progression [301]. In early‐stage TNBC, Meng et al. found that knockdown of ANCO1 resulted in enhanced aneuploidy, cellular senescence, and invasion in 3D stroma, suggesting that deletion of ANCO1 expression regulates chromatin accessibility and promotes progression of early‐stage TNBC [302]. Xu et al. identified a new cell subpopulation with abnormally high CXCL14 expression levels in positive lymph nodes of breast cancer patients by a combination of scRNA‐seq and scATAC‐seq assays to obtain chromatin accessibility profiles, suggesting that CXCL14 is a key regulator and marker of lymph node metastasis of breast cancer [303]. LaFave et al. proposed that epigenomic state transitions are characteristic of LUAD progression in mice, and they used single‐cell epigenomics to analyze chromatin state transitions in a LUAD mouse model characterized by activation of the RUNX transcription factor, which mediates extracellular matrix remodeling to promote metastasis [304]. These results suggest that dynamic changes in chromatin accessibility can serve as a real‐time monitoring indicator and biomarker of tumor aggressiveness.
In addition, in terms of predicting tumor prognosis and survival, some studies in recent years have also confirmed the role played by chromatin accessibility. In PDAC, transcription factors ZKSCAN1 and HNF1B exhibit differential chromatin accessibility patterns predictive of disease‐free survival [305]. NSCLC patients with ARID1B mutations show improved survival due to enhanced DNA damage response and cGAS–STING pathway activation, highlighting its prognostic utility [306]. In GBM, elevated chromatin accessibility at the GSTM1 locus predicts shorter survival, validated across multiple patient cohorts [307]. Through these studies, we can see that chromatin accessibility markers have multidimensional prognostic value and are gradually moving from basic research to clinical practice.
In conclusion, these findings demonstrate the ability of dynamic regulation of chromatin accessibility in revealing the mechanisms of tumor progression and metastasis and as a prognostic biomarker of tumors, which provides new prospects for precision medicine in tumors.
Chromatin Accessibility as a Therapeutic Target
4.2
Given that chromatin accessibility abnormalities are closely associated with tumorigenesis, progression, and treatment resistance, targeting key nodes of the chromatin accessibility regulatory network (e.g., DNA methylation, histone modification, chromatin remodeling complexes, etc.) has become an emerging strategy for tumor therapy. These mechanisms and clinical potentials are systematically described in terms of the following five aspects.
Targeting DNA Methylation
4.2.1
DNA methylation (e.g., CpG island hypermethylation) drives tumorigenesis by silencing oncogenes, and DNA methylation is dynamically regulated by DNMTs and demethylases, which have been identified as inhibitory targets for a variety of cancers.
DNMT inhibitor (DNMTi), such as azacitidine and decitabine, have been approved by the United States Food and Drug Administration (US FDA) for the treatment of myelodysplastic syndromes (MDS), AML, and chronic myelomonocytic leukemia [308, 309]. Beyond hematologic malignancies, DNMTis show promise in solid tumors [310]. For example, decitabine exhibits efficacy in preclinical models of castration‐resistant PC (CRPC) and NEPC, particularly in RB1‐deficient subtypes, suggesting biomarker‐driven strategies could improve outcomes [311]. Combining DNMTis with other therapies enhances efficacy. In OC, adenosine deaminase 1 (ADAR1) deletion synergizes with DNMTis to remodel the immune microenvironment, reducing tumor burden and extending survival [312]. Zebularine, another DNMTi, sensitizes tumors to immunotherapy by activating the cGAS–STING pathway [313].
In addition, combinations with other epigenetic drugs may provide better therapeutic effects. Rodems et al. showed that the DNMTis decitabine or guadecitabine (SGI‐110) enhanced antitumor immunity by increasing the accessibility and expression of HLA‐I in PC cells, when combining them with the HDAC inhibitor LBH‐589 (LBH) [314]. Zhang et al. tested the effects of the combination of DNMTi (5‐aza‐2′‐deoxycytidine) and the EZH2 inhibitor GSK126 on drug sensitivity, DNA methylation, nucleosome accessibility, and gene expression profiles in human HCC cell lines. An increase in the number of upregulated genes after combination therapy was associated with prolonged antiproliferative effects and increased nucleosome accessibility. And potential therapeutic targets were identified and provided a rationale for therapeutic efficacy in HCC patients [269].
Targeting DNA methylation modification factors provides a unique epigenetic intervention strategy for tumor therapy by remodeling chromatin accessibility. In the future, it is necessary to combine multiomics technologies to resolve methylation dynamics, develop highly selective inhibitors, and overcome drug resistance through combination therapy (e.g., immune checkpoint blockade, targeted metabolism) to ultimately achieve individualized epigenetic precision therapy.
Targeted Histone Modifications
4.2.2
Histone modification is a central mechanism of epigenetic regulation that affects tumorigenesis, progression, and treatment resistance by dynamically altering chromatin structure and gene expression [315]. In recent years, small molecule inhibitors targeting histone modifying enzymes have become an important strategy for cancer therapy. In this section, we will explore representative drugs targeting histone methylation and acetylation with their mechanisms of action and discuss their clinical translational potential.
Histone methylation is coregulated by methyltransferases (HMT) and demethylases, and HMT plays a crucial role in many cellular processes and has been associated with different types of cancer [316]. Xie et al. found that SETD2, an RNA polymerase II (Pol II)‐associated HMT, catalyzes the cotranscriptional methylation of H3K36me2, and they established a tumor‐suppressor gene model in which SETD2‐mediated activation of an enhancer by H3K36me3 deletion drove the oncogenic transcriptional output by regulating chromatin accessibility. And a mechanism‐based therapeutic strategy for SETD2‐deficient cancers was discovered by targeting specific histone chaperone complexes, including ASF1A/B and SPT16, providing unique therapeutic opportunities [239].
EZH2 is the catalytic subunit of PRC2, which functions as a HMT, and its dysregulation may promote cancer development [317]. Tazemetostat is a first‐in‐class oral EZH2 inhibitor approved by the US FDA for the treatment of follicular lymphoma and epithelioid sarcoma [318]. A recent study revealed that EZH2‐mediated modification of H3K27me3 histone trimethylation decreased chromatin accessibility and that the EZH2 inhibitor Tazemetostat dose‐dependently decreased cell viability and increased lipid peroxidation in HCC cells. This result reveals a novel epigenetic mechanism controlling lipid peroxidation and iron death susceptibility in HCC and provides a theoretical basis for exploring EZH2‐targeted therapies against this malignancy [255]. Other EZH2 inhibitors valemetostat, CPI‐1205, and PF‐06821497 are currently in clinical development [319]. Ku et al. demonstrated that furazamidine (FM) inhibits the PRMT1 and reduces the expression of genes related to H4R3me2 modification, chromatin accessibility, and glycolysis thereby reversing chemoresistance in pancreatic cancer [214]. In addition, histone lysine demethylase (KDM) controls and maintains epigenetic factors that influence chromatin structure and cellular properties, and its dysregulation is associated with a variety of diseases, including malignancies. Zhang et al. showed that demethylase inhibitors of KDM2–7 (e.g., CBA‐1, JDI‐16, PKF118‐310, etc.) play an important role in the treatment of cancer, but the selectivity and intracellular activity of these inhibitors among subfamilies remains to be developed [320]. Although resistance and selectivity challenges still need to be addressed, we cannot deny that drugs targeting histone methylation have become an important strategy for cancer therapy.
PTMs of histones may play a critical role in cancer development and progression by regulating gene transcription, chromatin remodeling, and nuclear structure. Histone acetylation is a well‐studied posttranslational histone modification controlled by the opposing activities of HATs and HDACs, and HDAC inhibitors capable of reestablishing acetylation homeostasis may be useful in cancer therapy [321]. HDAC inhibitors are potent drug molecules that induce histone acetylation at lysine residues and induce open chromatin conformation at TSG loci, leading to tumor suppression [322]. The US FDA approves HDAC inhibitors for the treatment of certain cancers (e.g., T‐cell lymphomas), such as vorinostat (SAHA), romidepsin (i.e., depsipeptide, a bicyclic peptide), and belinostat. Panobinostat (LBH 589) has been approved for the treatment of MM [323]. In a recent study, Wang et al. found that the glycolytic enolase 2 (ENO2) constitutes a useful predictive biomarker and therapeutic target for resistance to antiangiogenic therapy in CRC and revealed a previously undefined and metabolism independent role of the ENO2‐derived metabolite phosphoenolpyruvic acid in regulating resistance to antiangiogenic therapy through its role as an endogenous HDAC1 inhibitor [324]. Yang et al. showed that PCI‐34051, a selective HDAC8 inhibitor, enhanced the efficacy of antitumor immunity and immune checkpoint blockade in HCC in a mouse model [325]. Similarly, Mormino et al. found that HDAC8 regulates human and mouse glioma cell viability and tumor migration through α‐microtubulin acetylation and inhibits NK cell‐mediated cytotoxic activity. Inhibition of HDAC8 by the specific inhibitor PCI‐34051 reduced tumor volume in a mouse model of glioma [326]. Psilopatis et al. indicated that HDAC inhibitors (apicidin, tricosuppressor TSA, and pabinostat LBH589, among others) inhibited tumor growth in vitro and in vivo, enhanced transcription of silenced physiological genes and induced cell cycle arrest and apoptosis in endometrial cancer cells, which could be a promising therapeutic alternative for endometrial cancer [327]. Jia et al. found that LAQ824, a novel pan‐HDAC inhibitor, inhibits PDAC progression and suppresses immune escape by promoting antigen presentation, providing a new strategy for targeting PDAC between epigenetic regulation and immunogenicity [328].
In addition, CBP/P300 is the most well‐studied acyltransferase that mediates multiple types of acylation on histones and nonhistone proteins. Nguyen et al. have determined that acK13–HOXB13 mediated by the HAT CBP/p300 is synergistic with the lineage specificity of a key depot‐resistant CRPC target and with the tumor‐promoting SE of H3K27 acetylation, thus acting as an epigenetic regulator of tumor growth. PSMA‐targeting agents and (R)‐9b may be novel therapeutic modalities for targeting HOXB13–ACK1 axis‐regulated PCs [329]. Welsh et al. showed that the P300 inhibitor GNE‐781, by blocking SE accessibility and inhibiting Myc, IRF4 expression, sensitizing MM to immunomodulatory imide drugs [330]. The above studies suggest that anticancer drugs targeting histone acetylation exert anticancer effects through global modulation of chromatin accessibility, but optimization of selectivity and combination therapy regimens is required.
Through these studies, we can determine that drugs targeting histone modification factors provide a new paradigm for cancer therapy by precisely regulating chromatin accessibility and gene expression. Although EZH2 and HDAC inhibitors, among others, have been successfully translated to the clinic, there is still a need to overcome drug resistance, optimize targeting, and explore combination strategies. Future studies should focus on subtype‐selective inhibitors and epigenetic–immunologic synergistic therapies to expand the beneficiary population.
Targeted Chromatin Remodeling Complexes
4.2.3
Chromatin remodeling complexes can serve as emerging therapeutic targets by regulating chromatin structure and accessibility in an ATP‐dependent manner, affecting gene transcription, DNA repair, and cell fate decisions [331].
Targeting SWI/SNF Complexes
4.2.3.1
The SWI/SNF complex facilitates the exposure of gene regulatory regions by sliding nucleosomes. Notably, mutations in genes encoding subunits of the SCRC are present in more than 20% of human cancers [331]. ARID1A is a core component of the SWI/SNF complex, and Cui et al. showed through their study that the inhibitor of arachidonic acid metabolism, aspirin, selectively inhibits the growth of ARID1A‐deficient CRCs, sensitizing tumors deficient in ARID1A to immunotherapy, providing a promising therapeutic strategy [332]. ARID1B is another ARID1 subfamily member, and ARID1A proteins are mutually exclusive components of the SWI/SNF complex [333]. Zhu et al. found that mutations in the SWI/SNF gene ARID1B lead to impaired DNA damage response and repair as well as altered chromatin accessibility. Notably, NSCLC patients harboring ARID1B mutations exhibited better OS and progression‐free survival following immune checkpoint inhibitor (ICI) therapy, indicating a potential predictive role for ARID1B mutations in ICI response. These findings shed light on the biological and therapeutic significance of ARID1B in lung cancer, emphasizing its potential as a target for precision medicine and immunotherapeutic strategies [306].
All SWI/SNF complexes contain the SMARCA family as the catalytic subunit of the ATPase that drives nucleosome sliding and expulsion [334]. A study on bladder cancer showed that loss of the SWI/SNF complex subunit SMARCB1 increased chromatin accessibility of the STAT3 locus in vitro and drove disease progression in bladder cancer patients. pSTAT3‐selective inhibitor TTI‐101 reduced xenografts originating from the SMARCB1 KO cell line of origin and xenotransplantation originating from SMARCB1‐deficient patient tumor growth in the model and demonstrated in several preclinical models that targeting the IL6/JAK/STAT3 molecular pathway is a potential therapeutic approach for SMARCB1‐deficient bladder cancer [243]. Redin et al. identified a role for SMARCA4, the catalytic subunit of the SWI/SNF complex, as a regulator of metastasis in subtypes of SCLC, with altered chromatin accessibility and enhances NE programs. In addition, the SMARCA4 inhibitor FHD‐286 (Foghorn Therapeutics) drives ERBB pathway activation in SCLC and sensitizes SCLC tumors to afatinib. Ultimately, they designated SMARCA4 as a key regulator of NE state plasticity and identified novel therapeutic strategies for SCLC [335]. With regard to FHD‐286, another study found it to be an orally bioavailable and selective BRG1/BRM inhibitor, which reduced AML burden, improved survival, and attenuated the AML initiation potential of stem cell progenitor cells [336].
Other studies have found that PFI‐3, a recently developed bromodomain inhibitor specifically targeting SWI/SNF chromatin remodelers, effectively blocked chromatin binding of its target bromodomain and dissociated the corresponding SWI/SNF proteins from chromatin, sensitizing several human cancer cell lines to DNA damage induced by chemotherapeutic agents such as adriamycin [337]. Panditharatna et al. inhibited glioma progression by linking the core of the BRG1 ATPase inhibitor to phthalimide and creating a degrader, JQ‐dS‐4. A new strategy for epigenetically targeted therapy of pediatric gliomas targeting the lethal type of ATPase activity of the BAF complex is shown [338]. In conclusion, SWI/SNF subunits are potential therapeutic targets for a wide range of cancers, and further understanding of the exact role of SWI/SNF complex subunits in cancer is needed to further develop new strategies against drug resistance and targeting specificity and to address precision therapy in the context of complex mutations.
Targeting Other Complexes
4.2.3.2
In addition, targeting therapy regarding other chromatin remodeling complexes has been partially investigated in recent years. Chromatin structural domain deconjugating enzyme DNA‐binding protein 4 (CHD4) is a core member of the NuRD complex, and Oyama et al. noted that the dual SMARCA5/CHD4 inhibitor ED2–AD101 sensitized OC cells to cisplatin by decreasing the expression of multidrug resistance 1 (MDR1) [339]. Graca Marques et al. found that the NuRD subunit CHD4 serves as a therapeutic target for Ewing sarcoma, and that CHD4 inhibitors led to an overall increase in DNA accessibility and induction of spontaneous DNA damage, resulting in increased susceptibility to DNA damaging agents. This resulted in inhibition of tumor growth and improved OS [340]. Xu et al. found that BPTF (the largest subunit of the ISW1 complex) is a chromatin remodeling factor in NSCLC, and that an inhibitor of the bromodomain of BPTF called C620‐0696, which inhibits the progression of NSCLC primarily by inhibiting c‐Myc transcription [341]. Nano et al. indicated that sorafenib, a potent inhibitor of RUVBL1 and RUVBL2, which are important members of the chromatin remodeling complex INO80, can inhibit the ATPase activity of the RUVBL1/2 complex, which provides research support for targeting chromatin remodeling complexes [342].
Therefore, we believe that the strategy of targeting chromatin remodeling complexes provides a new dimension for cancer therapy by modulating chromatin accessibility. And more research is still needed in the future to drive the progress of this field toward personalized therapy.
Targeted Transcription Factors
4.2.4
Transcription factors play a crucial role in determining the fate of cells during development and cancer progression [343]. In recent years epigenetic drugs have become an emerging strategy for cancer therapy by targeting the functional domains of transcription factors or modulating the epigenetic modifications they bind.
Li et al. observed that CRC cells metastasized to the liver showed enrichment of HNF4A, a liver‐specific transcription factor belonging to the HNF family. The epigenetic shift favoring liver tropism and its effects on immune response, metabolism, and malignancy heterogeneity of liver metastatic CRC cells at single‐cell resolution were elucidated. These findings reveal a critical role for HNF4A, which may provide an important therapeutic target for liver metastasis and enhancement of immunotherapeutic response in CRC patients [240]. Wang et al. showed that the transcriptional repressor ZBTB18 is a potent chromatin modulator, and loss of its activity enhances chromatin accessibility and transcriptional adaptation, thereby promoting phenotypic changes required for metastasis. In contrast, restoration of ZBTB18 activity reduces chromatin accessibility to promoters of genes that drive metastasis, such as Tgfbr2, thereby preventing activation of the TGFβ1 pathway, which in turn reduces cell migration and invasion [344].
In terms of specific drugs, Holmes’ study led to the discovery of MYC as a transcription factor, and its inhibitor MYCi975 selectively altered the MYC and MAX cis‐groups and modulated the epigenomic landscape to regulate target gene expression, sensitizing drug‐resistant PC cells to ENZ, and estrogen‐receptor‐positive breast cancer cells to 4‐hydroxytamoxifen [345]. Chen et al. found that the master transcription factor ONECUT2 regulates gene expression by a mechanism that includes promoter binding, enhanced genome‐wide chromatin accessibility, and SE reprogramming. The OC2 inhibitor CSRM‐617 suppressed the genealogical plasticity reprogramming induced by the AR signaling inhibitor ENZ, and targeted inhibition of OC2 may play an important role in blocking or delaying the emergence of desmoplasma‐resistant PC [285]. In a similar study, Leppanen et al. found that the AR inhibitor ENZ increased chromatin accessibility and expression of the cognate frameshift transcription factor SIX2, which promotes stem‐like reprogramming, NE differentiation, and AR inhibitor resistance, suggesting that targeted SIX2 inhibition may represent a promising approach to overcoming the cellular plasticity mechanisms a promising therapeutic strategy; however, as a transcription factor, direct inhibition of SIX2 may be challenging [286].
Through these studies, we can easily see that epigenetic drugs targeting transcription factors provide new options for cancer therapy by interfering with transcription complex assembly or function and specifically modulating chromatin accessibility of oncogenes.
Targeting ncRNAs
4.2.5
Although ncRNAs do not encode proteins, they play a key role in epigenetic regulation. In recent years, epigenetic drugs targeting ncRNAs have provided new directions for precision cancer therapy by directly or indirectly modulating their functions. Based on the latest research advances, this section discusses the clinical applications of these drugs and the challenges associated with their translation.
Tang et al. reported that LINC01057 as lncRNA is a regulator of NF‐κB signaling, leads to effective chromatin accessibility at NF‐κB‐responsive promoters, promotes MES differentiation, and is a potential target for therapeutic intervention in the MES subtype of GBM. LINC01057 inhibition suppresses proliferation, invasion, and radioresistance of GBM cells in vitro and in vivo inhibited tumor growth [346]. Zhang et al. identified LINC01977 as a cancer testis lncRNA that was hijacked by SEs, which promoted proliferation and invasion in vitro and in vivo. In early‐stage LUAD, higher chromatin accessibility and high TGF‐β expression were observed in the SE region of LINC01977, and they suggested that LINC01977 hijacked by SE may be a valuable therapeutic target, especially for the treatment of early‐stage LUAD [231]. A study on breast cancer progression found that Malat1 lncRNA plays a key role in regulating breast cancer pathogenesis, and that treatment of Malat1‐specific ASO resulted in a dramatic shift of breast tumors to a highly differentiated, less invasive state, representing a potential therapy to inhibit breast cancer progression. It also indicates that future use of patient‐derived xenograft models as well as organoids can further investigate the efficacy of Malat1 ASO as a therapeutic agent to reduce tumor progression [347]. Ye et al. discovered a novel magnetic thermosensitive cationic liposome drug carrier for codelivery of oxaliplatin (OXA) and antisense lncRNA of MDC1 (MDC1‐AS) to cervical cancer cells, which enhances in vitro and in vivo inhibition of the growth of cervical cancer cells and serves as a novel targeted therapeutic system for cervical cancer [348]. Several other studies have also corroborated that circRNA vaccines and circRNA‐based therapeutic platforms have superior applications in the treatment of melanoma [349, 350, 351].
The prospect of targeting ncRNAs for the treatment of cancer is clear, although more research is needed in the future that needs to be further validated to drive the translation of this field to the clinic.
The above multiple studies on therapeutic targets suggest that epigenetic drugs targeting chromatin accessibility provide a multidimensional intervention strategy for cancer therapy by intervening in DNA methylation, histone modification, chromatin remodeling, transcription factors, and ncRNAs. Despite the challenges of selectivity and drug resistance, the development of precision editing technologies and the integration of multiomics analyses hold great promise for translating personalized epigenetic therapies based on chromatin dynamics into clinical practice in the future. This advancement is expected to drive cancer treatment into the “era of epigenetic precision.” Representative drugs targeting different targets are summarized in Table 1.
Chromatin Accessibility in Combination Therapy
4.3
Based on the above review, we can see that chromatin‐accessible targeted therapeutics have become an important strategy for cancer treatment by modulating epigenetic modifications to remodel chromatin structure. However, there are still challenges such as selectivity and drug resistance. Therefore, the research direction in recent years has also gradually favored its combination with chemotherapy, immunotherapy, targeted therapy, and radiotherapy to exert synergistic effects (Figure 4). We also summarized current clinical trials of different epigenetic drugs for combination therapy of cancer (Table 2).
Chromatin accessibility in cancer therapeutic targets and combination therapy strategies. (A) Targeting DNA methylation: Zebularine specifically sensitizes the cGAS–STING pathway and promotes CD8 T‐cell and NK‐cell infiltration, thereby enhancing antitumor immunity. (B) Targeting histone modification: The EZH2 inhibitor Tazemetostat enhances lipid ROS accumulation by epigenetically modulating genes associated with cysteine metabolism and ferroptosis suppression. (C) Targeting chromatin remodeling complexes: ARID1B deficiency leads to impaired DNA damage response and activation of the cGAS–STING pathway in NSCLC; an inhibitor targeting the BRG1–BAF complex reduces the level of H3K27M, which reduces the maintenance of glioma stem cells in an oligodendrocyte precursor cell‐like state. (D) Targeting transcription factors: (1) MYCi975 disrupts the dimerization structure of MYC and MAX, thereby reducing H3K27ac levels and target gene expression; (2) targeting inhibitors of SIX2 downregulates the Wnt/β‐catenin pathway and induces mesenchymal–epithelial transition (MET). These two mechanisms sensitize drug‐resistant prostate cancer to enzalutamide. (E) Targeting noncoding RNA: Inhibition of LINC01977 hijacked by a super‐enhancer reduces epithelial–mesenchymal transition by downregulating the TGF‐β/SMAD3 pathway. (F) Combination of epigenetic drugs with chemotherapy, immunotherapy, targeted therapy, and radiotherapy.
Combination Chemotherapy
4.3.1
In terms of combination chemotherapy, Gu et al. found that the PRMT1 controls chromatin accessibility related to cancer cell metabolism and is a key regulator of pancreatic cancer development. Combining its inhibitors (FM and TCE) with gemcitabine represents an effective therapeutic approach for patients with pancreatic cancer, particularly those with Gem resistance [214]. Following a study by Oyama et al. who found that CHD4 mediates platinum sensitization by modulating MDR1 expression in OC, the CHD4/SMARCA5 inhibitor ED2–AD101 and cisplatin showed synergistic effects, suggesting that CHD4 inhibition has the potential to be a novel therapeutic strategy in combination with platinum agents [339]. Brown et al. found that sequential treatment with DNMTi azacitidine and carboplatin slowed high‐grade plasmacytoid OC cell growth, as well as demethylated and upregulated pathways involved in the immune response, suggesting that this combination could be used to increase the response of patients who are resistant to multiple therapies, such as platinum‐based ICIs [378].
Combination Immunotherapy
4.3.2
In terms of combination immunotherapy, Weng et al. found that the DNMTi decitabine enhanced the formation of immune synapses between ex vivo expanded allogeneic γδ T cells and cancer cells and enhanced the antitumor immunity of γδ T cells. This result supports the potential of combining DNMTis with γδ T cell‐based immunotherapy for lung cancer treatment [379]. Li et al. used a mouse model to show that the combination of decitabine and anti‐PD‐L1 therapy was effective in reducing RCC tumor load, particularly in SETD2‐deficient renal cancers, which underscores the synergistic potential [380]. Similarly, Zhang et al. found that DNMTi zebularine stimulated cGAS–STING–NF‐κB/IFNβ signaling to enhance tumor cell immunogenicity, thereby promoting effective CD4+ and CD8+ T cell‐mediated tumor cell killing. This finding supports the use of combination regimens including DNMTi and immunotherapy for cancer treatment [381]. Bear found that treatment with decitabine and pembrolizumab in the neoadjuvant preadjuvant window sensitized breast cancer to standard NCT by recruiting TIL to tumor tissue and was well tolerated by treatment [382].
Combination Targeted Therapy
4.3.3
In terms of combination targeted therapy, Redin et al. found that the SMARCA4 inhibitor FHD‐286, a subunit of the SWI/SNF complex, in combination with afatinib, showed significant effects in slowing SCLC tumor growth compared with the two drugs alone, even in PDX originating from tumors after several lines of treatment, supporting the potential of this combination as a therapeutic strategy for SCLC [335]. Shim et al. evaluated a combination strategy of olaparib and DNMTi 5‐AZA in epithelial OC (EOC) cells with a significant antitumor effect, suggesting that this combination therapy may be a potential therapeutic strategy for EOC [383]. Lai et al. found that the selective DNMT3b inhibitor, nanomycin A, significantly increased the sensitivity of HCC cells to sorafenib, and that targeting DNMT3b showed synergistic effects with sorafenib in the treatment of sorafenib‐resistant HCC, providing a therapeutic strategy for patients with HCC that expresses cancer stemness traits [353]. After Gomez's study, it was found that the DNMTs and the ADAR1 coinhibition reduced tumor load and prolonged survival in an immunocompetent mouse model of OC. This suggests that combining epigenetically induced transposable element transcription with inhibition of RNA editing represents another novel therapeutic strategy [312].
Combination Radiotherapy
4.3.4
In terms of combination radiotherapy, Sun et al. showed that NMS‐P118, an inhibitor targeting METTL3, inhibited the growth of xenograft tumors in mice and exhibited synergistic effects with in vivo radiotherapy, establishing a novel radiotherapy combination therapy strategy by inactivating the PARP‐1–METTL3–LPAR5 axis [384]. Abbotts et al. found that DNMTis induced a BRCAness phenotype that sensitized NSCLC to PARP inhibitors and ionizing radiation [385]. Ullrich et al. found that decitabine alone and in combination with valproic acid VPA increased SSTR2 levels in pheochromocytomas as well as uptake of radiotracer in vitro, a study that demonstrated that epigenetic modifiers and peptide receptor radionuclide therapy combination has potential for clinical application [386].
Collectively, these findings indicate that combining chromatin accessibility‐targeted agents with conventional therapies markedly improves anticancer efficacy through multimechanistic synergy. Preclinical and early clinical trials have validated its potential, but further solutions are needed to address toxicity management, drug resistance mechanisms, and individualized treatment regimen design. Future research should focus on precise combination strategies and translational biomarkers to advance this field into clinical practice. Furthermore, we utilized the STRING database (https://string‐db.org/) to investigate the interactions between certain key proteins or molecules involved in regulating chromatin accessibility in this study (Figure 5).
Protein–protein interaction (PPI) network involving distinct proteins affecting chromatin accessibility. This protein interaction network was constructed based on the STRING database, encompassing 50 core proteins extensively studied to date (including those frequently cited herein). The roles of these proteins in regulating chromatin accessibility have been thoroughly investigated. Each node represents a protein; larger nodes typically denote hub proteins within the network, potentially possessing significant regulatory functions. Connecting lines symbolize interactions between proteins, with thicker lines indicating stronger and more reliable evidence supporting the interaction.
Therapeutic targeting of chromatin accessibility is rapidly advancing from preclinical validation toward clinical application. To comprehend the efficacy, limitations, and translational potential of these strategies, critical evaluations across diverse model systems are required. Mouse models (e.g., gene‐edited mice, PDX) have played a vital role in elucidating mechanisms and assessing in vivo efficacy, while emerging human models such as PDOs and 3D bioprinted tumors replicate human tumor biology and microenvironmental features with unprecedented precision. The table below synthesizes findings from these diverse models, highlighting therapeutic principles, species‐specific variations, and the critical role of advanced human models in mitigating clinical translation risks (Table 3).
Conclusion and Outlook
5
Summaries and Shortcomings
5.1
In this review, we systematically elaborate on the structural basis and dynamic regulatory mechanisms of chromatin accessibility. Existing studies have demonstrated that chromatin accessibility, acting as a “gateway” to epigenetic regulation, is modulated by a variety of epigenetic regulatory mechanisms. Herein, we summarize the mechanisms of DNA methylation, histone modification, ncRNA, chromatin remodeling complexes, transcription factors, and chromatin 3D structure that regulate chromatin accessibility and thus affect tumor cell fate. In addition, this review explores the multidimensional role of chromatin accessibility in tumorigenesis and progression, including tumorigenesis, progression, metabolic reprogramming, angiogenesis, tumor stemness, immunity and microenvironment, and tumor drug resistance. Notably, existing studies are mostly based on traditional tissue‐level high‐throughput sequencing technologies (e.g., ATAC‐seq), which are difficult to portray chromatin accessibility heterogeneity at the single‐cell level in the tumor microenvironment. Although single‐cell multiomics technologies (scATAC‐seq combined with scRNA‐seq) and spatial transcriptomics (e.g., Stereo‐seq) have been studied by some scholars but have not yet been popularized [406, 407, 408], it is difficult to reveal the epigenetic basis of cell‐to‐cell interactions in the tumor microenvironment. The integration of multiomics data can resolve the driving mechanisms of accessibility changes in specific tumor types and reveal cell subpopulation‐specific regulatory networks. In addition, the dynamic regulation mechanism of chromatin accessibility is ambiguous and lacks real‐time tracking tools. A combination of real‐time dynamic monitoring techniques, such as single‐molecule live‐cell RNA imaging using CRISPR–Csm [409] and time‐resolved epigenomics (CUT&Tag time‐series analysis) [410, 411], is needed to reveal the molecular mechanisms at a deeper level.
Moreover, this review provides a comprehensive discussion around chromatin‐accessible tumor markers, epigenetic drug development, targeted therapies and combination strategies. In terms of therapeutic strategies, drugs targeting epigenetic nodes such as DNA methylation (e.g. decitabine) [311], histone modification (e.g. the EZH2 inhibitor Tazemetostat) [255], chromatin remodeling complexes (e.g. the SWI/SNF inhibitor FHD‐286) [335], and ncRNAs (e.g. MALAT1 ASO) [347] and other epigenetic nodes are in clinical or preclinical studies. Combination chemotherapy, ICIs, and other regimens have shown synergistic potential [214, 335, 380]. Due to space constraints, this review does not address the side effects of epigenetic drugs.
However, the efficacy of epigenetic drugs (e.g., DNMT, HDAC inhibitors) is limited by the epigenetic plasticity of tumor cells. Considering that current studies are mostly based on cellular and animal models, we believe that the establishment of an organoid model will be helpful to gain a deeper understanding of the dynamic regulatory mechanisms of chromatin accessibility and to develop precise therapeutic strategies targeting the open regions of chromatin [412, 413]. In addition, although the excellent anticancer efficacy of epigenetic drugs has been demonstrated, the development of highly selective inhibitors, new strategies for drug resistance, and target specificity requires further investigation. Moreover, although some studies have been conducted using combination strategies with different target epigenetic drugs [269, 314], more research is needed to provide theoretical support at the mechanistic level and to provide more optimized combination strategies.
In addition, the current therapeutic hotspots focus on the combination of epigenetic drugs with conventional therapies (chemotherapy, immunotherapy, targeted therapy, and radiotherapy) [378, 382, 383, 384], which can significantly improve the anticancer efficacy through the synergy of multiple mechanisms. Preclinical and early clinical trials have validated its potential, but large‐sample cohort studies are still needed to validate it through both mechanistic studies and clinical trials. Future research should focus on precise combination strategies to advance this field toward clinical translation and promote cancer treatment into the “era of individualized epigenetic precision.”
Challenges and Prospects
5.2
As research advances, novel research strategies and techniques are now making more significant breakthroughs as research continues to progress.
Proteolysis‐targeting chimera (PROTAC), which enables functional studies of chromatin regulatory factors by inducing targeted protein degradation, has emerged as a valuable tool and garnered considerable attention from researchers in academia and the medical field [414]. PROTAC, as a pioneering novel therapeutic approach, redefines the principles of traditional drug discovery. It operates in an event‐driven manner, inducing ubiquitin modification for the clearance of pathogenic proteins through transient binding mechanisms based on the ubiquitin–proteasome system [415]. The PROTAC technology has been successfully used for the degradation of many oncogenic targets to overcome drug resistance, such as targeting AR in ENZ‐resistant PC [416], targeting ER in drug‐resistant breast cancer [417], targeting BTK in ibrutinib‐resistant lymphomas [418], and targeting proteins in BET denervation‐resistant PC [419, 420]. Moreover, combination therapy of PROTAC with epigenetic drugs has also shown promise, such as HDAC–PROTAC (SIRT2 degrader), which can provide higher selectivity and specificity while reducing off‐target effects [421]. Moreover, PROTAC technology opens a new chapter in precision therapy by transforming traditionally nondruggable proteins into interventional targets through a “catalytic degradation” mechanism [414].
However, PROTACs face substantial hurdles in becoming a successful drug discovery approach, primarily related to uncertainty in efficacy, technical challenges, and high development costs. Given the relatively small number of clinical candidates available, it remains to be seen whether PROTAC will become a clinically useful anticancer drug, and more in vitro data and pharmacokinetic studies are needed to pave the way for its application in the clinic. Therefore, the great potential of PROTAC technology in cancer therapy remains to be more fully explored and exploited.
In addition, in recent years, CRISPR and CRISPR‐associated proteins have become a revolutionary gene editing tool, and the CRISPR/Cas technology has also demonstrated great potential in tumor therapy by systematically targeting specific gene loci and regulating chromatin accessibility [422]. A team from Zhejiang University developed a cryo‐shock tumor cell delivery CRISPR–Cas system that triggered synthetic lethality and prolonged survival in mice by CDK4 ablation in KRAS‐mutant NSCLC [423]. Several studies have demonstrated that CRISPR/Cas gene editing can be used to overcome drug resistance in a variety of cancers and that different resistance mechanisms can be targeted using this technology [424, 425, 426, 427]. In addition, in vivo genome‐wide CRISPR screening can be used to identify targets that modulate drug resistance or immunotherapy sensitivity in a variety of cancers. using genome‐wide CRISPR screening, Cao's team found that CD28 in cancer cells promotes immune escape by stabilizing PD‐L1 mRNA, providing a novel target for overcoming anti‐PD‐1 resistance [428]. Belk et al. identified a new target for anti‐PD‐1 resistance through a series of in vitro and in vivo CRISPR–Cas9 screening systems to identify chromatin remodeling factors that limit T‐cell persistence and demonstrate that modulation of epigenetic status improves T‐cell responses in cancer immunotherapy.
But translating CRISPR/Cas to in vivo gene editing poses significant challenges that need to be addressed, including concerns about specificity, safety, and efficient delivery [429]. Based on several studies, Xu et al. suggest that nanotechnology‐based delivery of CRISPR/Cas9 for cancer gene editing and immunotherapy paves the way for its clinical translation, and that nanotechnology‐based delivery of gene editing using CRISPR/Cas9 is a new dawn in the field of cancer therapy [430]. Thus, although still in its infancy, the CRISPR system offers great promise in the discovery of important cancer genes and therapeutic targets and will continue to play an irreplaceable role in the future.
Nowadays, with the generation‐by‐generation development of artificial intelligence, the integration of single‐cell multiomics and artificial intelligence, interdisciplinary fusion, and big data‐driven are becoming increasingly prominent. The scBridge algorithm developed by Sichuan University effectively overcomes the interference of cellular heterogeneity on multiomics analysis by iteratively integrating scRNA‐seq and scATAC‐seq data [406]. The construction of AI prediction platforms, such as the training of deep learning models based on the The Cancer Genome Atlas and ICGC databases (e.g., XGraphCDS, scMultiomeGRN) [431, 432], has enabled the prediction of patient‐specific chromatin accessibility targets and drug sensitivity from genetic pathways, realizing a leap from “static typing” to “dynamic management” in cancer therapy.
PROTAC and CRISPR technologies are driving chromatin accessibility research from descriptive analysis to precision intervention, but their clinical translation is still limited by delivery efficiency, editing accuracy, and multiomics integration capability. In the future, it is necessary to break through the technical bottleneck through interdisciplinary cooperation, to realize the personalized epigenetic therapy of “time‐space‐target,” to promote the chromatin accessibility markers from scientific research to the clinical leap, and ultimately to rewrite the clinical practice of tumor treatment.
Conclusion
6
Chromatin accessibility, as a core hub of epigenetic regulation, has been gradually transformed from basic mechanism investigation to clinical application and is moving from static description to dynamic resolution and precise intervention. Despite the challenges of heterogeneity, technical resolution, and therapeutic safety, with the rapid development of single‐cell technology, PROTAC‐targeted therapy, and CRISPR gene editing tools, it is expected to realize a new paradigm of tumor therapy that emphasizes both spatio‐temporal regulation and individualized intervention. However, there are still many challenges in the complexity, technical bottleneck, and clinical translation of tumor epigenetic regulation. Multiomics integration, artificial intelligence, and interdisciplinary innovation will be the key to break through the current bottlenecks, which is expected to reveal the panoramic regulatory network of chromatin accessibility in tumorigenesis and development, and ultimately promote the epigenetic‐based precision tumor therapy to enter into a new stage of comprehensive leap (Figure 6).
Future perspectives and clinical translation of chromatin accessibility researches. Prognostic modeling based on chromatin accessibility, future perspectives on chromatin accessibility research through multiomics integration and emerging technologies, and its translation into clinical trials and prognostic analysis.
Author Contributions
Wentao Xia, Kun Ding, and Yansu Chen drafted the manuscript and prepared the figures. Min Jiang and Yefei Huang revised the manuscript text. All authors have read and approved the manuscript.
Funding
This study was supported in part by the National Natural Science Foundation of China (82473588), the 2023 Qing Lan Project (Chen Yansu), the Graduate Research Innovation Plan of Jiangsu Province (KYCX23_2964), the Natural Science Foundation of Jiangsu Province (BK20241038), the Natural Science Fund for Colleges and Universities in Jiangsu Province (24KJD320005), and the General Program of Basic Science (Natural Science) Research for Colleges and Universities in Jiangsu Province (24KJB330010).
Conflicts of Interest
The authors declare no conflicts of interest.
Ethics Statement
The authors have nothing to report.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1P. A. Jones and S. B. Baylin , “The Epigenomics of Cancer,” Cell 128, no. 4 (2007): 683–692.17320506 10.1016/j.cell.2007.01.029PMC 3894624 · doi ↗ · pubmed ↗
- 2S. Zhou , A. E. Treloar , and M. Lupien , “Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations,” Cancer Discovery 6, no. 11 (2016): 1215–1229.27807102 10.1158/2159-8290.CD-16-0745 PMC 5117653 · doi ↗ · pubmed ↗
- 3T. Chiba , H. Marusawa , and T. Ushijima , “Inflammation‐associated Cancer Development in Digestive Organs: Mechanisms and Roles for Genetic and Epigenetic Modulation,” Gastroenterology 143, no. 3 (2012): 550–563.22796521 10.1053/j.gastro.2012.07.009 · doi ↗ · pubmed ↗
- 4C. H. Waddington , “The Epigenotype. 1942,” International Journal of Epidemiology 41, no. 1 (2012): 10–13.22186258 10.1093/ije/dyr 184 · doi ↗ · pubmed ↗
- 5T. B. Toh , J. J. Lim , and E. K. Chow , “Epigenetics in Cancer Stem Cells,” Molecular Cancer 16, no. 1 (2017): 29.28148257 10.1186/s 12943-017-0596-9PMC 5286794 · doi ↗ · pubmed ↗
- 6A. Zhao , H. Zhou , J. Yang , M. Li , and T. Niu , “Epigenetic Regulation in Hematopoiesis and Its Implications in the Targeted Therapy of Hematologic Malignancies,” Signal Transduct Target Ther 8, no. 1 (2023): 71.36797244 10.1038/s 41392-023-01342-6PMC 9935927 · doi ↗ · pubmed ↗
- 7V. Davalos and M. Esteller , “Cancer Epigenetics in Clinical Practice,” CA: A Cancer Journal for Clinicians 73, no. 4 (2023): 376–424.36512337 10.3322/caac.21765 · doi ↗ · pubmed ↗
- 8S. L. Klemm , Z. Shipony , and W. J. Greenleaf , “Chromatin Accessibility and the Regulatory Epigenome,” Nature Reviews Genetics 20, no. 4 (2019): 207–220.