First high-quality genome assembly of Umbelopsis nana isolated from forest soil
Ryuka Iizuka, Tomohiro Suzuki, Nozomu Watanabe, Javier F Tabima, David Hibbett, Yoko Katayama, Makoto Yoshida

TL;DR
This paper presents the first high-quality genome assembly of the soil fungus Umbelopsis nana, offering insights into its ecological and biotechnological potential.
Contribution
The study provides the first publicly available genome sequence for Umbelopsis nana, filling a gap in the genus' phylogeny.
Findings
The genome assembly consists of 18 contigs totaling 27.2 Mb with an N50 of 1.77 Mb.
16 contigs were flanked by telomeric repeats, and the assembly showed 98.1% complete Benchmarking Universal Single-Copy Orthologs.
Functional annotation predicted 9,068 genes, offering a foundation for studying the genus' evolutionary and functional diversity.
Abstract
Species of the genus Umbelopsis are ubiquitous soil saprotrophic fungi known for their role in ecological nutrient cycling and biotechnological potential in lipid production. Here, we present the genome assembly of Umbelopsis sp. strain THIF13, which is characterized by its capability to produce carbonyl sulfide. Using PacBio Sequel long-read data, a high-quality genome assembly was generated comprising 18 contigs totaling 27.2 Mb, which is larger than other Umbelopsis genomes, with an N50 of 1.77 Mb. Notably, 16 contigs were flanked by telomeric repeats at both ends. The assembly showed high completeness, with 98.1% complete Benchmarking Universal Single-Copy Orthologs and 11.15% repetitive sequences. Multilocus phylogenetic analysis clearly identified the strain as Umbelopsis nana; thus, this study reports the first publicly available genome sequence for this species. Functional…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
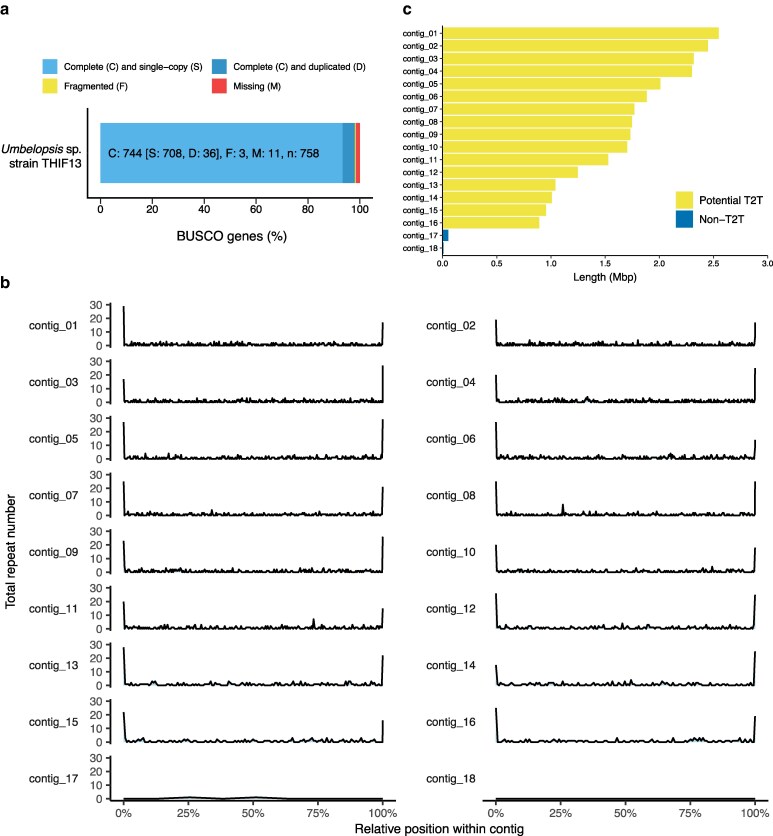 Fig. 1
Fig. 1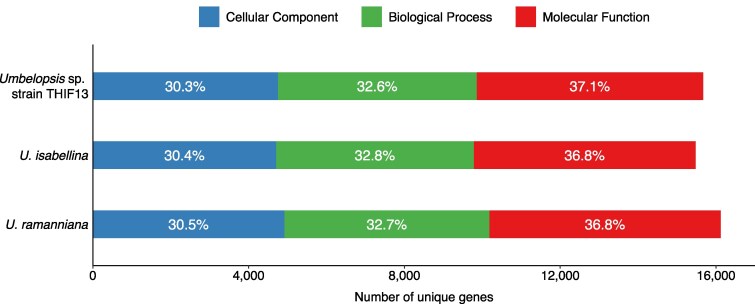 Fig. 2
Fig. 2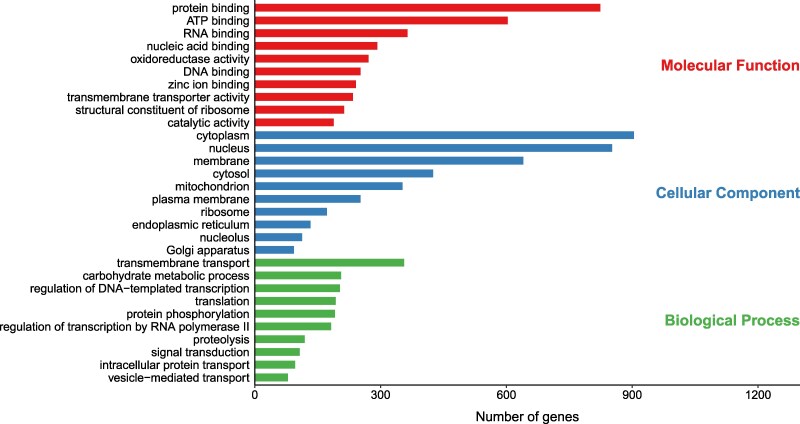 Fig. 3
Fig. 3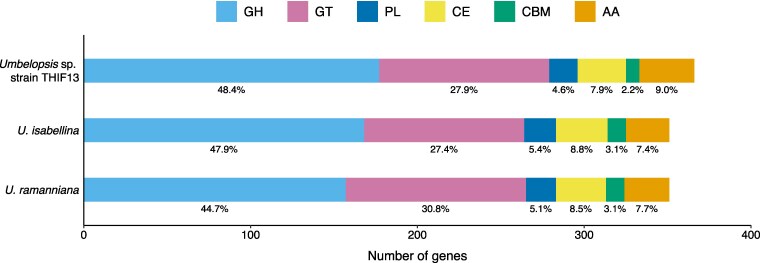 Fig. 4
Fig. 4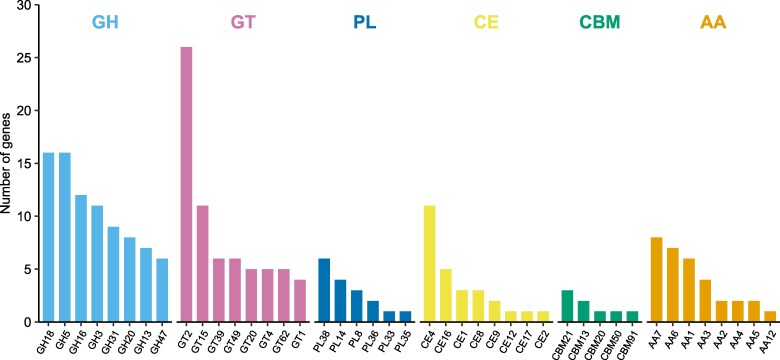 Fig. 5
Fig. 5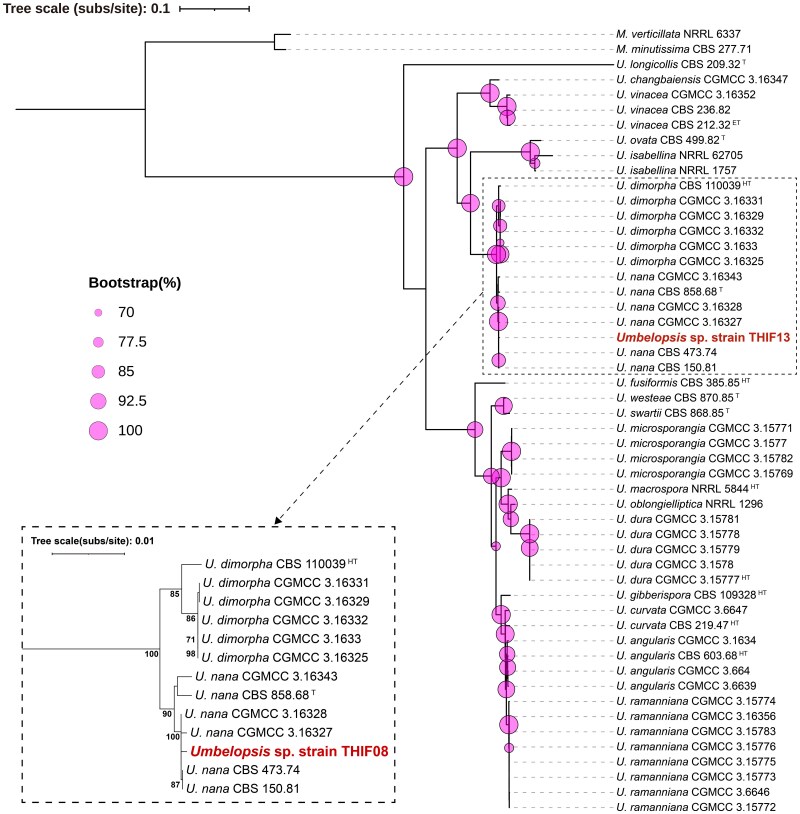 Fig. 6
Fig. 6| Species | Strains | SSU | ITS | LSU |
|
|
|
|---|---|---|---|---|---|---|---|
|
| CBS 277.71 | … | … | ||||
|
| NRRL 6337 | … | … | ||||
| U. angularis | CGMCC 3.664 | ||||||
|
| CGMCC 3.6639 | ||||||
|
| CGMCC 3.1634 | ||||||
|
| CBS 603.68HT | ||||||
|
| CGMCC 3.16347 | ||||||
|
| CGMCC 3.6647 | ||||||
|
| CBS 219.47HT | ||||||
|
| CBS 110039HT | ||||||
|
| CGMCC 3.16325 | ||||||
|
| CGMCC 3.16329 | ||||||
|
| CGMCC 3.1633 | ||||||
|
| CGMCC 3.16331 | ||||||
|
| CGMCC 3.16332 | ||||||
|
| CGMCC 3.15777HT | ||||||
|
| CGMCC 3.15778 | ||||||
|
| CGMCC 3.15779 | ||||||
|
| CGMCC 3.1578 | ||||||
|
| CGMCC 3.15781 | ||||||
|
| CBS 385.85HT | ||||||
|
| CBS 109328HT | ||||||
|
| NRRL 62705 | ||||||
|
| NRRL 1757 | ||||||
|
| CBS 209.32T | ||||||
|
| NRRL 5844HT | ||||||
|
| CGMCC 3.15769 | ||||||
|
| CGMCC 3.1577 | ||||||
|
| CGMCC 3.15771 | ||||||
|
| CGMCC 3.15782 | ||||||
|
| CBS 858.68T | ||||||
|
| CBS 150.81 | ||||||
|
| CBS 473.74 | ||||||
|
| CGMCC 3.16327 | ||||||
|
| CGMCC 3.16328 | ||||||
|
| CGMCC 3.16343 | ||||||
|
| NRRL 1296 | ||||||
|
| CBS 499.82T | ||||||
|
| CGMCC 3.15772 | ||||||
|
| CGMCC 3.15773 | ||||||
|
| CGMCC 3.15774 | ||||||
|
| CGMCC 3.15775 | ||||||
|
| CGMCC 3.15776 | ||||||
|
| CGMCC 3.6646 | ||||||
|
| CGMCC 3.16356 | ||||||
|
| CGMCC 3.15783 | ||||||
|
| CBS 868.85T | ||||||
|
| CGMCC 3.16352 | ||||||
|
| CBS 212.32ET | ||||||
|
| CBS 236.82 | ||||||
|
| CBS 870.85T | ||||||
|
| THIF13 |
| Value | |
|---|---|
| Total contigs | 18 |
| Total assembly size (Mb) | 27.2 |
| Average sequence length (Mb) | 1.51 |
| Median sequence length | 1.72 |
| N50 length (Mb) | 1.77 |
| N25/N75/N90/N95 length (Mb) | 2.32/1.70/1.01/0.95 |
| L50 | 7 |
| GC content (%) | 44.16 |
| Ambiguous bases (Ns) (%) | 0.00 |
| Total repetitive content (%) | 11.15 |
| Genome coverage | 30× |
| Category |
|
|
| |||
|---|---|---|---|---|---|---|
| InterProScan-annotated proteins | 7,635 | (84.2%) | 7,482 | (84.6%) | 7,838 | (85.1%) |
| eggNOG-annotated proteins | 8,129 | (89.6%) | 7,909 | (89.5%) | 8,311 | (90.2%) |
| GO-term-assigned genes | 6,753 | (74.5%) | 6,647 | (75.2%) | 6,950 | (75.4%) |
| Pfam-annotated genes | 6,946 | (76.6%) | 6,877 | (77.8%) | 7,241 | (78.6%) |
| CAZyme-annotated genes | 356 | (3.9%) | 340 | (3.8%) | 343 | (3.7%) |
| MEROPS-annotated genes | 317 | (3.5%) | 319 | (3.6%) | 320 | (3.5%) |
| BUSCOs identified in annotation | 1,291 | (14.2%) | 1,287 | (14.6%) | 1,356 | (14.7%) |
| Total genes | 9,068 | … | 8,839 | … | 9,213 | … |
| KEGG pathway (level B) |
|
|
| |||
|---|---|---|---|---|---|---|
| Signal transduction | 1,057 | (17.49%) | 1,005 | (17.17%) | 1017 | (16.9%) |
| Transport and catabolism | 551 | (9.11%) | 550 | (9.4%) | 570 | (9.47%) |
| Carbohydrate metabolism | 490 | (8.11%) | 443 | (7.57%) | 441 | (7.33%) |
| Cell growth and death | 475 | (7.86%) | 478 | (8.17%) | 463 | (7.69%) |
| Translation | 436 | (7.21%) | 423 | (7.23%) | 446 | (7.41%) |
| Amino acid metabolism | 405 | (6.7%) | 386 | (6.6%) | 411 | (6.83%) |
| Folding, sorting, and degradation | 323 | (5.34%) | 319 | (5.45%) | 326 | (5.42%) |
| Lipid metabolism | 306 | (5.06%) | 300 | (5.13%) | 339 | (5.63%) |
| Glycan biosynthesis and metabolism | 246 | (4.07%) | 225 | (3.84%) | 239 | (3.97%) |
| Metabolism of cofactors and vitamins | 223 | (3.69%) | 207 | (3.54%) | 224 | (3.72%) |
| Replication and repair | 214 | (3.54%) | 232 | (3.96%) | 227 | (3.77%) |
| Energy metabolism | 190 | (3.14%) | 182 | (3.11%) | 181 | (3.01%) |
| Cellular community—eukaryotes | 165 | (2.73%) | 147 | (2.51%) | 150 | (2.49%) |
| Transcription | 158 | (2.61%) | 161 | (2.75%) | 161 | (2.68%) |
| Xenobiotics biodegradation and metabolism | 128 | (2.12%) | 146 | (2.49%) | 149 | (2.48%) |
| Cell motility | 123 | (2.03%) | 111 | (1.9%) | 119 | (1.98%) |
| Metabolism of other amino acids | 118 | (1.95%) | 113 | (1.93%) | 113 | (1.88%) |
| Nucleotide metabolism | 103 | (1.7%) | 105 | (1.79%) | 106 | (1.76%) |
| Biosynthesis of other secondary metabolites | 82 | (1.36%) | 78 | (1.33%) | 78 | (1.3%) |
| Chromosome | 63 | (1.04%) | 60 | (1.03%) | 59 | (0.98%) |
| Metabolism of terpenoids and polyketides | 59 | (0.98%) | 59 | (1.01%) | 59 | (0.98%) |
| Signaling molecules and interaction | 50 | (0.83%) | 48 | (0.82%) | 60 | (1%) |
| Information processing in viruses | 34 | (0.56%) | 35 | (0.6%) | 38 | (0.63%) |
| Cellular community—prokaryotes | 33 | (0.55%) | 27 | (0.46%) | 29 | (0.48%) |
| Membrane transport | 13 | (0.22%) | 12 | (0.21%) | 13 | (0.22%) |
| Total annotated genes | 6,045 | 5,852 | 6,018 | |||
- —JSPS KAKENHI
- —JST10.13039/501100008868
- —Institute of Global Innovation Research in TUAT10.13039/501100009483
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Plant Pathogens and Fungal Diseases · Mycorrhizal Fungi and Plant Interactions
Introduction
Species of the genus Umbelopsis (order Umbelopsidales, subphylum Mucoromycotina) are ubiquitous soil saprotrophs that contribute to lignocellulosic biomass decomposition and nutrient cycling in forest ecosystems. These fungi inhabit terrestrial environments and are primarily isolated from forest soil, but they can also be isolated from the fruiting bodies of mushrooms (Parshikov et al. 1999; Watanabe et al. 2001; Summerbell 2005). Although their ecological role as decomposers in forest ecosystems is well recognized, certain species within Umbelopsis, such as U. isabellina and U. ramanniana, have garnered significant interest for their biotechnological potential, particularly in lipid production (Kamisaka et al. 1993; Pillai et al. 1998; Chatzifragkou et al. 2010; Papanikolaou and Aggelis 2019). The lipids they accumulate are rich in polyunsaturated fatty acids (Gardeli et al. 2017; Hou et al. 2024), making them attractive candidates for sustainable biodiesel and nutritional applications.
Umbelopsis previously included species assigned to Mortierella or Micromucor (Mortierellales) but is now classified in its own order, Umbelopsidales (Amos and Barnett 1966; Yip 1986; Spatafora et al. 2016). Historically, species delimitation in Umbelopsis largely depended on morphological traits such as sporangial structure and colony appearance. However, the subtle variations and often overlapping characteristics among species made it difficult to achieve clear identification based on morphology alone (Meyer and Gams 2003; Sugiyama et al. 2003). Early molecular investigations in Umbelopsis primarily utilized nuclear ribosomal DNA loci, including the internal transcribed spacer (ITS) region and large subunit (LSU) rRNA genes (Meyer and Gams 2003; Sugiyama et al. 2003). However, subsequent research increasingly demonstrated that relying on a single ribosomal DNA marker, such as ITS alone, often lacked the resolution needed for reliable species-level identification and conflicted with morphological data in Umbelopsis (Ogawa et al. 2005, 2011). To address these limitations, protein-coding genes such as actin (ACT) were added to multilocus sequence analyses. The combined use of ribosomal and protein-coding markers (ITS-LSU-ACT) significantly improved resolution, enabling clearer species boundaries and facilitating the description of new taxa (Wang et al. 2014, 2015; Crous et al. 2017; Hou et al. 2024). Moreover, a comprehensive 6-gene phylogeny combining ribosomal DNA with protein-coding loci such as minichromosome maintenance complex component 7 (MCM7) and cytochrome c oxidase subunit I (COX1) proved instrumental in resolving the cryptic U. ramanniana sensu lato species complex (Wang et al. 2022).
Despite their recognized ecological importance, diverse application potential, and advances in phylogenetic understanding, comprehensive genomic resources for Umbelopsis species remain limited. To date, genome sequences have been reported for only a few representative Umbelopsis species (mainly U. isabellina and U. ramanniana), while biological and genomic information on other valid species, such as Umbelopsis nana, remains limited, focusing mainly on morphological and phylogenetic descriptions (e.g. Wang et al. 2015). In this study, we focused on Umbelopsis sp. strain THIF13, which was originally isolated from forest soil and characterized by its capability to produce carbonyl sulfide (COS), a major sulfur-containing gas in the atmosphere (Masaki et al. 2016). However, its precise species identity remained unresolved because initial identification relied solely on the ITS region, which offers insufficient resolution for differentiating closely related species within the genus. Therefore, a high-quality genome assembly was generated using the PacBio Sequel system and multilocus phylogenetic analysis was performed. These analyses identified the strain as U. nana; thus, this study provides the first genomic insight into this species, directly addressing the knowledge gap regarding its evolutionary and functional diversity.
Materials and methods
Mycelium cultivation and DNA sequencing
Umbelopsis sp. strain THIF13 (deposited in the NITE Biological Resource Center (NBRC), National Institute of Technology and Evaluation (NITE), under the accession number NBRC 117090) was originally isolated from forest soil collected in the Karasawa-yama area, Field Museum, Tokyo University of Agriculture and Technology, Tochigi, Japan, and subsequently established via single-spore isolation (Masaki et al. 2016). The strain was cultivated in potato dextrose broth (Becton, Dickinson and Company, Franklin Lakes, NJ) at 25 °C with shaking at 150 rpm for 12 d. Mycelial biomass was harvested by vacuum filtration through filter paper, frozen in liquid nitrogen, and lyophilized. Genomic DNA was extracted using a cetyltrimethylammonium bromide extraction protocol (Doyle and Doyle 1987). Residual RNA was removed by incubation with RNase A (Qiagen, Hilden, Germany). A genomic DNA library was constructed using the [3.0] PacBio Microbial Library kit (PacBio, Menlo Park, CA) and HiFi sequencing was performed on a PacBio Sequel IIe system (PacBio).
Genome assembly, filtering, and evaluation
To evaluate the heterozygosity of the sequenced strain, a k-mer analysis was performed. The PacBio HiFi reads were used to count 21-mers using Jellyfish v2.3.1 (Marçais and Kingsford 2011), and the genome characteristics, including heterozygosity, were estimated using GenomeScope v2.0 (Vurture et al. 2017; Ranallo-Benavidez et al. 2020).
De novo assembly was performed with hifiasm v0.25.0-r726 (Cheng et al. 2021) (Supplementary File 1). Circular contigs were detected and redundant sequences removed using Circlator v1.5.5 (Hunt et al. 2015) (Supplementary File 2). These circular contigs were identified as the putative mitochondrial genome via BLAST searches and were separated from the nuclear assembly. To remove redundant contigs representing putative haplotigs or assembly artifacts, whole-contig alignment was first performed with Mauve v1.1.3 (Darling et al. 2004) to identify containment relationships. Subsequently, redundancy was quantified by contig-vs-contig BLASTN searches using BLAST+ v2.15.0 (Camacho et al. 2009). Contigs showing >99.9% identity over 90% of their length were excluded. Filtered contigs were then scanned for telomeric repeats using tidk v0.2.65 (Brown et al. 2025) within a 6,000 bp window at the contig ends (Supplementary File 3). Assembly continuity metrics, including N50, were calculated with N50Stat.pl from the NGS QC Toolkit v2.3 (Patel and Jain 2012) and assembly completeness was evaluated with Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.7.1 against the fungi_odb10 dataset (Simão et al. 2015).
Repeat sequence prediction
A species-specific repeat library was generated using RepeatModeler2 v2.0.6 (Flynn et al. 2020), which included the LTR structural discovery pipeline (-LTRStruct flag). The resulting repeat library was used with RepeatMasker v4.1.9 (Smit et al. 2015), utilizing RMBlast 2.14.1+ (Camacho et al. 2009) as the search engine to identify and soft-mask repetitive elements throughout the genome, employing the -xsmall option to report only the most significant matches (Supplementary File 4).
Gene prediction and functional annotation
Gene models were predicted using the Funannotate v1.8.17 pipeline (Palmer and Stajich 2020) (Supplementary File 5). First, the genome assembly, previously soft-masked for repetitive elements, was further soft-masked with Tantan v50 (Frith 2011) to optimize gene prediction. Conserved single-copy orthologs identified by BUSCO v5.7.1 (fungi_odb10 dataset) (Simão et al. 2015) were employed to train the ab initio predictors Augustus v3.3.2 (Stanke and Morgenstern 2005), GlimmerHMM v3.0.4 (Majoros et al. 2004), and SNAP v2006-07-28 (Korf 2004), while GeneMark-ES v4.72 (Ter-Hovhannisyan et al. 2008) was self-trained on the assembly. Transcript and protein evidence from U. ramanniana strain AG (Amses et al. 2022), comprising RNA-seq-derived transcripts and annotated protein models downloaded from the JGI MycoCosm portal (Grigoriev et al. 2012, 2014), were aligned to the masked genome. The transcripts were aligned using Minimap2 v2.28-r1209 (Li 2018) to generate an exon-intron hints file. Protein evidence was incorporated by mapping the protein set to the genome, first with a rapid search using DIAMOND v2.1.8 (Buchfink et al. 2015) followed by a more precise alignment with Exonerate v2.4.0 (Slater and Birney 2005) to refine gene structure predictions. Augustus was then run in evidence-aware mode using the hints file, with its parameters further optimized using the –optimize_augustus flag, while GeneMark-ES, GlimmerHMM and SNAP performed pure ab initio predictions. Finally, all evidence-based and ab initio outputs were merged into a nonredundant consensus gene set using EvidenceModeler v1.1.1 (Haas et al. 2008).
The consensus protein set was annotated with InterProScan v5.69–101.0 (Jones et al. 2014) to assign domains, families, and Gene Ontology terms, and with eggNOG-mapper v2.1.12 (Cantalapiedra et al. 2021) using the eggNOG 5.0 database (Huerta-Cepas et al. 2019) via DIAMOND blastp for orthology (Buchfink et al. 2015), COG categories, and pathway mappings. These annotations were imported into Funannotate's annotate module together with the predicted gene models. Funannotate then performed HMMER searches against Pfam v37.0 (Mistry et al. 2021) and dbCAN v12.0 (Zhang et al. 2018), DIAMOND blastp searches against UniProt release 2024_04 (UniProt Consortium 2025) and MEROPS v12.5 (Rawlings et al. 2018), and BUSCO Dikarya model scans, and it standardized gene and product names with Gene2Product v1.94. Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation was retrieved from the eggNOG-mapper v2.1.12 results.
Comparative analyses
For comparative analyses, genome and protein sequence data for U. isabellina strain WA0000067209 (BioProject: PRJNA668042) (Muszewska et al. 2021), and genome, transcript, and protein sequence data for U. ramanniana strain AG (BioProject: PRJNA196032) (Amses et al. 2022) were downloaded from the National Center for Biotechnology Information (NCBI) GenBank database. To ensure consistent annotation standards across all genomes, the repeat prediction workflow and the Funannotate pipeline described above were applied to these datasets.
Phylogenetic analyses
6 loci (SSU rDNA, ITS, LSU rDNA, ACT, MCM7, and COX1) were retrieved from GenBank for 49 Umbelopsis strains. These sequences were concatenated with the sequence of Umbelopsis sp. strain THIF13, resulting in a dataset comprising 50 Umbelopsis strains for analysis. Two Mortierella species were added as outgroups (Table 1). For Umbelopsis sp. strain THIF13, the corresponding sequences for these 6 loci were extracted from its genome assembly by BLASTN searches using BLAST + v2.15.0 (Camacho et al. 2009), as whole-genome data were unavailable for most reference strains. Each locus was aligned independently with MAFFT v7.481 using the L-INS-i algorithm (Katoh and Standley 2013) (Supplementary File 6). The resulting alignments were then subjected to trimming with trimAl v1.4.rev15 using the -gappyout method to remove gappy regions based on automated thresholds (Capella-Gutiérrez et al. 2009). The 6 alignments were concatenated, and a RAxML-style partition file defining locus boundaries was generated. Maximum-likelihood inference (ML) was carried out with IQ-TREE 2 v2.4.0 (Minh et al. 2020), using optimal models for each partition as determined by ModelFinder (Kalyaanamoorthy et al. 2017): TN + F + R2 for SSU, HKY + F + R2 for COX1, HKY + F + I + G4 for ACT, TIM2 + F + G4 for MCM7, TIM3 + F + I + G4 for LSU, and TIM2 + F + G4 for ITS. Node support was evaluated via 1,000 ultrafast bootstrap replicates (Minh et al. 2013; Hoang et al. 2018) and 1,000 SH-like approximate likelihood ratio tests (Anisimova et al. 2011). During each replicate the tree was further optimized by likelihood-based nearest neighbor interchange to refine branch lengths and reduce artefactual support.
Additionally, a Bayesian inference (BI) analysis was conducted using MrBayes v3.2.7a (Ronquist et al. 2012) to corroborate the ML topology. The same concatenated alignment and partition file were used. The GTR + I + G model was applied, with all parameters unlinked across the 6 partitions. Two independent MCMC runs were performed for 10 million generations, sampling every 1,000 generations. Convergence was confirmed when the average standard deviation of split frequencies (Lakner et al. 2008) fell below 0.01 and the potential scale reduction factor (Gelman and Rubin 1992) for all parameters approached 1.0. The first 25% of samples (2,500 trees) were discarded as burn-in, and a 50% majority-rule consensus tree was generated from the remaining trees to obtain posterior probabilities. The resulting phylogenies (both ML and BI) were visualized in iTOL v7 (Letunic and Bork 2024).
Results and discussion
Genome assembly, filtering, and evaluation
Whole-genome sequencing was performed using the PacBio Sequel IIe system. A total of 113,886 HiFi reads were generated, with a mean read length of 7,752 bp and a read N50 of 9,012 bp. All HiFi reads were used for de novo assembly. Prior to assembly, a k-mer analysis was performed on these PacBio HiFi reads to evaluate genome characteristics, particularly heterozygosity. The 21-mer frequency spectrum showed a single, sharp peak (Supplementary Fig. 1a), which is characteristic of a haploid or homozygous genome. The estimated heterozygosity rate was 0 to 0.033% (Supplementary Fig. 1b). This result strongly indicates that Umbelopsis sp. strain THIF13 was monokaryotic, or at least highly homozygous.
To generate a high-quality draft genome of Umbelopsis sp. strain THIF13, PacBio HiFi reads were assembled with hifiasm, yielding 25 contigs. Nonchromosomal and redundant sequences were removed through a 2-step filtering process. In the first step, Circlator detected 2 circular contigs (putative mitochondrial genomes) and 2 additional contigs whose entire sequences were contained within other contigs, all of which were subsequently excluded. In the second step, the remaining 21 contigs were aligned with Mauve and subjected to contig–contig BLASTN search to detect sequences sharing > 99.9% identity. Three contigs identified as redundant were discarded, resulting in a final assembly of 18 contigs. BUSCO analysis against the fungi_odb10 dataset revealed 744 complete BUSCOs out of 758 groups (98.1%) both before and after filtering, indicating high assembly completeness and confirming that only duplicate sequences had been removed (Fig. 1a). Scanning with tidk identified the heptameric repeat AAACCCT. Visual inspection of the repeat density (Fig. 1b) revealed distinct peaks concentrated at both ends of the 16 contigs. This distinct localization pattern suggests high contiguity and potential chromosome completeness (Fig. 1c). However, definitive confirmation of the chromosomal structure requires further validation using complementary methods, such as Hi-C sequencing or karyotype analysis. Interestingly, while the hexamer TTAGGG telomere motif is commonly observed in filamentous fungi (Podlevsky et al. 2008), the AAACCCT repeat observed in Umbelopsis represents a lineage-specific divergence in telomere sequence. The final Umbelopsis sp. strain THIF13 assembly comprises 18 contigs totaling 27.2 Mb, with an N50 of 1.77 Mb and a median contig length of 1.72 Mb, indicative of a highly contiguous assembly (Table 2). Notably, the genome size of Umbelopsis sp. strain THIF13 is larger than previously reported Umbelopsis genomes, such as U. ramanniana AG (23.1 Mb) (Amses et al. 2022), U. isabellina (22.1 Mb), U. vinacea (23.2 Mb) (Muszewska et al. 2021), and Umbelopsis sp. WA50703 (22.7 Mb) (Dziurzyński et al. 2025).
Evaluation of the Umbelopsis sp. strain THIF13 draft genome assembly. a) BUSCO completeness based on the fungi_odb10 dataset. b) Occurrence of the telomeric repeat “AAACCCT” across the 18 genomic contigs. Each panel shows an individual contig. The x-axis represents the normalized position within each contig, while the y-axis denotes the frequency of the identified repeat. c) Length distribution of the filtered contigs. Contigs with telomeric repeats detected at both ends (Potential telomere-to-telomere, Potential T2T) are shown in yellow; contigs lacking telomeric repeats (Non-T2T) are shown in blue.
Repeat sequence annotation
Analysis of the filtered genome assembly revealed that 11.15% (3,032,892 bp) of the assembly was annotated as repetitive sequences. The majority of these were interspersed repeats (10.44%), predominantly composed of DNA transposons (4.59%) and unclassified elements (4.80%). Within DNA transposons, the Tc1-IS630-Pogo superfamily was the most prevalent, dominating the repeat landscape in Umbelopsis sp. strain THIF13. Retroelements comprised 1.05% of the genome, with LTR elements (0.82%, mainly Gypsy/DIRS1) and LINEs (0.23%, mainly L1/CIN4) being identified. Other repeat categories included simple repeats (0.51%), low complexity regions (0.08%), and small RNA-related sequences (0.12%).
To investigate the larger genome size (27.2 Mb) of strain THIF13, a comparative analysis was performed with U. ramanniana and U. isabellina. The reference species exhibited markedly lower repeat contents (1.65% and 0.90%, respectively) compared with Umbelopsis sp. strain THIF13 (11.15%). These results suggest that the genomic expansion in Umbelopsis sp. strain THIF13 is likely attributable to the accumulation of repetitive elements and the associated increase in noncoding intergenic regions, given that the number of functional genes remains conserved.
Functional annotation
Gene prediction and functional annotation were performed for the Umbelopsis sp. strain THIF13 genome and the annotation metrics were compared with 2 representative Umbelopsis species, U. isabellina and U. ramanniana (Table 3). The Umbelopsis sp. strain THIF13 genome was predicted to contain a total of 9,068 genes. While this is slightly higher than U. isabellina (8,839 genes), it is lower than U. ramanniana (9,213 genes). Overall, a substantial proportion of genes were successfully annotated across all 3 Umbelopsis genomes, with 84.2% annotated by InterProScan, 89.6% by eggNOG, 74.5% by GO-term, and 76.6% by Pfam for Umbelopsis sp. strain THIF13. Regarding specific functional categories, 3.9% of Umbelopsis sp. strain THIF13 genes were annotated as carbohydrate-active enzymes (CAZymes) and 3.5% as MEROPS peptidases. Such variations may also be influenced by differences in sequencing technologies and assembly approaches.
The comparable proportions of CAZymes and MEROPS peptidases, along with the overall similarity in annotation metrics across the 3 Umbelopsis species, suggest a largely conserved core gene content and functional repertoire within this genus. A substantial percentage of BUSCOs were identified within the annotated gene sets of all 3 Umbelopsis species, further confirming the completeness and reliability of the predicted gene models.
GO analysis
To functionally categorize the predicted genes, GO analysis (Ashburner et al. 2000; Gene Ontology Consortium 2021) was performed for the Umbelopsis sp. strain THIF13 genome and compared with other species. The overall distribution of genes across the 3 main GO domains showed strong consistency among all 3 Umbelopsis species (Fig. 2). Further detailed analysis of the top 10 most abundant GO terms within each domain for Umbelopsis sp. strain THIF13 is presented in Fig. 3. Within the Molecular Function domain, highly represented terms included “protein binding,” “ATP binding,” and “RNA binding.” For Cellular Component, prominent terms comprised “cytoplasm,” “nucleus,” and “membrane,” indicating key cellular structures. In the Biological Process domain, “transmembrane transport,” “carbohydrate metabolic process,” and “regulation of transcription by RNA polymerase” were among the most frequently assigned terms. This strong conservation across major GO categories suggests a highly similar and fundamental functional repertoire among these Umbelopsis species, likely reflecting their shared core cellular processes and structures essential for fungal life.
Distribution of gene ontology categories in Umbelopsis sp. strain THIF13, U. isabellina, and U. ramanniana: molecular function (red), cellular component (blue), and biological process (green).
Distribution of GO categories in Umbelopsis sp. strain THIF13. The horizontal bar chart shows the number of genes assigned to GO terms within the 3 main GO domains. For each domain, the top 10 most abundant GO terms based on gene count are displayed.
Analysis of CAZyme
The genes annotated as CAZymes (Cantarel et al. 2009; Lombard et al. 2014) were compared in the 3 Umbelopsis genomes (Fig. 4). Their CAZyme complements are highly conserved. Glycoside hydrolases (GHs) were the most abundant CAZyme class in all 3 species, with Umbelopsis sp. strain THIF13 having a slightly higher proportion, followed by Glycosyltransferases (GTs). Other CAZyme classes identified included auxiliary activities (AAs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), and carbohydrate-binding modules (CBMs).
Distribution of CAZyme family repertoires in Umbelopsis sp. strain THIF13, U. isabellina, and U. ramanniana. Each bar is segmented by CAZyme class: glycoside hydrolases (GH, light blue), glycosyl transferases (GT, pink), polysaccharide lyases (PL, dark blue), carbohydrate esterases (CE, yellow), carbohydrate-binding modules (CBM, teal), and auxiliary activities (AA, orange).
Delving into the specific CAZyme families (Fig. 5), the GH profile includes numerous families associated with deconstructing the polysaccharide components of plant cell walls and fungal biomass, such as GH5 (cellulases/hemicellulases) and GH18 (chitinases). Additionally, the genome contains other GH families, such as GH16 (comprising 12 enzymes in subfamilies 2, 3, 4, 18, 19, and 23; e.g. endo-β-1,3-glucanases and cell wall transglycosylases) (Viborg et al. 2019) and GH3 (e.g. β-glucosidases), typically involved in the hydrolysis of polysaccharides into assimilable mono- or oligo-saccharides. The genome also encodes enzymes known for the degradation of cellulose, including a GH6 family cellulase and an AA14 family lytic polysaccharide monooxygenase. The AA class is also well-represented, featuring AA1 (laccases), which are associated with the modification of lignin and other aromatic compounds, as well as AA3 (glucose–methanol–choline oxidoreductases), AA7 (oligosaccharide oxidases), and 2 AA2 ascorbate peroxidases (Levasseur et al. 2013). Additionally, GTs represent a major class, notably GT2 (e.g. chitin and cellulose synthases), known for its role in cell wall integrity and biosynthesis, which is essential for fungal growth. Finally, various PLs, CEs, and CBMs were also identified in the genome.
The bar chart displays the number of genes within individual CAZyme families, grouped by their broader classes. For each class, up to the 8 most abundant families are shown. Classes with fewer than 8 identified families display all detected families.
The prevalence of these enzymes, particularly GHs and GTs, suggests a strategy focused on polysaccharide degradation and cell wall synthesis, consistent with a saprotrophic lifestyle (Kubicek et al. 2014). Furthermore, the comprehensive CAZyme repertoire identified in Umbelopsis sp. strain THIF13, characterized by a broad array of families involved in the degradation of complex plant carbohydrates, supports its saprotrophic lifestyle and ability to thrive in lignocellulose-rich forest environments, consistent with the known ecology of Umbelopsis species (Osono 2011).
KEGG pathway analysis
To further understand the metabolic and cellular capabilities of Umbelopsis sp. strain THIF13, its predicted genes were analyzed using the KEGG pathway database (Ogata et al. 1999; Kanehisa and Goto 2000; Du et al. 2014). The distribution of genes across Level B pathways, specifically those categorized within the major KEGG functional categories of “metabolism,” “genetic information processing,” “environmental information processing,” and “cellular processes,” was then compared with that of U. isabellina and U. ramanniana (Table 4). The KEGG Level B pathway profiles were highly similar across the 3 Umbelopsis species, reflecting conserved core metabolic and cellular functions. Among the various Level B pathways, “signal transduction” was the most highly represented category, indicating a substantial genetic investment in environmental sensing and cellular communication pathways, which are critical for fungal adaptation to diverse environments. Other prominent pathways included “carbohydrate metabolism,” “amino acid metabolism,” “transport and catabolism,” “lipid metabolism,” and “folding, sorting, and degradation.” The high representation of these pathways is consistent with their saprotrophic lifestyle, requiring robust machinery for nutrient acquisition, energy generation, and protein homeostasis from complex organic matter (Osono 2011; Kubicek et al. 2014). Furthermore, this metabolic potential likely supports lipid accumulation, a characteristic trait of Umbelopsis species, by providing essential precursors and energy.
Phylogenetic analysis
In the previous study, initial ITS region sequencing and BLAST homology searches against GenBank failed to resolve the species identity of Umbelopsis sp. strain THIF13 (Masaki et al. 2016). Therefore, in this study, following Wang et al. (2022), 6 loci were selected for phylogenetic inference (SSU rDNA, ITS, LSU rDNA, ACT, MCM7, and COX1) (Table 1). Sequences from 50 Umbelopsis strains and 2 Mortierella species as outgroups were concatenated into a 5,121 bp matrix (ITS: 605 bp, LSU: 1,020 bp, SSU: 1,097 bp, ACT: 868 bp, MCM7: 983 bp, and COX1: 548 bp) and analyzed by maximum likelihood in IQ-TREE 2. In the resulting tree (Fig. 6), the overall tree topology and the robust support for major clades were largely consistent with previous multilocus phylogenetic analyses of Umbelopsis (Wang et al. 2022; Hou et al. 2024). Notably, Umbelopsis sp. strain THIF13 was clearly resolved within the Umbelopsis nana clade, a placement strongly supported by both ultrafast bootstrap (UFBoot = 90%) and SH-aLRT (93%) tests. This clade also contains the ex-type strain of U. nana (CBS 858.68, originally described as Mortierella alba) (Von Arx 1982). Pairwise p-distances among the 7 U. nana strains, including strain THIF13, were below the detection threshold (0 to 1 × 10⁻⁶), indicating near-identity across the concatenated loci. The split separating U. nana from U. dimorpha received 100% ultrafast bootstrap support. The BI analysis (Supplementary Fig. 2) yielded a congruent topology for all major clades, confirming the placement of Umbelopsis sp. strain THIF13 within the U. nana clade with the highest posterior probability (PP = 1.00). However, the branching order among closely related strains within the U. nana clade differed significantly between the ML and BI analyses. While the ML tree resolved the clade via sequential bifurcations, the BI tree defined this region as a polytomy where 4 branches diverge from a single node. This inconsistency confirms that the low genetic divergence (P-distances 0) among these U. nana strains prevents robust resolution of the internal topology. These results collectively confirm the assignment of strain THIF13 to U. nana. Following this identification, the strain has been deposited in the NBRC culture collection under the accession number U. nana NBRC 117090. Hereafter, we refer to this organism as U. nana strain THIF13 throughout the remainder of this manuscript. This genome represents the first publicly available genome sequence for U. nana.
Phylogenetic tree of 49 Umbelopsis isolates and 2 Mortierella outgroups inferred from a concatenated alignment of 6 loci (SSU rDNA, ITS, LSU rDNA, ACT, MCM7, and COX1; 5,121 bp). The tree was reconstructed by maximum likelihood in IQ-TREE 2. Node support was assessed with 1,000 ultrafast bootstrap replicates; values on the main tree are represented by scaled circles, while values on the inset are shown as percentages. The inset shows a magnified view of the clade containing Umbelopsis sp. strain THIF13 (highlighted in red). Scale bars indicate substitutions per site. The isolate U. dura CGMCC 3.1578 was excluded because its sequence was identical to that of U. dura CGMCC 3.15777. HT, ex-holotype strain; ET, ex-epitype strain; T, ex-type strain.
Conclusion
In this study, the first high-quality genome sequence of Umbelopsis nana strain THIF13 (NBRC 117090), whose species identity was clearly established through multilocus phylogenetic analysis, was successfully generated and characterized. Comprehensive functional annotation, including comparative analyses of Gene Ontology terms, CAZymes, and KEGG pathways against other Umbelopsis species, revealed a robust and versatile metabolic repertoire, which is broadly conserved among Umbelopsis species. The genomic resource presented herein substantially expands the currently limited genomic data for the Umbelopsis genus. This provides a critical foundation for future research, enabling fine-scale comparative genomic studies to identify specific genetic determinants of unique biological traits of U. nana (such as COS production by strain THIF13) that are not captured by broad functional annotations, as well as the exploration of its biotechnological potential.
Supplementary Material
jkag022_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Amos RE, Barnett HL. 1966. Umbelopsis versiformis, a new genus and species of the imperfects. Mycologia. 58:805. 10.2307/3756856. · doi ↗
- 2Amses KR et al 2022. Diploid-dominant life cycles characterize the early evolution of Fungi. Proc Natl Acad Sci U S A. 119:e 2116841119. 10.1073/pnas.2116841119.36037379 PMC 9457484 · doi ↗ · pubmed ↗
- 3Anisimova M, Gil M, Dufayard J-F, Dessimoz C, Gascuel O. 2011. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol. 60:685–699. 10.1093/sysbio/syr 041.21540409 PMC 3158332 · doi ↗ · pubmed ↗
- 4Ashburner M et al 2000. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 25:25–29. 10.1038/75556.10802651 PMC 3037419 · doi ↗ · pubmed ↗
- 5Brown MR, Manuel Gonzalez de La Rosa P, Blaxter M. 2025. Tidk: a toolkit to rapidly identify telomeric repeats from genomic datasets. Bioinformatics. 41:btaf 049. 10.1093/bioinformatics/btaf 049.39891350 PMC 11814493 · doi ↗ · pubmed ↗
- 6Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12:59–60. 10.1038/nmeth.3176.25402007 · doi ↗ · pubmed ↗
- 7Camacho C et al 2009. BLAST+: architecture and applications. BMC Bioinformatics. 10:421. 10.1186/1471-2105-10-421.20003500 PMC 2803857 · doi ↗ · pubmed ↗
- 8Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. 2021. Egg NOG-mapper v 2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol. 38:5825–5829. 10.1093/molbev/msab 293.34597405 PMC 8662613 · doi ↗ · pubmed ↗