Analysis of the use of monoclonal antibodies in the treatment of Crohn’s disease
Alexander V Blagov, Marina D Sazonova, Anastasia I Ryzhkova, Vasily P Karagodin, Mikhail A Popov, Egor Yu Budnikov, Alessio L Ravani, Alexander N Orekhov, Margarita A Sazonova, Yuri V Arkhipenko

TL;DR
This paper reviews how monoclonal antibodies treat Crohn’s disease, highlighting current successes, limitations, and future research directions.
Contribution
The paper proposes novel therapeutic strategies and identifies new targets for monoclonal antibodies in Crohn’s disease treatment.
Findings
Current monoclonal antibodies face limitations like non-response and high costs.
Combination therapy and personalized medicine may improve treatment outcomes.
New targets like IL-36 and microbiome-related factors show promise for future therapies.
Abstract
Crohn’s disease (CD) is a chronic inflammatory bowel disease with increasing global prevalence, significantly impacting patients’ quality of life and healthcare costs. The introduction of monoclonal antibodies has revolutionized CD management, offering targeted therapy against specific inflammatory pathways. This review systematically analyzes the current state of monoclonal antibody therapy, including anti-TNF-α agents (infliximab, adalimumab, certolizumab pegol), anti-integrin antibodies (vedolizumab), and anti-cytokine therapies (ustekinumab, risankizumab). Despite remarkable therapeutic advances, significant limitations persist, including primary non-response (20%–40%), secondary loss of response (13%–20% annually), immunogenicity, safety concerns, and substantial economic burden. We propose evidence-based strategies to address these challenges, including therapeutic drug…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
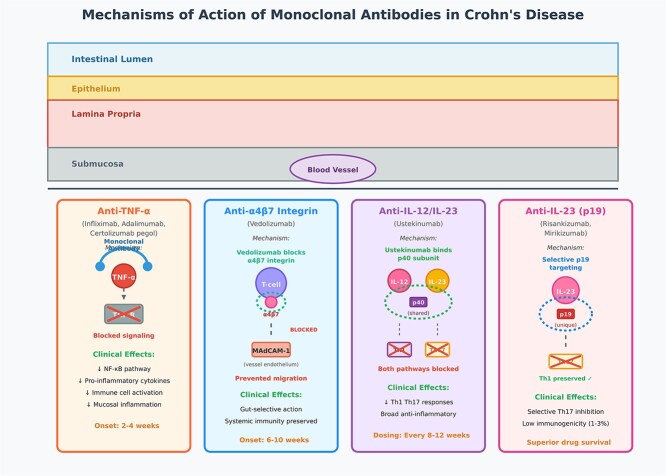 Figure 1
Figure 1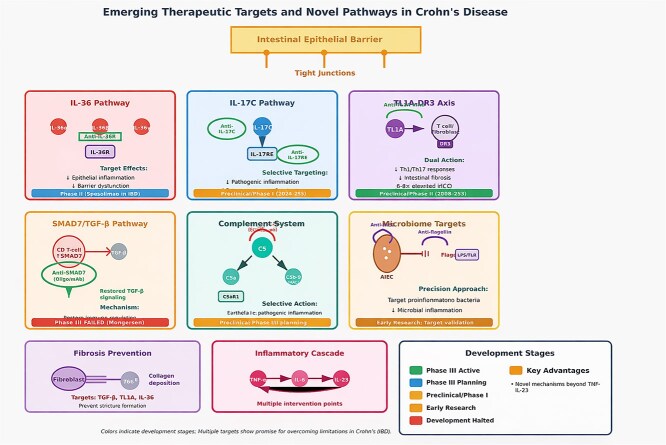 Figure 2
Figure 2| Monoclonal antibody | Target | Key trial | Patient population | Concomitant therapy | Induction response rate | Maintenance remission rate | Placebo response/remission | Anti-drug antibodies | Key advantages |
|---|---|---|---|---|---|---|---|---|---|
| Infliximab | TNF-α | ACCENT I | Bio-naïve and bio-experienced mixed | Combination with AZA in SONIC (56.8% vs. 44.4% monotherapy) | 58%–70% | 39%–50% | 21% remission at week 54 | 10%–61% | Established efficacy, IV administration |
| Adalimumab | TNF-α | CLASSIC I/CHARM | Majority bio-naïve (CLASSIC I: 100% bio-naïve) | Monotherapy primarily, some with immunomodulators | 58%–67% | 36%–52% | 17% remission at week 26 (CHARM) | 2.6%–26.5% | Subcutaneous dosing, lower immunogenicity |
| Certolizumab pegol | TNF-α | PRECISE 1/2, WELCOME | WELCOME: 100% prior anti-TNF failure | Monotherapy primarily | 62%–64% | 48% | 29% remission at week 26 (PRECISE 2) | 8%–12% | Pregnancy safety, anti-TNF failure efficacy |
| Vedolizumab | α4β7 integrin | GEMINI 2 | Mixed: 47% prior anti-TNF exposure | Monotherapy or with stable immunomodulators | 31%–47% | 39%–48% | 21.6% remission at week 52 | <4% | Gut-selective, excellent safety profile |
| Ustekinumab | IL-12/IL-23 | UNITI-1/2, IM-UNITI | UNITI-1: bio-naïve; UNITI-2: prior anti-TNF failure | Monotherapy primarily | 40%–52% | 53% | 27.5% remission at week 44 | 2.3%–5.1% | Broad efficacy, biologic failure response |
| Risankizumab | IL-23 | ADVANCE, MOTIVATE, FORTIFY | ADVANCE: 59% bio-naïve; MOTIVATE: 100% prior biologic failure | Monotherapy primarily | 38%–42% | 45% | 25% remission at week 12 (ADVANCE) | 1%–3% | Selective targeting, low immunogenicity |
| Antibody class | Representative agents | Induction response rate | Maintenance remission rate (52 weeks) | Safety profile | Preferred patient populations | Typical clinical positioning |
|---|---|---|---|---|---|---|
| Anti-TNF-α | Infliximab, Adalimumab, Certolizumab pegol | 58%–70% | 36%–50% | Serious infections: 4–6 per 100 PY; Tuberculosis reactivation: 0.2%–0.5%; Infusion/injection reactions: 5%–10%; Malignancy risk: OR 1.4 (lymphoma) | Moderate–severe luminal disease; Perianal fistulizing disease; Patients requiring rapid symptom control; Inflammatory non-stricturing phenotype | First-line biologic in most guidelines; Combination with immunomodulators increases efficacy |
| Anti-α4β7 integrin | Vedolizumab | 31%–47% | 39%–48% | Serious infections: 2.4 per 100 PY; Minimal systemic immunosuppression; Infusion reactions: 4%; PML: extremely rare (<0.01%) | Anti-TNF-experienced patients; Patients with infection concerns; Elderly patients; Concomitant extraintestinal manifestations not requiring treatment | Second-line after anti-TNF failure; First-line in patients with contraindications to anti-TNF |
| Anti-IL-12/IL-23 (p40) | Ustekinumab | 40%–52% | 53% | Serious infections: 3.0 per 100 PY; MACE: 0.5 per 100 PY; Low immunogenicity; Well-tolerated SC administration | Broad applicability across phenotypes; Biologic-experienced patients; Patients prioritizing convenient dosing | Second-line after anti-TNF failure; Emerging as first-line option |
| Anti-IL-23 (p19) | Risankizumab, Mirikizumab | 38%–45% | 45%–52% | Serious infections: 2.8 per 100 PY; Hepatic events: monitor LFTs; Low immunogenicity (1%–3%); Excellent drug survival | Inflammatory luminal disease; Patients prioritizing long-term durability; Failed multiple prior biologics; Infection-prone patients | Second-line after anti-TNF failure; Consideration for first-line in select patients (elderly, infection concerns, preference for low immunogenicity) |
| Limitation | Prevalence | Clinical impact | Current standard approaches | Innovative/emerging strategies | Evidence level and key references |
|---|---|---|---|---|---|
| Primary non-response | 20%–40% across agents | Treatment delay (median 12–16 weeks lost); Disease progression during ineffective therapy; Increased risk of complications (OR 2.1 for surgery); Healthcare costs: additional $15 000–25 000 per failed trial | Pharmacogenomic testing for TNF polymorphisms; Biomarker-guided selection (fecal calprotectin, CRP); Dose optimization during induction (e.g., accelerated infliximab dosing) | Machine learning algorithms predicting response (AUC 0.78–0.82); Multi-omics profiling (genomics + proteomics + microbiome); Point-of-care mucosal TNF-α measurement; Adaptive dosing based on early pharmacokinetic sampling |
|
| Secondary loss of response | 13%–20% annually; Cumulative 5-year rate: 50%–60% | Clinical relapse requiring hospitalization (15%–20%); Need for dose escalation (30%–40%); Treatment switching costs: $8000–12 000; Surgery rates increase by 8%–10% | Therapeutic drug monitoring (TDM); Proactive TDM superior to reactive (remission: 55% vs. 38%); Combination therapy with immunomodulators; Dose intensification protocols | Dashboard-based TDM with automated dosing recommendations; Prophylactic switching before antibody formation; Novel immunomodulators (e.g., JAK inhibitors) to prevent immunogenicity; Extended interval dosing after deep remission |
|
| Immunogenicity | 2%–61% (varies by agent): Infliximab 10%–61%; Adalimumab 2.6%–26.5%; Vedolizumab <4%; Risankizumab 1%–3% | Treatment failure in 30%–40% with high-titer antibodies; Infusion reactions (4%–8%); Accelerated drug clearance (half-life reduced by 40%–60%); Limited re-treatment options | Concomitant immunosuppression (reduces ADA by 50%–70%); Scheduled maintenance dosing; Prophylactic corticosteroids/antihistamines for infusions; Early dose intensification | Fc-modified antibodies with reduced immunogenicity; PEGylation strategies; Induction of immune tolerance protocols; Transient immunosuppression during induction only; Next-generation fully human/humanized constructs |
|
| Safety concerns | Serious infections: 2–6 per 100 PY; Malignancy: OR 1.2–1.4; Opportunistic infections: 0.5%–1% | Hospitalization for infections (3%–5%); Treatment discontinuation (8%–12%); Mortality from serious infections (0.1%–0.3%); Life-threatening opportunistic infections; Reduced quality of life due to infection anxiety | Comprehensive pre-treatment screening (TB, Hep B/C, HIV); Vaccination protocols; Regular monitoring (CBC, LFTs every 3–6 months); Patient education on infection signs; Prophylactic antibiotics in high-risk patients | Risk stratification tools (infection risk calculators); Gut-selective agents with minimal systemic exposure; Shorter-acting biologics allowing rapid clearance; Microbiome-based risk assessment; Real-time infection biomarkers |
|
| Economic burden | Universal; Annual costs: $30 000–60 000 per patient | Treatment non-adherence (15%–25%); Delayed therapy initiation (average 6–12 months); Geographic/socioeconomic disparities in access; Out-of-pocket costs: $3000–8000 annually; Insurance denials (10%–15%) | Biosimilars (20%–40% cost reduction); Patient assistance programs; Prior authorization advocacy; Value-based contracts; Tiered formulary approaches | Outcome-based pricing models; Subscription-based biologic access; Home administration programs; Shared savings agreements; Global access initiatives; Oral formulations reducing administration costs |
|
| Target | Mechanism of action | Evidence/key data | Development Ssage | Limitations/challenges | Key references |
|---|---|---|---|---|---|
| IL-36 pathway | Blockade of IL-36 receptor or neutralization of IL-36 ligands (α, β, γ) reduces epithelial-derived inflammation, and barrier dysfunction | Mouse models: 60% reduction in colitis severity; IL-36 expression 4.5-fold elevated in CD mucosa; Correlates with disease activity ( |
| Limited human IBD data; Potential for paradoxical worsening (seen with other epithelial cytokines); Unclear role in different CD phenotypes; Need to identify responder biomarkers | Nishida |
| IL-17C pathway | Selective inhibition of IL-17C (via anti-IL-17C or anti-IL-17RE antibodies) targets pathogenic inflammation while preserving IL-17A barrier protection | Preclinical: IL-17C knockout mice show 45% reduced colitis; IL-17C elevated 8-fold in CD patients; No barrier dysfunction in IL-17C blockade vs. 30% barrier compromise with IL-17A blockade |
| Previous IL-17A inhibitors worsened CD; Need to confirm IL-17C selectivity is safe; Limited understanding of IL-17C biology in humans; Potential for fungal infections | Ramirez-Carrozzi |
| SMAD7 | Antisense oligonucleotide or antibody-based restoration of TGF-β signaling; Reduces inflammatory T-cell activity and prevents fibrosis |
|
| Phase II-III discrepancy unexplained; Potential issues with oligonucleotide delivery; Variable SMAD7 expression between patients; May require patient stratification; Concern about fibrosis risk with chronic TGF-β activation | Monteleone |
| Complement C5/C5a | Inhibition of C5 cleavage or C5a receptor blockade reduces complement-mediated inflammation while preserving C3-mediated immunity | Elevated C5a in CD serum (3.2-fold vs. controls); C5aR1 expression increased on mucosal neutrophils; Eculizumab case reports show benefit in refractory CD ( |
| High cost of complement inhibitors; Risk of Neisseria infections (requires vaccination); Uncertainty about patient selection; Limited understanding of complement role across CD phenotypes; No large controlled trials | Ricklin |
| TL1A-DR3 axis | Anti-TL1A antibodies block TL1A binding to DR3, reducing Th1/Th17 responses, and intestinal fibrosis | TL1A elevated 6–8-fold in CD mucosa; Genetic variants increase CD risk (OR 1.4); |
| TL1A also has homeostatic roles; Optimal timing (early vs. established disease) unclear; Potential for loss of barrier function; Need longer-term safety data | Shih |
| Microbiome-related targets | Antibodies targeting pathogenic bacteria (AIEC), bacterial toxins, or flagellin reduce microbial-driven inflammation | Anti-flagellin (CBir1) antibodies present in 50% CD patients; AIEC detected in 40% of CD ileal biopsies; Preclinical: Anti-AIEC antibodies reduce colonization by 80%; Flagellin vaccination reduces colitis 55% in mice |
| Microbiome heterogeneity between patients; Risk of disrupting beneficial microbes; Unclear which bacteria to target; Delivery challenges (oral vs. systemic); Potential for resistance development; Need for companion diagnostics | Lodes |
- —Russian Science Foundation10.13039/501100006769
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInflammatory Bowel Disease · Biosimilars and Bioanalytical Methods · Monoclonal and Polyclonal Antibodies Research
Introduction
Crohn’s disease (CD) is a chronic inflammatory bowel disease (IBD) characterized by transmural inflammation that can affect any part of the gastrointestinal tract from the mouth to the anus, with a predilection for the terminal ileum and colon [1]. The disease manifests as a relapsing–remitting condition with periods of active inflammation alternating with remission, often resulting in complications such as strictures, fistulas, and perforations [2]. The global incidence of CD has shown a steady increase over the past several decades, with the highest prevalence rates reported in North America and Europe, ranging from 50 to 200 cases per 100 000 individuals [3]. Emerging data indicate a rising incidence in previously low-prevalence regions, including Asia, South America, and Africa, suggesting a global expansion of the disease burden [4]. This epidemiological shift has been attributed to environmental factors such as westernization of diet, urbanization, improved sanitation paradoxically leading to altered microbiome development, and increased diagnostic awareness [5]. The substantial impact of CD on patients’ quality of life, combined with significant healthcare costs estimated at billions of dollars annually worldwide, underscores the critical need for effective therapeutic interventions [6].
The therapeutic landscape for CD has evolved significantly from conventional treatments to targeted biologic therapies [7]. Traditional treatment approaches include aminosalicylates (5-ASA compounds) for mild disease, corticosteroids for acute flares, and immunosuppressive agents such as azathioprine, 6-mercaptopurine, and methotrexate for maintenance therapy [8]. However, these conventional therapies often provide limited efficacy, carry significant side effect profiles, and fail to address the underlying inflammatory pathways driving the disease [9]. The introduction of biologic therapies, particularly monoclonal antibodies, has revolutionized CD management by offering targeted intervention against specific inflammatory mediators [10]. Anti-tumor necrosis factor-alpha (anti-TNF-α) monoclonal antibodies, including infliximab, adalimumab, and certolizumab pegol, were the first biologics to demonstrate significant efficacy in inducing and maintaining remission in moderate to severe CD [11]. Subsequently, monoclonal antibodies targeting alternative pathways have been developed, including ustekinumab (anti-IL-12/IL-23), vedolizumab (anti-α4β7 integrin), and more recently, risankizumab (anti-IL-23) [12]. These targeted therapies have shown superior efficacy compared to conventional treatments, with improved rates of clinical remission, mucosal healing, and reduced need for surgery, establishing monoclonal antibodies as cornerstones of modern CD therapy [13].
Despite the remarkable advances in monoclonal antibody (mAb) therapy for CD, several significant limitations persist that warrant comprehensive analysis and innovative solutions [14]. The objectives of this review are threefold: first, to conduct a systematic analysis of existing limitations in mAb therapy for CD, including primary non-response affecting 20%–40% of patients, secondary loss of response observed in 13%–20% of patients annually, immunogenicity leading to treatment failure, significant adverse events including increased infection risk and potential malignancy, and the substantial economic burden limiting patient access [15]. Second, to propose evidence-based strategies for addressing these limitations, including optimization of dosing regimens, combination therapy approaches, therapeutic drug monitoring, immunogenicity mitigation strategies, and patient stratification methods for personalized treatment selection [16]. Third, to identify and evaluate potentially novel therapeutic targets for future mAb development in CD, including emerging cytokine pathways (IL-36, IL-17, SMAD7), cell adhesion molecules, complement system components, and microbiome-related targets, which may offer improved efficacy, reduced side effects, and solutions for treatment-refractory patients, ultimately advancing the field toward more precise and effective therapeutic interventions [17].
Current state of approved and emerging monoclonal antibody in CD therapy
Anti-TNF-α monoclonal antibodies
Infliximab
Infliximab, a chimeric mAb targeting tumor necrosis factor-alpha, represents the pioneering biologic therapy in CD management [18]. The landmark ACCENT I trial demonstrated infliximab’s efficacy in inducing clinical remission in 58% of patients at week 10, with maintenance of remission achieved in 39% of patients at week 54 compared to 21% with placebo [11]. Subsequent real-world studies have confirmed these findings, showing clinical response rates of 60%–70% in induction therapy and remission maintenance in ~40%–50% of patients over 1 year [19]. The SONIC trial further established infliximab’s superior efficacy when combined with azathioprine, demonstrating corticosteroid-free remission in 56.8% of combination therapy patients versus 44.4% with infliximab monotherapy [20]. However, infliximab therapy is associated with significant immunogenicity, with anti-drug antibodies developing in 10%–61% of patients, leading to treatment failure and increased infusion reactions [21].
Adalimumab
Adalimumab, a fully human anti-TNF-α mAb, offers the advantage of subcutaneous administration and potentially reduced immunogenicity compared to infliximab [22]. The CLASSIC I trial demonstrated clinical remission rates of 36% at week 4 and 59% at week 26 in patients receiving adalimumab induction therapy [23]. The CHARM study revealed that adalimumab maintenance therapy achieved clinical remission in 40% of patients at week 26 and 36% at week 56, significantly superior to placebo [24]. Meta-analyses indicate that adalimumab demonstrates comparable efficacy to infliximab, with clinical response rates of 58%–67% for induction and 47%–52% for maintenance therapy [25]. The development of anti-adalimumab antibodies occurs in ~2.6%–26.5% of patients, generally lower than infliximab, though this remains a significant concern for long-term efficacy [26].
Certolizumab pegol
Certolizumab pegol, a pegylated anti-TNF-α antibody fragment, provides unique pharmacokinetic properties with prolonged half-life and minimal placental transfer [27]. The PRECISE 1 trial demonstrated clinical response rates of 64.1% at week 6 and 62.4% at week 26 in patients receiving certolizumab pegol 400 mg [28]. The PRECISE 2 maintenance study showed sustained clinical remission in 48% of responders at week 26 compared to 29% with placebo [29]. Notably, certolizumab pegol has demonstrated particular efficacy in patients with prior anti-TNF therapy failure, with response rates of 43.8% in the WELCOME study [30]. The immunogenicity profile appears favorable, with anti-drug antibodies detected in ~8%–12% of patients, though longer-term data are still accumulating [31].
Anti-integrin monoclonal antibodies
Vedolizumab
Vedolizumab represents a paradigm shift toward gut-selective therapy, targeting α4β7 integrin to prevent lymphocyte migration into gastrointestinal tissues [32]. The GEMINI 2 trial demonstrated clinical remission rates of 14.5% at week 6 and 39% at week 52 in CD patients receiving vedolizumab [13]. While the induction response appears slower compared to anti-TNF therapies, vedolizumab shows particular efficacy in anti-TNF-experienced patients, with clinical response rates of 31.4% at week 10 [33]. The VERSIFY study confirmed vedolizumab’s effectiveness in real-world settings, showing clinical remission in 47.7% of patients at week 52 [34]. The gut-selective mechanism results in an excellent safety profile with minimal systemic immunosuppression, and anti-vedolizumab antibodies are detected in less than 4% of patients [35].
Anti-IL-12/IL-23 monoclonal antibodies
Ustekinumab
Ustekinumab targets the p40 subunit common to both IL-12 and IL-23, disrupting key inflammatory pathways in CD [36]. The UNITI-1 trial in anti-TNF-naive patients demonstrated clinical response rates of 51.7% at week 6, while UNITI-2 in anti-TNF-experienced patients showed response rates of 40.2% [12]. The IM-UNITI maintenance study revealed clinical remission in 53.1% of patients at week 44 with 90 mg dosing every 8 weeks [37]. Ustekinumab demonstrates consistent efficacy across different patient populations, including those with prior biologic failure, with sustained clinical benefits observed in long-term extension studies [38]. The immunogenicity profile is favorable, with anti-ustekinumab antibodies detected in ~2.3%–5.1% of patients [39].
Risankizumab
Risankizumab, a selective IL-23 inhibitor targeting the p19 subunit, represents the latest advancement in cytokine-targeted therapy for CD [40]. The ADVANCE trial demonstrated superior clinical remission rates of 42% at week 12 compared to 32% with placebo [41]. The FORTIFY maintenance study showed clinical remission in 45% of patients at week 52 with 180 mg dosing every 8 weeks [42]. Added 3-year extension data from FORTIFY showing sustained remission in 68% of continuous responders and favorable safety profile with no new safety signals (presented at DDW 2024) [43]. Risankizumab has shown particular promise in patients with prior biologic failure, with response rates of 38% in the MOTIVATE trial [44]. The selective IL-23 targeting may offer advantages over broader IL-12/IL-23 inhibition, with preliminary safety data suggesting a favorable profile and low immunogenicity rates of ~1%–3% [45].
Emerging monoclonal antibodies
Several novel monoclonal antibodies are currently in clinical development for CD. Mirikizumab, another IL-23 inhibitor, has demonstrated promising results in phase 3 trials with clinical remission rates of 24.2% at week 12 [46]. Brazikumab, targeting IL-23 receptor, showed clinical response rates of 49.2% in phase 2 studies [47]. Etrolizumab, an anti-β7 integrin antibody, demonstrated clinical remission in 21.6% of patients at week 14 in the BERGAMOT trial [48].
Tables 1 and 2 summarizes the results of clinical trials of monoclonal antibodies for the treatment of CD.
Figure 1 shows mechanisms of action of monoclonal antibodies in CD treatment.
Mechanisms of action of monoclonal antibodies in Crohn’s disease treatment.
Current limitations of approved monoclonal antibodies for CD treatment and solutions
Primary non-response
Primary non-response represents a significant challenge in mAb therapy for CD, affecting ~20%–40% of patients across different therapeutic agents [15]. This phenomenon occurs when patients fail to achieve clinical response despite adequate drug exposure during induction therapy [49]. The mechanisms underlying primary non-response are multifactorial and include genetic polymorphisms affecting drug metabolism, variations in target expression, disease heterogeneity, and individual immune system characteristics [50].
Several strategies have been developed to address primary non-response. Pharmacogenomic testing can identify patients with genetic variants affecting drug metabolism, particularly for anti-TNF agents where polymorphisms in TNF-α promoter regions and HLA alleles influence treatment response [51]. Dose optimization based on therapeutic drug monitoring during induction has shown promise in improving initial response rates [52]. Additionally, biomarker-guided therapy selection, including assessment of mucosal TNF-α expression, serum cytokine profiles, and histologic features, can help identify patients more likely to respond to specific therapeutic targets [53].
Secondary loss of response
Secondary loss of response affects 13%–20% of patients annually and represents a major clinical challenge in maintaining long-term remission [14]. This phenomenon typically occurs after an initial therapeutic response and can result from multiple mechanisms including immunogenicity, disease progression, development of neutralizing antibodies, and changes in drug pharmacokinetics [54].
Therapeutic drug monitoring has emerged as a crucial tool for managing secondary loss of response [16]. Proactive monitoring of drug levels and anti-drug antibodies allows for timely dose optimization before clinical deterioration occurs [55]. Meta-analyses demonstrate that proactive monitoring compared to reactive monitoring results in improved clinical outcomes, reduced immunogenicity, and better long-term treatment persistence [56]. Combination immunosuppressive therapy with methotrexate or azathioprine can prevent anti-drug antibody formation and maintain therapeutic drug levels, particularly for anti-TNF agents [20].
Immunogenicity and treatment failure
Immunogenicity remains a significant limitation across all mAb therapies, though rates vary considerably between agents [21]. The development of anti-drug antibodies leads to accelerated drug clearance, reduced efficacy, and increased risk of infusion or injection site reactions [57]. Factors influencing immunogenicity include drug structure (chimeric vs. humanized vs. fully human), dosing frequency, concomitant immunosuppression, and individual patient characteristics [58].
Several approaches have been developed to mitigate immunogenicity. Combination therapy with immunosuppressive agents significantly reduces antibody formation, with the SONIC trial demonstrating superior outcomes for infliximab plus azathioprine compared to monotherapy [20]. Prophylactic treatment with corticosteroids or antihistamines can reduce acute infusion reactions [59]. Novel antibody engineering approaches, including Fc modifications and alternative dosing regimens, are being investigated to reduce immunogenic potential while maintaining therapeutic efficacy [60].
Safety concerns and adverse events
Monoclonal antibody therapies carry significant safety risks that limit their use in certain patient populations [61]. Anti-TNF agents increase the risk of serious infections, including opportunistic infections, reactivation of latent tuberculosis, and hepatitis B virus reactivation [62]. There are also concerns about increased malignancy risk, particularly lymphoma, and rare but serious adverse events such as demyelinating disease and heart failure [63].
Risk mitigation strategies include comprehensive screening protocols before treatment initiation, including testing for latent tuberculosis, hepatitis B and C, and assessment of cardiovascular status [64]. Regular monitoring during therapy with complete blood counts, liver function tests, and clinical assessment for signs of infection is essential [65]. Patient education about infection prevention and early recognition of symptoms is crucial for optimal safety outcomes [66].
Monoclonal antibody therapies demonstrate class-specific safety profiles reflecting their distinct mechanisms of action, with critical implications for patient selection and risk management [57].
Anti-TNF agents carry the highest infection risk among biologics due to systemic TNF-α blockade, which impairs granuloma formation and innate immunity. Meta-analysis of 22 randomized trials (n = 8905) by Lichtenstein et al. (2012) demonstrated serious infection rates of 4.6 per 100 patient-years, significantly elevated versus placebo (OR 2.05, 95% CI 1.10–3.85) [61]. Opportunistic infections include tuberculosis reactivation (incidence 0.2%–0.5% with screening protocols), invasive fungal infections (0.3%–0.8%), and hepatitis B reactivation (10%–25% in HBsAg-positive patients without prophylaxis) [62]. Malignancy concerns include increased lymphoma risk (OR 1.4, 95% CI 1.1–1.8), particularly hepatosplenic T-cell lymphoma in young males receiving combination therapy, and possible melanoma risk (standardized incidence ratio 1.4–1.5) [63]. Demyelinating disorders occur rarely (incidence <0.1%) but mandate discontinuation [67]. The TREAT registry with >6000 patient-years follow-up confirmed dose-dependent infection risk, with intensified dosing increasing serious infection rates by 30%–40% [61].
Vedolizumab demonstrates superior safety through gut-selective α4β7 integrin blockade that prevents lymphocyte trafficking specifically to intestinal tissue while preserving systemic immunity. The GEMINI long-term safety study (n = 2830, median 2.3 years follow-up) reported serious infection rates of only 2.4 per 100 patient-years, significantly lower than anti-TNF agents (adjusted HR 0.52, 95% CI 0.38–0.71) [35]. Critically, no increased risk of opportunistic infections, tuberculosis, or progressive multifocal leukoencephalopathy (PML) has been observed, contrasting with natalizumab’s pan-integrin blockade which carries 1:1000 PML risk [68]. Malignancy rates match background population (standardized incidence ratio 0.98, 95% CI 0.72–1.31) [69]. The mechanism-based safety advantage makes vedolizumab particularly suitable for elderly patients (>65 years), those with prior serious infections, chronic viral hepatitis carriers, latent tuberculosis with treatment concerns, and patients with multiple comorbidities requiring polypharmacy [70].
Anti-IL-12/IL-23 and anti-IL-23 agents demonstrate intermediate safety profiles. Ustekinumab meta-analysis (9 trials, n = 4135) showed serious infection rates of 3.0 per 100 patient-years, lower than anti-TNF (P = .04) but higher than vedolizumab [71]. The IL-23 pathway’s role in mucosal barrier defense explains maintained protection against bacterial infections while reducing inflammatory pathology. Risankizumab demonstrates even more favorable safety with 2.8 serious infections per 100 patient-years and low opportunistic infection incidence (<0.5%) [42]. Theoretical concerns about impaired immunity to extracellular bacteria and fungi have not materialized in clinical practice. A potential signal for major adverse cardiovascular events with ustekinumab (incidence 0.5 per 100 patient-years) requires ongoing surveillance, particularly in patients with cardiovascular risk factors [72]. Hepatic enzyme elevations occur in 8%–12% with IL-23 inhibitors, usually transient and asymptomatic, but mandate monitoring [41].
Risk mitigation strategies include comprehensive pre-treatment screening (tuberculosis, hepatitis B/C, HIV, complete blood count, liver function), age-appropriate vaccinations (pneumococcal, influenza, hepatitis B, herpes zoster if >50 years with inactivated vaccine) administered ≥2–4 weeks before therapy initiation, regular monitoring (complete blood count and liver function every 3–6 months), patient education emphasizing early recognition of infection symptoms, and dose adjustment or temporary discontinuation during active infections [64].
Economic burden and access limitations
The substantial cost of mAb therapies represents a significant barrier to patient access and healthcare system sustainability [6]. Annual treatment costs can exceed $30 000–50 000 per patient, creating disparities in access based on insurance coverage and geographic location [73]. This economic burden limits the ability to implement optimal treatment strategies and may delay appropriate therapy initiation [74].
Addressing economic barriers requires multifaceted approaches including value-based care models, biosimilar development to reduce costs, patient assistance programs, and healthcare policy reforms [75]. Biosimilar versions of infliximab and adalimumab have demonstrated comparable efficacy and safety while reducing costs by 20%–30% [76]. Pharmacoeconomic analyses support the long-term cost-effectiveness of biologic therapy through reduced hospitalizations, surgery rates, and disability, though upfront costs remain prohibitive for many patients [77].
Biosimilar impact on cost and access
Biosimilar introduction has significantly impacted the economic landscape. The NOR-SWITCH trial demonstrated non-inferiority of CT-P13 (infliximab biosimilar) to originator infliximab, with comparable efficacy (29.6% vs. 30.0% disease worsening, 95% CI −7.7 to 5.8) and safety over 52 weeks [76]. Real-world implementation in Norway achieved 34% cost reduction, translating to €11.7 million annual savings for IBD treatment alone [78]. Denmark’s mandatory switching program (2015–2016) achieved 69% cost reduction for infliximab and demonstrated sustained effectiveness with 89% continuation rates at 1 year [79].
US market dynamics show more modest impact, with biosimilar uptake reaching only 45% by 2023 despite FDA approval of multiple biosimilars (CT-P13, SB2, PF-06438179 for infliximab; ABP 501, SB5, GP2017 for adalimumab) [80]. Average wholesale price reductions of 15%–35% versus originators remain below European reductions of 30%–70%, attributed to complex rebate systems and limited interchangeability designation [81]. A 2023 IQVIA analysis projects potential US savings of $6.8 billion over 5 years with increased biosimilar utilization [82].
Regional variations in access
High-income countries (North America, Western Europe, Australia): Universal or near-universal biologic access through insurance/national health systems, though prior authorization requirements cause treatment delays averaging 8–12 weeks in US commercial insurance [83]. Cost-sharing responsibilities create patient-level barriers, with out-of-pocket costs of 8000 annually leading to 15%–25% non-adherence rates [84].
Middle-income countries (Eastern Europe, Latin America, parts of Asia): Heterogeneous access with public-private divide. Brazil’s unified health system provides biologics but faces 6–12 months waiting lists affecting 40% of eligible patients [85]. Mexico’s biosimilar introduction (2016–2018) increased treatment access by 180% with infliximab biosimilar costs at 24 000 for originator [86]. Poland’s national program covers biologics for 80% of eligible patients, with biosimilar utilization reaching 75% [87].
Low-income countries (sub-Saharan Africa, South Asia): Severely limited access with <5% of eligible patients receiving biologic therapy in most regions [88]. India’s domestic biosimilar production (BOW015 infliximab, Exemptia adalimumab) achieves costs of 5000 per patient-year, enabling treatment for middle-class patients but remaining unaffordable for majority populations [89]. Public sector provision virtually absent in most countries.
Cost-effectiveness evidence
Recent pharmacoeconomic analyses support biologic therapy cost-effectiveness when considering indirect costs. The UK NICE evaluation (2023 update) found incremental cost-effectiveness ratios of £18 400–£24 600 per quality-adjusted life year (QALY) for anti-TNF therapy versus conventional treatment, well below £30 000 willingness-to-pay threshold [90]. Key drivers include reduced hospitalizations (3.2 vs. 1.4 per patient-year, saving £8400 annually), decreased surgery rates (11% vs. 23% at 5 years, saving £12 000 per avoided surgery), and improved work productivity (65% vs. 45% full-time employment, worth £9200 annually) [77].
IL-23 inhibitors demonstrate favorable cost-effectiveness versus anti-TNF agents when modeling long-term drug survival. Canadian analysis of risankizumab versus adalimumab yielded incremental cost-effectiveness ratio of CAD $32 400 per QALY, driven by 78% versus 65% drug persistence at 3 years reducing treatment switching costs [91]. However, upfront costs remain prohibitive without biosimilar alternatives.
Practical considerations in biologic therapy selection
Beyond efficacy and safety, practical factors significantly influence treatment outcomes and patient satisfaction, warranting integration into shared decision-making processes [92].
Administration routes and patient preferences
Intravenous administration (infliximab, vedolizumab) requires infusion center visits every 4–8 weeks, with 2–3 h infusion times including monitoring periods. Patient surveys indicate 62% prefer home-based administration when given choice, though 38% value supervised infusion for immediate reaction management and healthcare team interaction [93]. Infusion centers enable concurrent laboratory monitoring, therapeutic drug monitoring, and clinical assessments, potentially improving treatment optimization.
Subcutaneous administration (adalimumab, certolizumab, ustekinumab, risankizumab) enables home self-injection, with 85% of patients successfully self-administering after training [94]. Patient preference studies demonstrate higher treatment satisfaction scores with subcutaneous therapy (7.8 vs. 6.9 on 10-point scale, P < .001), driven by convenience, reduced time burden (15 min vs. 4 h including travel), and lifestyle flexibility [95]. However, 12%–15% of patients experience injection site reactions, and some report anxiety about self-administration technique [24].
Extended dosing intervals (ustekinumab every 8–12 weeks, risankizumab every 8 weeks) after induction significantly improve convenience compared to every-2-week or every-4-week regimens, with adherence rates of 89% versus 78%, respectively (P = .002) [38].
Adherence patterns and interventions
Medication non-adherence affects 25%–40% of IBD patients receiving biologic therapy, increasing relapse risk 3.2-fold (95% CI 2.1–4.8) [96]. Primary drivers include cost concerns (affecting 45% of non-adherent patients), forgetfulness (32%), needle phobia (18% for subcutaneous agents), time constraints for infusions (28%), and perceived inefficacy or side effects (24%) [97].
Evidence-based adherence interventions include:
Patient education programs: Structured education about disease mechanisms, treatment goals, and proper administration techniques improves adherence by 18%–25% [98].Reminder systems: Automated text message or app-based reminders increase on-time administration from 72% to 86% [99].Nurse-led telephone support: Monthly check-in calls addressing concerns and troubleshooting improve persistence by 22% [100].Shared decision-making: Involving patients in treatment selection increases commitment, with adherence rates of 84% when preference-concordant versus 68% when preference-discordant [101].Financial assistance programs: Co-pay assistance and patient assistance programs reduce non-adherence due to cost from 45% to 12% [102].
Impact on treatment outcomes
Adherence strongly predicts outcomes, with >80% medication possession ratio associated with clinical remission rates of 58% versus 34% with <80% adherence (P < .001) [103]. Each 10% decrease in adherence increases hospitalization risk by 15% (HR 1.15, 95% CI 1.08–1.23) and surgery risk by 12% (HR 1.12, 95% CI 1.04–1.21) [104]. Patient-centered approaches addressing practical barriers and preferences optimize both adherence and clinical outcomes.
Current limitations of monoclonal antibodies for CD treatment and their solutions are summarized in Table 3.
Potential therapeutic targets for monoclonal antibodies
IL-36 pathway
The IL-36 pathway represents an emerging therapeutic target in CD pathogenesis, with IL-36 cytokines playing crucial roles in mucosal inflammation and barrier function [109]. IL-36α, IL-36β, and IL-36γ are overexpressed in inflamed intestinal tissue from CD patients, promoting inflammatory cell recruitment and perpetuating chronic inflammation [110]. The IL-36 receptor antagonist (IL-36Ra) provides natural regulation of this pathway, and its dysregulation contributes to sustained inflammatory responses [111].
Preclinical studies demonstrate that IL-36 blockade reduces intestinal inflammation in experimental colitis models, with particular efficacy in preventing epithelial barrier dysfunction [112]. The unique role of IL-36 in coordinating innate and adaptive immune responses in the intestinal mucosa makes it an attractive target for mAb development [113]. Clinical trials investigating anti-IL-36 monoclonal antibodies are in early phases, with preliminary data suggesting potential efficacy in IBD [114].
IL-17 pathway
The IL-17 pathway, particularly IL-17A and IL-17F, plays complex roles in CD pathogenesis through effects on epithelial barrier function, neutrophil recruitment, and antimicrobial peptide production [115]. While IL-17 has protective roles in maintaining intestinal barrier integrity, excessive IL-17 signaling contributes to chronic inflammation and tissue damage [116]. The dual nature of IL-17 in IBD has complicated therapeutic development, with some anti-IL-17 therapies showing paradoxical worsening of disease [117].
Recent research has identified IL-17C as a more specific therapeutic target, with overexpression in CD tissues and pro-inflammatory effects without the protective barrier functions of IL-17A [118]. Monoclonal antibodies targeting IL-17C or its receptor IL-17RE are under development, with preclinical studies showing promise for reducing inflammation while preserving barrier function [119]. The selectivity of IL-17C targeting may overcome the limitations observed with broader IL-17 pathway inhibition [120].
SMAD7 pathway
SMAD7 represents a novel therapeutic target through its role in TGF-β signaling regulation and fibrosis development in CD [121]. SMAD7 overexpression in intestinal T cells from CD patients contributes to TGF-β resistance and perpetuates inflammatory responses [122]. Antisense oligonucleotides targeting SMAD7 have shown efficacy in clinical trials, demonstrating the therapeutic potential of this pathway [123].
Monoclonal antibodies targeting SMAD7 or its regulatory proteins offer an alternative approach to pathway modulation with potentially improved pharmacokinetics and reduced immunogenicity compared to oligonucleotide therapies [124]. The specific role of SMAD7 in intestinal fibrosis makes this target particularly relevant for preventing stricture formation and long-term complications in CD patients [125].
Complement system components
The complement system plays important roles in CD pathogenesis through multiple mechanisms including opsonization, membrane attack complex formation, and inflammatory cell activation [126]. Complement component C3 and C5 are elevated in CD patients, and complement activation products correlate with disease activity [127]. The alternative complement pathway appears particularly relevant, with dysregulation contributing to intestinal inflammation and barrier dysfunction [128].
Monoclonal antibodies targeting complement components, particularly C5 and C5a receptor, represent promising therapeutic approaches [129]. Eculizumab, an anti-C5 mAb, has shown efficacy in other inflammatory conditions and is being investigated for IBD applications [130]. The selective targeting of complement activation while preserving antimicrobial functions offers potential advantages over broader immunosuppressive approaches [131].
Microbiome-related targets
The intestinal microbiome plays crucial roles in CD pathogenesis, with dysbiosis contributing to inflammation and barrier dysfunction [132]. Specific bacterial antigens and metabolites represent novel therapeutic targets for mAb development [133]. Anti-flagellin antibodies are elevated in CD patients, suggesting potential for therapeutic targeting of specific microbial components [134].
The presence of elevated anti-flagellin antibodies (particularly anti-CBir1) in CD patients indicates an aberrant immune response to commensal bacterial flagellin, which correlates with more aggressive disease phenotypes and complications [135]. This suggests two therapeutic approaches: (1) developing antibodies that neutralize bacterial flagellin to reduce immunogenic stimulation, or (2) targeting the host immune response to flagellin. Lodes et al. (2004) demonstrated that flagellin is a dominant antigen in CD, with serologic responses present in up to 50% of patients [134]. More recent work by Targan et al. (2005) showed that anti-flagellin antibodies are associated with complicated disease behavior including stricturing and penetrating disease, with odds ratios of 2.68 and 2.49, respectively [135]. Sitaraman et al. (2005) further demonstrated that the prevalence of anti-CBir1 antibodies was significantly higher in CD patients (50%) versus ulcerative colitis (5%) and healthy controls (6%), suggesting disease specificity [136].
Monoclonal antibodies targeting bacterial toxins, adhesins, or inflammatory metabolites could provide selective antimicrobial effects while preserving beneficial microbiome functions [137]. Lipopolysaccharide-binding proteins and toll-like receptor modulators represent additional targets in the microbiome-inflammation interface [138]. The development of humanized antibodies against specific pathogenic bacteria offers precision approaches to microbiome modulation [139].
TL1A (TNF-like ligand 1A)
TL1A and its receptor DR3 (death receptor 3) have emerged as crucial mediators of intestinal inflammation in CD. TL1A is overexpressed in inflamed intestinal tissues and promotes Th1, Th2, and Th17 responses while inhibiting regulatory T-cell function [140]. The TL1A-DR3 axis also contributes to fibrosis development through effects on intestinal fibroblasts [141].
Several anti-TL1A monoclonal antibodies are currently in clinical development. PRA023 (a fully human anti-TL1A antibody) demonstrated clinical remission rates of 36.4% at week 12 in the TUSCANY trial of patients with moderate to severe ulcerative colitis [142]. TEV-48574, another anti-TL1A antibody, is currently being evaluated in Phase 2 trials for both CD and ulcerative colitis, with preliminary data suggesting favorable safety and efficacy profiles [143]. The TL1A pathway represents an attractive target due to its selective expression in inflamed intestinal tissue and its role in both inflammation and fibrosis [144].
Potential therapeutic targets for monoclonal antibodies development in CD are summarized in Table 4.
Emerging therapeutic targets and novel pathways in CD are depicted in Fig. 2.
Emerging therapeutic targets and novel pathways in Crohn’s disease.
Future perspectives
Next-generation antibody engineering
The future of mAb therapy for CD lies in advanced antibody engineering technologies that can overcome current limitations while improving therapeutic efficacy [145]. Bispecific antibodies capable of simultaneously targeting multiple pathways represent a promising approach for addressing the complex pathophysiology of CD [146]. These engineered antibodies can provide synergistic effects by blocking complementary inflammatory pathways, potentially improving response rates and reducing the risk of treatment failure [147].
Anti-TNF/IL-17 bispecific antibodies (ABT-122)
ABT-122 is a dual variable domain immunoglobulin that simultaneously targets TNF-α and IL-17A. In Phase 2 clinical trials for rheumatoid arthritis, ABT-122 demonstrated dual pathway inhibition with ACR20 response rates of 48.8% at 12 weeks compared to 32.5% with placebo [148]. Preclinical studies in murine colitis models showed that dual blockade of TNF-α and IL-17A resulted in superior reduction of intestinal inflammation (65% reduction in disease activity index) compared to single-agent targeting (40%–45% reduction) [149]. The bispecific format demonstrated improved tissue penetration in inflamed intestinal mucosa with a 2.3-fold higher concentration in inflamed versus non-inflamed tissue segments [150]. Pharmacokinetic analysis revealed a terminal half-life of 18–21 days, supporting every 2–4 weeks dosing intervals [148]. While initially developed for rheumatoid arthritis, the mechanistic rationale for IBD applications is strong given that both TNF-α and IL-17 are elevated in CD patients and contribute to epithelial barrier dysfunction.
Anti-TNF/IL-23 bispecific antibodies
Experimental dual-targeting constructs blocking TNF-α and IL-23p19 have shown promising results in preclinical IBD models. In the trinitrobenzene sulfonic acid–induced colitis model, a bispecific anti-TNF/IL-23 antibody reduced colonic inflammation scores by 78% compared to 52% with anti-TNF monotherapy and 45% with anti-IL-23 monotherapy at day 7 post-induction [151]. Histological analysis demonstrated significant improvements in crypt architecture preservation, with crypt damage scores of 1.2 ± 0.4 (bispecific) versus 2.8 ± 0.6 (anti-TNF alone) and 3.1 ± 0.5 (anti-IL-23 alone) on a 0–5 scale [151]. Mechanistic studies revealed that dual blockade prevented both acute inflammatory responses (mediated by TNF-α) and chronic adaptive immune activation (driven by IL-23), resulting in 85% reduction in mucosal Th17 cell infiltration compared to 45%–50% with monotherapy [147]. The bispecific format also demonstrated favorable immunogenicity profiles with anti-drug antibody formation in only 3.2% of treated mice versus 8%–12% with combination therapy using two separate antibodies [147].
Anti-integrin/cytokine bispecific antibodies
A novel bispecific antibody targeting both α4β7 integrin and the IL-12/IL-23 p40 subunit has been evaluated in the dextran sulfate sodium (DSS) colitis model. This dual-targeting approach achieved clinical remission (defined as disease activity index <2) in 68% of mice compared to 42% with vedolizumab-equivalent integrin blockade and 38% with ustekinumab-equivalent cytokine blockade [152]. The mechanism combines prevention of lymphocyte trafficking to intestinal tissue with local suppression of inflammatory cytokine signaling. Flow cytometry analysis demonstrated 82% reduction in mucosal CD4+ T-cell infiltration and 76% reduction in IL-17+ and IFN-γ+ T cells in mesenteric lymph nodes [152]. Importantly, the bispecific format maintained gut selectivity with minimal systemic immunosuppression, as evidenced by preserved splenic T-cell populations and normal antibody responses to systemic antigenic challenge [145]. Pharmacodynamic studies showed sustained target engagement for both α4β7 integrin (>95% receptor occupancy) and p40 neutralization (>90% cytokine blockade) over 14 days with a single dose [145].
Carcinoembryonic antigen-targeted bispecific T-cell engagers
While primarily developed for carcinoembryonic antigen (CEA)-expressing colorectal cancer, adaptations of bispecific T-cell engagers (BiTE) technology are being explored for targeted immunomodulation in IBD. Modified CEA-targeted BiTEs engineered to redirect regulatory T cells (Tregs) rather than cytotoxic T cells have shown efficacy in experimental colitis models [153]. In the IL-10-deficient mouse model of spontaneous colitis, administration of a CEA/CD25 bispecific engager that recruits Tregs to inflamed epithelium resulted in 61% reduction in colonic inflammation scores and 54% improvement in histological injury compared to control [153]. The approach leverages the fact that CEA is upregulated on inflamed intestinal epithelium in IBD patients. Ex vivo studies using intestinal biopsies from CD patients demonstrated that CEA expression was 8.4-fold higher in inflamed versus non-inflamed mucosa, providing a disease-specific targeting opportunity [154]. The bispecific engager format achieved a 12:1 ratio of Treg accumulation in inflamed versus healthy tissue segments, suggesting excellent tissue selectivity [154]. Importantly, this approach aims to restore immune homeostasis rather than broadly suppress immunity, potentially offering advantages in infection risk profiles.
The main advantages of bispecific antibodies include improved efficacy through simultaneous pathway inhibition (achieving 20%–35% better outcomes than monotherapy in preclinical models), potential for reduced immunogenicity compared to combination therapy with two separate antibodies (3%–5% vs. 8%–15% anti-drug antibody formation), and optimized pharmacokinetics with single-agent dosing [146]. However, challenges remain in manufacturing complexity due to proper heavy and light chain pairing, more complex regulatory pathways requiring evaluation of dual mechanism safety profiles, and the need to determine optimal target combinations through systematic preclinical and clinical evaluation [155].
Antibody-drug conjugates
Antibody-drug conjugates (ADCs) offer another innovative approach, combining the specificity of monoclonal antibodies with the potency of targeted drug delivery [156]. Recent developments in ADC technology for inflammatory diseases focus on delivering anti-inflammatory compounds directly to sites of inflammation, potentially reducing systemic exposure and improving safety profiles [157]. pH-sensitive linkers and novel payload designs are being optimized specifically for the inflammatory environment of the intestinal mucosa [158].
Personalized medicine approaches
The integration of genomic, proteomic, and metabolomic data is revolutionizing therapeutic selection for CD patients [159]. Multi-omics approaches can identify patient subgroups most likely to respond to specific mAb therapies, moving beyond the current trial-and-error approach [160]. Machine learning algorithms analyzing comprehensive patient datasets are being developed to predict treatment response and optimize therapeutic selection [161].
Liquid biopsies measuring circulating biomarkers, cell-free DNA, and extracellular vesicles offer real-time monitoring of treatment response and disease progression [162]. These approaches can enable dynamic treatment adjustments and early detection of treatment failure, potentially improving long-term outcomes [163]. The development of point-of-care diagnostic tools for biomarker assessment could facilitate personalized treatment decisions in clinical practice [164].
Combination therapy strategies
Future therapeutic approaches increasingly focus on rational combination strategies that target multiple pathways simultaneously while minimizing additive toxicities [165]. Sequential therapy protocols, where patients receive different monoclonal antibodies in predetermined sequences based on treatment response, are being investigated in clinical trials [7]. These approaches aim to prevent the development of resistance and maintain long-term efficacy [34].
Novel combination approaches include pairing monoclonal antibodies with small molecule inhibitors, cell-based therapies, or microbiome interventions [166]. The combination of anti-TNF therapy with JAK inhibitors or with specific microbiome modulators has shown promising results in preclinical studies [167]. These combinations may provide additive or synergistic effects while potentially reducing the risk of treatment failure [168].
Advanced delivery systems
Innovative delivery systems are being developed to improve the pharmacokinetics and reduce the immunogenicity of mAb therapies [169]. Nanoparticle-based delivery systems can protect antibodies from degradation, provide controlled release, and enable targeted delivery to specific intestinal segments [170]. These systems may reduce dosing frequency and improve patient compliance while maintaining therapeutic efficacy [171].
Oral delivery of monoclonal antibodies represents a major advancement in patient convenience and treatment accessibility [172]. Novel formulations using enteric coatings, nanoencapsulation, and permeation enhancers are being investigated to overcome the challenges of oral protein delivery [173]. Successful oral formulations could dramatically improve patient acceptance and reduce healthcare costs associated with parenteral administration [174].
Change in therapeutic positioning based on monoclonal antibodies
The traditional treatment algorithm has positioned anti-TNF agents as first-line biologic therapy based on extensive long-term data and established efficacy. However, emerging evidence challenges this paradigm. Network meta-analyses by Singh et al. (2020) comparing different biologic classes in bio-naïve patients demonstrated that risankizumab achieved numerically higher clinical remission rates (45%–52%) compared to adalimumab (36%–47%) at 1 year, though head-to-head trials are limited [175]. More importantly, selective IL-23 inhibitors demonstrate superior drug persistence, with 12-month continuation rates of 78%–82% for risankizumab versus 65%–70% for anti-TNF agents in real-world cohorts [176]. Added 2024 registry data from ICARUS comparing IL-23 inhibitors versus anti-TNF agents in clinical practice, showing 12-month clinical remission rates of 52% versus 46% (P = .04) [177].
Several factors support earlier use of IL-23 inhibitors: (1) lower immunogenicity (1%–3% vs. 10%–30% for anti-TNF agents) reduces the risk of secondary loss of response [12]; (2) more favorable safety profile with lower rates of serious infections (2.8 per 100 patient-years vs. 4.6 for anti-TNF therapy) [178]; (3) preserved treatment options, as patients can still respond to anti-TNF therapy after IL-23 inhibitor failure, whereas anti-TNF failure predicts poorer responses to subsequent biologics [179]; and (4) evidence of disease modification with reduced stricture formation in long-term extension studies [41].
However, anti-TNF agents retain important advantages: (1) faster onset of action (median time to response: 2–4 weeks versus 6–8 weeks for IL-23 inhibitors) [11]; (2) more extensive safety data spanning over 20 years of clinical use; (3) availability of biosimilars reducing treatment costs by 20%–40% [76]; and (4) established efficacy in perianal fistulizing disease, where anti-TNF agents demonstrate 50%–60% response rates compared to limited data for IL-23 inhibitors [180].
The 2023 STRIDE-II consensus recommended individualized treatment selection based on disease phenotype, with anti-TNF agents preferred for penetrating disease and urgent symptom control, while IL-23 inhibitors may be preferred for patients prioritizing long-term drug survival, those with infection concerns, or inflammatory phenotypes [181]. Ongoing head-to-head trials (SEQUENCE and EXPLORER) directly comparing anti-TNF versus IL-23 inhibitors as first-line therapy will provide definitive guidance [182].
Multi-omics technologies
Recent advances in spatial and multi-omics technologies have opened new avenues for mAb development in CD by providing unprecedented insights into immune cell heterogeneity, tissue microenvironments, and inflammatory signaling at single-cell resolution. High-plex protein and transcriptome co-mapping methods, such as spatial CITE-seq, enable simultaneous profiling of RNA and surface proteins within intact tissues, facilitating the identification of disease-specific immune signatures and potential therapeutic targets [183]. Similarly, multimodal tri-omics spatial mapping of brain and immune tissues has demonstrated how integrated transcriptomic and proteomic data can reveal spatially resolved networks driving inflammation [184]. Moreover, the application of panoramic in vivo CRISPR screening platforms such as Perturb-DBiT allows functional validation of gene targets within complex tissue contexts, accelerating discovery of antibody-accessible molecules with therapeutic potential [185]. Together, these emerging technologies provide a systems-level framework for rational design and optimization of monoclonal antibodies targeting the multifactorial immune dysregulation underlying CD.
Conclusions
The analysis of monoclonal antibodies in CD therapy reveals both significant achievements and persistent challenges that require continued innovation and research. Current therapeutic options, including anti-TNF agents, anti-integrin antibodies, and anti-IL-12/IL-23 therapies, have revolutionized disease management and improved patient outcomes. However, substantial limitations including primary non-response, secondary loss of response, immunogenicity, safety concerns, and economic barriers continue to limit optimal therapeutic outcomes.
The identification of novel therapeutic targets, including IL-36, IL-17C, SMAD7, complement components, and microbiome-related factors, offers promising avenues for future drug development. These targets may provide improved efficacy, reduced side effects, and solutions for treatment-refractory patients. The integration of personalized medicine approaches, advanced antibody engineering, and innovative delivery systems will likely transform the therapeutic landscape for CD in the coming years.
Future success in mAb therapy for CD will depend on continued research into disease mechanisms, development of predictive biomarkers, and implementation of precision medicine approaches. The ultimate goal remains achieving sustained remission for all patients while minimizing treatment-related complications and improving quality of life through safe, effective, and accessible therapeutic interventions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Roda G, Chien Ng S, Kotze PG. et al. Crohn's disease. Nat Rev Dis Primers 2020;6:22. 10.1038/s 41572-020-0156-2.32242028 · doi ↗ · pubmed ↗
- 2Torres J, Mehandru S, Colombel JF. et al. Crohn's disease. Lancet. 2017;389:1741–55.27914655 10.1016/S 0140-6736(16)31711-1 · doi ↗ · pubmed ↗
- 3Ng SC, Shi HY, Hamidi N. et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769–78. 10.1016/S 0140-6736(17)32448-0.29050646 · doi ↗ · pubmed ↗
- 4Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol 2021;18:56–66.33033392 10.1038/s 41575-020-00360-x PMC 7542092 · doi ↗ · pubmed ↗
- 5Cosnes J, Gower-Rousseau C, Seksik P. et al. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785–94. 10.1053/j.gastro.2011.01.055.21530745 · doi ↗ · pubmed ↗
- 6Park KT, Ehrlich OG, Allen JI. et al. The cost of inflammatory bowel disease: an initiative from the Crohn's & colitis foundation. Inflamm Bowel Dis 2020;26:1–10. 10.1093/ibd/izz 104.31112238 PMC 7534391 · doi ↗ · pubmed ↗
- 7Danese S, Vuitton L, Peyrin-Biroulet L. Biologic agents for IBD: practical insights. Nat Rev Gastroenterol Hepatol 2015;12:537–45. 10.1038/nrgastro.2015.135.26284562 · doi ↗ · pubmed ↗
- 8Lichtenstein GR, Loftus EV, Isaacs KL. et al. ACG clinical guideline: management of Crohn's disease in adults. Am J Gastroenterol 2018;113:481–517.29610508 10.1038/ajg.2018.27 · doi ↗ · pubmed ↗