Proximity-ligation metagenomics reveals disease-specific mobilome dynamics in disrupted gut ecosystems
Andrew J Sommer, Benjamin Auch, Alexander Khoruts, Jasmohan S Bajaj

TL;DR
This study uses a new metagenomic method to show how gut microbiome disruptions in diseases like C. difficile infection and cirrhosis affect antimicrobial resistance and virulence gene spread.
Contribution
The novel use of proximity ligation metagenomics reveals distinct mobilome dynamics in gut diseases.
Findings
Both rCDI and cirrhosis show increased chromosomal antibiotic resistance determinants.
Plasmid-mediated resistance amplification is more prominent in rCDI.
Ecological pressures from disease and antibiotics shape mobilome dynamics differently.
Abstract
Distinct ecological pressures shape accumulation of antimicrobial resistance and virulence genes in the gut microbiome. Using proximity ligation shotgun metagenomics to resolve host–mobilome relationships, we analyzed microbiomes from two patient cohorts: recurrent Clostridioides difficile infection (rCDI) and cirrhosis. While rCDI reflects antibiotic-driven disruption, cirrhosis-driven microbiome changes result from altered gut physiology. We found increased chromosomal determinants of antibiotic resistance in both, but plasmid-mediated amplification was more evident in rCDI.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
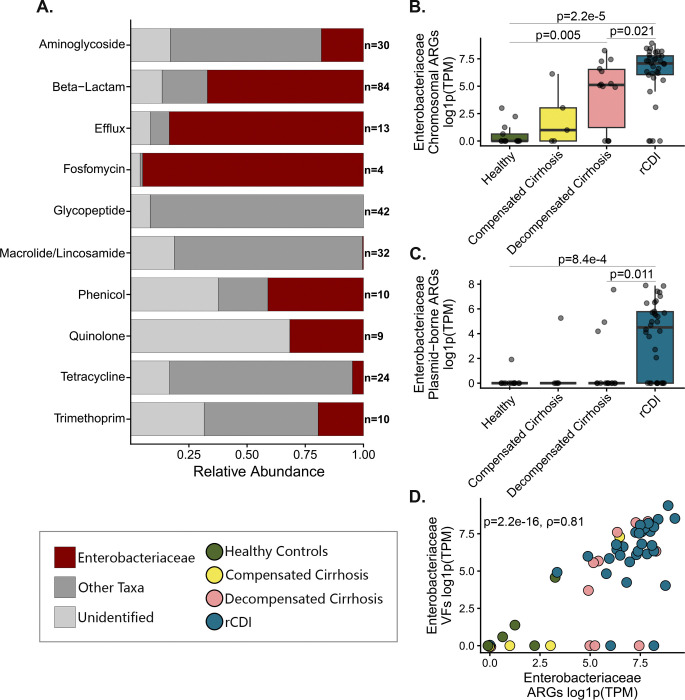 Figure 1
Figure 1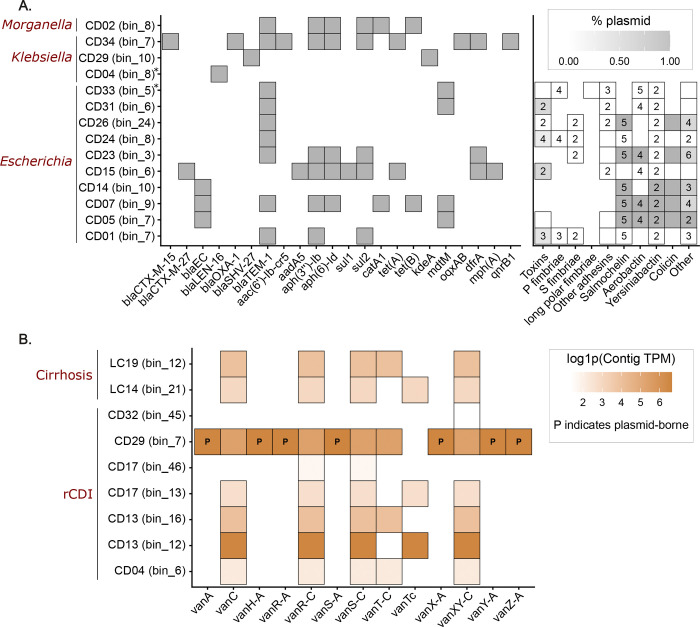 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsClostridium difficile and Clostridium perfringens research · Gut microbiota and health · Bacteriophages and microbial interactions
The human gut microbiome plays a central role in host physiology and colonization resistance against pathogens^1^, yet it also represents a major reservoir of antimicrobial resistance genes (ARGs) and virulence factors (VFs)^2^. High bacterial density and spatial organization within the gut create favorable conditions for horizontal gene transfer (HGT). These processes allow resistance and virulence traits to disseminate rapidly across taxa and contribute to the emergence of multidrug-resistant opportunistic pathogens^2,3^.
Members of the Enterobacteriaceae and Enterococcaceae families are of particular concern because they frequently cause bloodstream, intra-abdominal and urinary tract infections in medically complex patients. The surveillance of plasmid-borne extended spectrum β-lactamases (ESBLs) and vancomycin resistance genes (VRGs) within pathogenic lineages is of urgent clinical relevance; however, conventional shotgun metagenomics cannot reliably assign mobile genetic elements or ARGs to bacterial hosts^4^. Proximity ligation shotgun metagenomics (PLSM), also referred to as Hi-C metagenomics, addresses this limitation by cross-linking DNA molecules that are co-localized within intact cells prior to sequencing, allowing plasmids and bacteriophages to be assigned directly to host genomes and enables direct reconstruction of host-mobilome interactions^5,6^
We applied PLSM to fecal microbiomes from healthy individuals and two disease states associated with contrasting disrupted gut ecosystems: recurrent Clostridioides difficile infection (rCDI) and cirrhosis. rCDI microbiomes experience prolonged antibiotic exposure and inflammatory perturbations, whereas cirrhosis is characterized by liver and immune dysfunction, altered gut physiology, and impaired barrier integrity, typically before extensive antibiotic use^7–9^. Comparing these cohorts therefore provides an opportunity to distinguish antibiotic-driven from physiologically-driven influences on mobilome evolution.
We analyzed 66 stool samples [34 rCDI, 19 cirrhosis (5 compensated/14 decompensated) and 13 healthy, extended tables/text]. rCDI patients had prolonged antibiotic exposure for recurrent infection (6.9 ± 3.9 months). In contrast, we only included cirrhosis patients without absorbable antibiotic exposure, although half of decompensated patients received rifaximin for hepatic encephalopathy (HE) prophylaxis. PLSM generated an average of ~61 million reads per sample and enabled assembly of 2,070 bacterial metagenome-assembled genomes (MAGs) with ≥70% completeness and ≤5% contamination.
Healthy individuals exhibited microbiomes dominated by obligate anaerobic Clostridia, including Lachnospirales and Oscillospirales (extended Data Fig.1). In contrast, both rCDI and decompensated cirrhosis microbiomes showed increased abundance of Enterobacteriaceae and Enterococcaceae. Community disruption was most pronounced in rCDI samples, which also contained MAGs absent from healthy individuals, including Veillonellaceae and Fusobacteriaceae. Cirrhosis samples displayed a less pronounced shift toward Enterobacteriaceae, with a visual gradient between compensated and decompensated disease. These findings suggest that host physiological changes alone can create ecological conditions favoring facultative anaerobes.
Across all samples we assembled 705 plasmids and 2,521 bacteriophages with ≥50% completeness and ≤5% contamination. Although total plasmid abundance did not differ significantly between cohorts, plasmid host associations differed markedly (extended Data Fig.2). In healthy individuals, plasmids were primarily linked to Bacteroidaceae. In contrast, both rCDI and cirrhosis microbiomes exhibited increased plasmid associations with Enterobacteriaceae. Enterobacteriaceae-associated plasmids were significantly enriched in rCDI compared with both cirrhosis and healthy cohorts, whereas Bacteroides-associated plasmids declined. Thus, rCDI was characterized not simply by increased plasmid abundance but by a shift toward plasmids carried by clinically high-risk taxa. Notably, substantial inter-individual variability was observed, indicating that mobilome composition can differ markedly even within similar clinical contexts.
Enterobacteriaceae carried the largest ARG burden across all cohorts (Fig.1a). Both rCDI and cirrhosis microbiomes exhibited increased chromosomally encoded ARGs relative to healthy controls, consistent with expansion of Enterobacteriaceae taxa harboring intrinsic resistance determinants such as efflux pumps and β-lactamases (Fig.1b). In contrast, plasmid-borne ARGs showed a striking disease-specific pattern. Plasmid-encoded β-lactamases, including blaTEM-1, blaOXA-1, blaSHV-27 and blaCTX-M, were detected in rCDI microbiomes and were absent from cirrhosis and healthy samples despite cirrhosis-associated Enterobacteriaceae expansion and rifaximin use (Fig.1c extended data Fig.3). These findings suggest that intense antibiotic exposure in rCDI creates ecological conditions that favor plasmid-mediated horizontal gene transfer and maintenance of resistance elements, whereas physiological disruption in cirrhosis without absorbable antibiotics does not drive widespread plasmid-mediated ARG amplification.
ARG and VF abundance within Enterobacteriaceae showed a strong correlation (ρ = 0.81), driven largely by Escherichia MAGs (Fig.1d and extended data Fig.4). In rCDI samples, MAGs encoding plasmid-borne β-lactamases frequently co-encoded plasmid-borne siderophore systems, adhesins and colicin genes characteristic of extraintestinal pathogenic E. coli (ExPEC) (Fig.2a)^10,11^. In contrast, Klebsiella MAGs encoding plasmid β-lactamases rarely carried additional acquired virulence determinants, possibly reflecting the importance of variable capsular polysaccharide and lipopolysaccharide loci in the K. pneumoniae core genome^12^. These observations suggest that E. coli may act as a key mobilome hub linking resistance and virulence traits within gut reservoirs.
While VRGs were absent from healthy microbiomes, Enterococcus MAGs encoding VRGs were identified in several rCDI and cirrhosis patients (Fig.2b and extended data Fig.5). A single rCDI sample also contained a C. difficile MAG carrying VanG-type resistance genes, potentially enabling survival during ongoing vancomycin therapy. VRGs were also detected in cirrhosis patients without prior vancomycin exposure, reflecting a shift towards gram-positive taxa changes in cirrhosis and suggesting that ecological disruption alone may permit colonization by resistant Enterococcus populations^13^.
Despite enrichment of ARGs and VFs in disease cohorts, we observed no consistent relationship between resistance burden and conventional healthcare exposure metrics such as antibiotic duration, care intensity, or patient demographics (extended data Fig.6 and Fig.7). Some rCDI patients with modest healthcare exposure carried high ARG/VF loads, whereas others with extensive treatment histories did not. Similarly, rifaximin use, which has been linked to increased resistance to the last-resort antibiotic daptomycin^14^, was not significantly associated with increased Enterobacteriaceae ARG or VF abundance in cirrhosis patients (extended data Fig.7). When we compared the 10 rifaximin users (3 rCDI+7 decompensated cirrhosis) to non-users (decompensated and rCDI), we found no statistical differences in Enterobacteriaceae VF, Enterobacteriaceae ARG, and VRG abundance across samples (extended data Fig.8). These findings indicate that accumulation of resistance and virulence genes reflects complex interactions among microbial community structure, host physiology, environmental exposure, and mobile genetic element dynamics rather than simple antibiotic burden alone.
Limitations of this study include modest sizes of patient cohorts, clinical presentations potentially reflecting center-specific practices, enrolling of cirrhosis patients with minimal systemic antibiotic exposure, and the sequencing of a single fecal sample time point per patient. Nonetheless these findings demonstrate that distinct ecological pressures shape mobilome evolution in disrupted gut microbiomes. rCDI microbiomes, subjected to sustained antibiotic exposure, exhibit strong plasmid-mediated amplification of resistance genes and coupling of resistance and virulence traits within E. coli. In contrast, cirrhosis microbiomes accumulate genomic ARGs despite relatively limited absorbable antibiotics exposure, indicating that host-driven ecological disruption can prime microbiomes for resistance acquisition.
By resolving host–mobile element relationships at scale, PLSM provides a powerful tool for studying the evolutionary processes that drive the emergence of resistant and virulent pathobionts within the human microbiome. These results from two distinct but common conditions, rCDI and cirrhosis, both associated with disrupted gut ecosystems, highlight the importance of ecological context in shaping resistance reservoirs and underscore the need for microbiome-preserving therapeutic strategies.
Supplementary Material
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
• ExtendedDataMethods31626.docx
• Extendeddataresultstext31626.docx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sommer F. C Bäckhed F. Nat. Rev. Microbiol. 11, 227–238 (2013).23435359 10.1038/nrmicro 2974 · doi ↗ · pubmed ↗
- 2Anthony W. E. Infect. Dis. 223, S 209–S 213 (2021).
- 3Smillie C. S. Nature 480, 241–244 (2011).22037308 10.1038/nature 10571 · doi ↗ · pubmed ↗
- 4Murray C. J. L. The Lancet 3GG, 629–655 (2022).
- 5Bryson S. Gut Microbes 17, 2559019 (2025).40948444 10.1080/19490976.2025.2559019 PMC 12439552 · doi ↗ · pubmed ↗
- 6Beitel C. W. Peer J 2, e 415 (2014).24918035 10.7717/peerj.415PMC 4045339 · doi ↗ · pubmed ↗
- 7Feuerstadt P., Theriault N. C Tillotson G. BMC Infect. Dis. 23, 132 (2023).36882700 10.1186/s 12879-023-08096-0PMC 9990004 · doi ↗ · pubmed ↗
- 8Bajaj J. S. N. Engl. J. Med. 384, 2317–2330 (2021).34133861 10.1056/NEJ Mra 2021808 · doi ↗ · pubmed ↗