PaREx: an open-source pipeline for the automated analysis of Pseudomonas aeruginosa resistomes from whole-genome sequences
Carla López-Causapé, Matias Bonet, Biel Taltavull, Paola Medina-Retiga, Miquel A. Sastre-Femenía, Sara Cortés-Lara, María A. Gomis-Font, Fernando Gomez-Romano, Antonio Oliver

TL;DR
PaREx is a new automated tool for analyzing antibiotic resistance in Pseudomonas aeruginosa using whole-genome sequencing data.
Contribution
PaREx introduces an open-source pipeline and web tool for accurate resistome profiling in P. aeruginosa, including detection of mutation-driven resistance and AmpC variants.
Findings
PaREx accurately analyzes P. aeruginosa resistomes using 221 chromosomal genes and horizontally acquired resistance mechanisms.
The PDC analyzer web tool effectively designates PDCs and detects resistance to novel β-lactam antibiotics across thousands of genomes.
PaREx's performance matches manual analysis when calculating genotypic resistance scores from 260 isolates.
Abstract
Whole-genome sequencing (WGS) has become a valuable tool for bacterial typing and resistome analysis. However, most existing antibiotic resistance databases and bioinformatics tools are not useful for P. aeruginosa resistome profiling as they fail to detect mutation-driven antibiotic resistance mechanisms. In addition, the increasing diversity of AmpC variants (Pseudomonas-derived cephalosporinases [PDCs]) in recent years adds complexity to P. aeruginosa resistome profiling. To address these challenges, we have developed PaREx, an open-source automated pipeline specifically designed for the analysis of P. aeruginosa mutation-driven (221 chromosomal genes) and horizontally acquired resistome from whole-genome sequences, along with the PDC analyzer web tool, a user-friendly platform that designates PDCs and detects resistance to novel antipseudomonal β-lactams. The utility of Pseudomonas…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
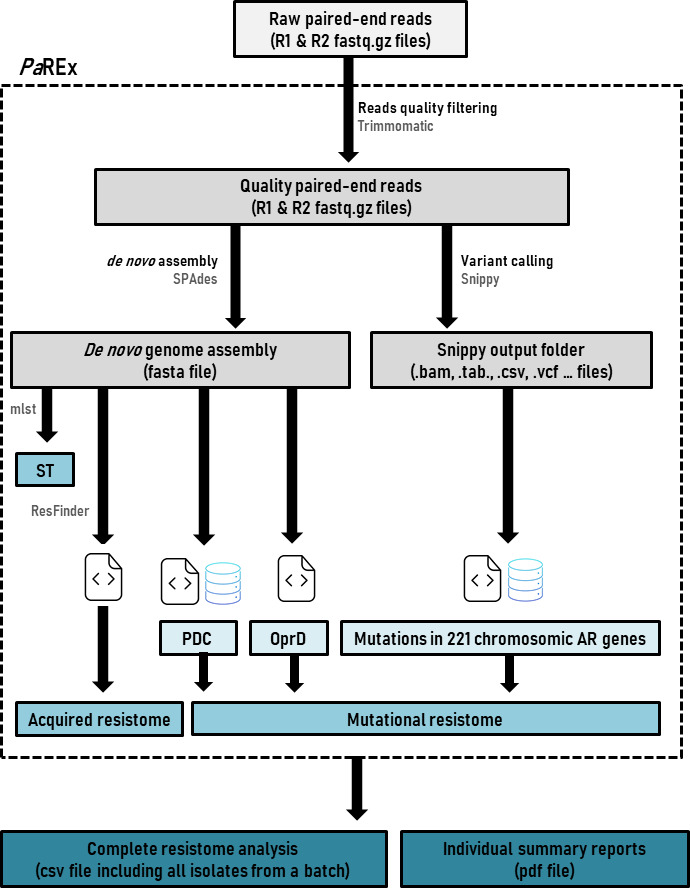 Fig 1
Fig 1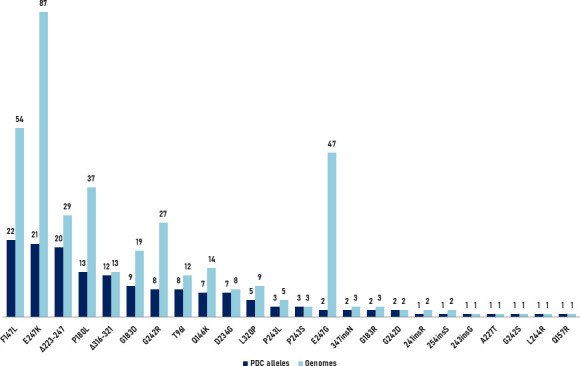 Fig 2
Fig 2| Name and version | Description |
|---|---|
| Trimmomatic v0.39 | A flexible read trimming tool for Illumina reads |
| SPAdes v3.15.3 | A |
| Snippy | A tool for rapid haploid variant calling and core genome alignment |
| mlst | A tool to scan contig files against PubMLST typing schemes |
| ResFinder | A tool (v4.5.0) and database (v2.1.0) for the identification of acquired antimicrobial resistance genes |
| Operation name | Running time | Output | |
|---|---|---|---|
| Decompress fastq.gz files | Unzip | 2 h 35 min | 854.2 GB |
| Read quality filtering | Trimmomatic | 6 h 17 min | 567.2 GB |
| SPAdes | 1d 1h 13 min | 1.8 GB | |
| Multilocus sequence typing (MLST), resistome characterization, and summary reports generation | Resistome | 15 h 6 min | 112.7 GB |
| Antibiotic | Indeterminate score values (%) | CA (%) | mE(%) | ME (%) | VME (%) |
|---|---|---|---|---|---|
| CAZ | 13.8 | 91.2 | – | 4.4 | 4.4 |
| TOL/TZ | 7.3 | 98.3 | – | 0 | 1.7 |
| MER | – | 75.0 | 21.1 | 2.7 | 1.1 |
| CIP | 10.4 | 91.8 | – | 1.3 | 6.9 |
| TOB | 10.4 | 98.7 | – | 0.9 | 0.4 |
- —Instituto de Salud Carlos IIIhttp://dx.doi.org/10.13039/501100004587
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Bacterial biofilms and quorum sensing · Genomics and Phylogenetic Studies
INTRODUCTION
Pseudomonas aeruginosa is one of the most frequent and severe causes of nosocomial infections, being the first cause of ventilator-associated pneumonia and particularly affecting intensive care units and immunocompromised patients (1). Likewise, P. aeruginosa is the most frequent driver of chronic respiratory infections in individuals with cystic fibrosis or patients with other chronic pulmonary conditions (2, 3).
Moreover, in recent years, the global prevalence of infections caused by multidrug-resistant (MDR) and extensively drug-resistant (XDR) P. aeruginosa strains has increased worldwide, significantly compromising and limiting the selection of effective antimicrobial therapies (1). This global threat results from a complex interplay in which the extraordinary ability of P. aeruginosa to develop resistance to nearly all available antimicrobials through the acquisition of chromosomal mutations, along with an increasing prevalence of horizontally acquired resistance determinants in this pathogen, and the nosocomial spread of certain P. aeruginosa high-risk clones worldwide play a major role (4).
Despite novel antipseudomonal agents being recently introduced into clinical practice contributing to mitigate this problem, carbapenem-resistant P. aeruginosa remains a major concern for public health and has been classified as a high-priority pathogen in the World Health Organization’s Bacterial Pathogen Priority lists (5). In this context, the development of robust tools for accurate characterization of P. aeruginosa resistomes would be very helpful for the design of therapeutic strategies, for monitoring and preventing resistance development, and for establishing effective infection control measures.
As high-throughput sequencing technologies have become increasingly affordable, many clinical and research microbiology laboratories have introduced their use in routine practice, making both bacterial typing and characterization of bacterial resistomes possible in a single assay. However, the analysis of whole-genome sequences (WGS) requires advanced bioinformatics skills and the use of different open-source software, databases, and tools. To facilitate these analyses, bioinformatic pipelines, such as BacPipe or Bactopia that integrates different parts of the analysis (6, 7), or even web platforms such as the Center for Genomic Epidemiology (https://genepi.dk/), which provides user-friendly tools such as ResFinder (8), have been developed in recent years. However, these pipelines and platforms are not pathogen-specific and use generic antibiotic resistance databases such as the ResFinder (9, 10), the Comprehensive Antibiotic Resistance Database CARD (11), or the NCBI AMRFinderPlus (12) databases, limiting resistome characterization of bacterial isolates to the detection of acquired resistance genes and some specific chromosomal resistance mutations. Unfortunately, these tools are not useful for the characterization of P. aeruginosa resistomes as in this pathogen, mutation-driven mechanisms play a major role in antibiotic resistance. Thus, the investigation of P. aeruginosa resistomes from whole-genome sequencing data requires highly time-consuming manual analysis and expertise on P. aeruginosa resistance mechanisms and genomics (13). Moreover, in recent years, and largely due to the introduction of novel beta-lactam/beta-lactamase inhibitor combinations into the clinical practice, there is a growing diversity and emerging role in resistance of AmpC variants (known as Pseudomonas-derived cephalosporinases [PDCs]) (14). Thus, an automated pipeline specifically designed for establishing P. aeruginosa antibiotic resistance genotypes, including PDC designation, is needed.
In this work, we introduce Pseudomonas aeruginosa Resistome Explorer (PaREx), an open-source pipeline for the automated analysis of P. aeruginosa resistomes from whole-genome sequences, which benefits from the use of different open-source and public bioinformatics tools, software, and databases along with custom-built databases and tools, enabling an accurate resistance genotype profiling of P. aeruginosa isolates.
RESULTS AND DISCUSSION
PaREx design and implementation
The PaREx is an open-source Python-based customizable pipeline that has been specifically designed for the automated analysis of P. aeruginosa resistomes from Illumina paired-end reads. PaREx uses different open-source bioinformatics tools, software, and publicly available databases along with custom-built databases, scripts, and tools and is composed of two main components: the PaREx pipeline and the PaREx databases. An overview of PaREx is represented in Fig. 1.
Overview of the PaREx pipeline.
As shown, PaREx comprises different steps that have been further detailed in the Methods and Materials section. Third-party open-source tools used by the PaREx pipeline are listed in Table 1 with their individual version numbers.
Validation of PaREx functionalities
A total of 260 P. aeruginosa clinical isolates were analyzed for the validation of PaREx functionalities using the batch mode. The csv summary file containing the compiled results for all the isolates and some of the individual pdf summary reports have been included in the Data S1 and S2 as examples of PaREx’ final reports.
The analysis of the 260 isolates took about 49 h with 8 CPUs, processing 188.7 GB of compressed input data (854.2 GB uncompressed) and generating a total output of 1.5 TB. Table 2 summarizes the detailed run times and output size for each step of the PaREx pipeline.
PaREx was able to assign a sequence type (ST) to 92% (239/260) of the isolates. Those for which an ST was not assigned harbored a new MLST allele (n = 11) that was not included in the database, represented a new combination of the MLST alleles (n = 3), or contained some MLST allele that was not completely covered (n = 7). Within the whole collection, 151 different STs were detected, being ST175 (n = 14; 10 XDR, 1 MDR, and 3 modR), ST244 (n = 11; 1 MDR, 3 modR, and 7 S), and ST253 (n = 10; 1 XDR, 1 MDR, 1 modR, and 7 S), detected in 10 or more isolates.
P. aeruginosa XDR ST175 is a high-risk clone widely distributed in Spain and is characterized by several genetic resistance markers, including the acquisition of the aminoglycoside-modifying enzyme ant(2’’)-Ia (aadB) and the presence of specific resistance mutations in its chromosomal genes gyrA (T83I and D87N), parC (S87W and L168Q), and mexZ (G195E) (15–17). PaREx reported the presence of the ant(2’’)-Ia aminoglycoside-modifying enzyme (100% identity) and parC mutations in all 10 XDR ST175 isolates as well as the characteristic mutations in the quinolone resistance-determining region of GyrA (T83I: 10/10 and D87N: 9/10) and MexZ (9/10) in almost all. Other resistance mutations previously reported among XDR ST175 Spanish isolates, such as the Q142X-inactivating mutation in the OprD porin and the G154R gain-of-function mutation in the ampC cephalosporinase regulator AmpR, were detected in 2 of them (15–17). Moreover, in 2 XDR ST175 isolates, PaREx reported the presence of blaVIM-2 (n = 2) along with either a blaOXA-2 or a blaOXA-210 gene, determinants also previously described in specific lineages of this high-risk clone (17).
Among the other 11 XDR isolates included in the collection used for PaREx validation, 4 belong to high-risk clone ST235 and 1 to high-risk clone ST111, these clones being characterized by the frequent horizontal acquisition of resistance gene determinants (4, 18). Accordingly, in all XDR ST235 and ST111 isolates, PaREx reported the presence of a horizontally acquired beta-lactamase along with aminoglycoside-acquired resistance genes.
Of note, for isolates exhibiting a susceptible phenotype (n = 165), PaREx did not detect any acquired beta-lactamase gene and any acquired aminoglycoside resistance genes. Likewise, on average, PaREx reported a minor number of resistance mutations within the basic resistome chromosomal genes for these isolates compared to those exhibiting multidrug-resistant and extensively drug-resistant phenotypes: 2.3 (S) vs 5.3 (MDR) and 7.7 (XDR).
Moreover, in previous studies, we developed and validated a genotypic scoring system based on the manual analysis of acquired resistance determinants and mutations within 40 chromosomal genes from whole-genome sequences with the aim to predict P. aeruginosa phenotypic susceptibility to ceftazidime (CAZ), ceftolozane/tazobactam (TOL/TZ), meropenem (MER), ciprofloxacin (CIP), and tobramycin (TOB) (13, 19, 20). Score values under 0.5 points were intended to predict phenotypic susceptibility, specifically, susceptibility at increased exposure for CAZ and CIP, and susceptibility at standard dosing for TOB, TOL/TZ, and MER. Conversely, score values equal to or above 1 were intended to predict a resistance phenotype, whereas intermediate score values (0.5 to <1) were considered indeterminate, with the exception of MER, for which these values should predict susceptibility at increased exposure. As manual resistome analysis in P. aeruginosa is very time-consuming and requires expertise on P. aeruginosa resistance mechanisms and genomics, its automatization can significantly reduce the turn-around time of the scoring system. Therefore, to demonstrate the potential utility of PaREx for this purpose, the PaREx final report was used for calculating the genotypic scores (Data S3). Table 3 summarizes the categorical agreement (CA) and the minor (mE), major (ME), and very major (VME) errors rates obtained for each antibiotic.
As shown, the major error rate for ceftazidime was >3%, and very major errors rates for ceftazidime, ceftolozane/tazobactam, and ciprofloxacin were >1.5% and thus would fall out of the limits established by ISO 20776-2:2021 (21). These numbers are, however, quite similar to those extracted from the original study reporting the scoring system (CAZ: indeterminate 20.6%, CA 93.1%, ME 5%, and VME 1.9%; TOL/TZ: indeterminate 9.3% and CA 100%; CIP: indeterminate 11.3%, CA 95%, ME 2.8%, and VME 2.2%) (13) and thus confirm the utility of PaREx for the automatic analysis of P. aeruginosa resistomes. Indeed, genotype-phenotype discrepancies might be attributed to different contributing factors. First, since validation of the genotypic scores was not the primary aim of this work, there exists a potential risk that discrepancies may result from experimental errors as minimum inhibitory concentrations (MICs) were determined only once per isolate as well as whole-genome sequencing. In addition, some of the observed discrepancies may be linked to limitations in the PaREx approach, as, for instance, the current version does not investigate the presence of large deletions that can impact phenotypic resistance. Moreover, in PaREx, the detection of resistance mutations relies on a variant calling analysis, and the inclusion of bioinformatics analysis based on the de novo assemblies could perhaps increase the detection of resistance mutations in some instances. Moreover, encountered sequence variants are filtered using a large list of natural polymorphisms, but since it may still not be fully saturated, variants not linked to antibiotic resistance could still be present. Finally, significant gaps remain in the understanding of P. aeruginosa resistance mechanisms and its complex interplay. Thus, further and continuous refinement of the genotypic score system is needed to improve its predictive accuracy. Despite these discrepancies, the PaREx pipeline constitutes a significant advancement by considerably reducing times and the expertise required for the analysis of P. aeruginosa resistomes from whole-genome sequences that can eventually be applied for this or other purposes such as resistance surveillance.
The PDC analyzer web tool
In addition to PaREx, we have developed the PDC analyzer web tool, which is available at: https://arpbigidisba.com. This is a user-friendly web tool based on the PDC designation module included in the PaREx pipeline. In addition, to inform users about the detected PDC allele variant and the amino acid variations encountered relative to the PAO1 reference cephalosporinase sequence (PDC-1), the PDC analyzer web tool additionally provides a warning message if a mutation associated with resistance and/or that may contribute to resistance to novel antipseudomonal cephalosporins, such as ceftolozane-tazobactam, ceftazidime-avibactam, and/or cefiderocol, is detected (Data S4). Conversely, the PDC analyzer web tool also alerts when a deleted (<90% coverage) or nonfunctional PDC is detected (premature stop codons or frameshift mutations).
Validation of the PDC analyzer web tool
Mack and collaborators (22) recently explored the presence of β-lactamase alleles in a global collection of 30,452 P. aeruginosa genomes, encountering that blaPDC was the most common bla gene family (30,842 genes). In that work, a known PDC allele variant was assigned in 29,369 isolates (254 distinct alleles), whereas in 1,144, an unknown PDC allele variant was detected (622 distinct alleles). To validate the PDC analyzer web tool and to determine the global prevalence of mutations linked to resistance to the novel antipseudomonal combinations ceftolozane/tazobactam, ceftazidime/avibactam, and cefiderocol in P. aeruginosa, we analyzed the 863 distinct PDC allele variants for which an accession number was available at the National Center for Biotechnology Information database.
The PDC analyzer web tool reported 593 alleles as new types (68.7%). Among the 270 alleles included in the PDC analyzer database, 67 alleles with mutations associated with resistance and 21 alleles with mutations that may contribute to resistance against novel antipseudomonal cephalosporins were detected. Likewise, 77 alleles with mutations associated with resistance and 39 alleles with mutations that may contribute to resistance were detected among the new allele types. Of note, in 14 PDC alleles, more than 1 resistance mutation was detected.
For each mutation associated with resistance identified with the PDC analyzer web tool, the corresponding counts of PDC alleles and genomes carrying each mutation are represented in Fig. 2.
PDC alleles and total genome counts per resistance mutation detected with the PDC analyzer web tool.
The PDC analyzer tool identified 24 different resistance mutations, with E247K/G, F147L, and P180L substitutions being the most frequently detected, along with mutations affecting the omega- (223–247) and the R2-loops (316–321). Of note, although the E247G mutation was present in 47 isolates, it was only associated with 2 different PDC alleles, suggesting that it may be related to the clonal dissemination of some specific ST.
In addition, 2 mutations that may contribute to resistance were detected: V239A and R126H; of note, V239A was identified in 57 different alleles and 250 genomes. These mutations were excluded from Fig. 2 because of the scale difference.
Based on this analysis, we can estimate that the global prevalence of reported genomes showing PDC resistance mutations to the novel antipseudomonal cephalosporins in P. aeruginosa is 1.1% (1.8% if the V239A and R126H mutations are also included). Remarkably, we found that 42.5% of the genomes for which a resistance mutation was identified contained a new type PDC allele; therefore, the approach applied by the PDC analyzer web tool, which focuses on detecting specific AmpC mutations rather than detecting PDC allele variants, offers a major resolution in WGS-based resistance prediction to the new antipseudomonal cephalosporins.
Concluding remarks
The implementation of whole-genome sequencing technologies has significantly contributed to our understanding of P. aeruginosa mutation-driven antibiotic resistance mechanisms. As P. aeruginosa antibiotic resistance genotypes should correlate with resistance phenotypes, a comprehensive and automated analysis of P. aeruginosa resistomes from whole-genome sequencing data would be of great benefit, for instance, for guiding therapeutic strategies and to develop new tools for antibiotic susceptibility prediction. In this work, we have introduced PaREx, a pipeline specifically designed for this purpose, providing an automated and user-friendly solution that facilitates its implementation despite limited expertise on bioinformatics or in P. aeruginosa resistance mechanisms. In contrast to other software and databases developed for the detection of resistance markers in which a few resistance mutations are detected, PaREx is able to analyze the presence of mutations in up to 221 chromosomal genes, including genes known to be involved in resistance to novel agents such as ceftolozane/tazobactam, ceftazidime/tazobactam, and/or cefiderocol. Moreover, in PaREx, the detection of resistance mutations consists in identifying all variants and then filtering the natural polymorphisms. This is a potential advantage of PaREx compared with other developed pipelines in which a list of mutations previously linked to antibiotic resistance is used for this purpose (23), thus avoiding the detection of novel resistance mutations and their use for other purposes such as genomic surveillance. Moreover, to our knowledge, the PaREx pipeline is the only one including chromosomal genes linked to novel agents such as cefiderocol.
Although the PaREx pipeline still has some margin for improvement, it represents a substantial advancement in the automated analysis of P. aeruginosa resistomes. In future versions, complementary bioinformatics approaches such as the analysis of resistance mutations directly on the de novo assemblies as well as the detection of gene absence and large genomic deletions will be integrated as it could be of some benefit for some particular scenarios and strains.
Along with PaREx, we have developed a web tool for the detection of mutations in the P. aeruginosa chromosomal cephalosporinase AmpC, which have been demonstrated to compromise susceptibility to cefiderocol, ceftolozane/tazobactam, and/or ceftazidime/avibactam. This user-friendly tool represents a valuable resource for guiding therapies and for the monitoring of resistance to these last-resource antipseudomonal agents in settings with limited bioinformatic resources. Of note, a web tool for calculating the genotypic scores, and thus providing a genomic antibiogram, from the PaREx final reports is currently under development.
MATERIALS AND METHODS
The PaREx pipeline and databases
Instructions for setting up the PaREx pipeline are available in the following repository: https://github.com/ARPBIGIDISBA/PaREx. As shown in Fig. 1, PaREx comprises different steps detailed in the following subsections.
Read quality filtering
The first step of the PaREx pipeline is quality filtering of raw Illumina paired-end reads using the open-source Java-based tool Trimmomatic v0.39 (24). By default, the following parameters are applied: LEADING:10, TRAILING:15, SLIDINGWINDOW:4:20, and MINLEN:100, parameters that aim to balance stringency with read retention, ensuring trimming of low-quality bases while preserving read length for PaREx downstream analysis.
De novo genome assembly
Quality paired-end reads are then *de novo-*assembled using the St. Petersburg genome assembler algorithm SPAdes v3.15.5 (25). By default, PaREx applies a multi-k-mer size strategy (-k 33, 55, 77, 99) to enhance assembly continuity and accuracy.
Multi-locus sequence typing
In the PaREx pipeline, MLST is performed using the de novo genome assemblies with the mlst open-source software (https://github.com/tseemann/mlst), which uses the publicly available PubMLST scheme and database (https://pubmlst.org/). Both the Sequence Type and the MLST allelic profile (acsA, aroE, guaA, nuoD, mutL, ppsA, and trpE) are included in the PaREx final reports along with the resistance genotype.
Acquired and mutational resistome profiling
As shown in Fig. 1, de novo assemblies are used for MLST and other additional downstream analyses, including the following: (1) detection of horizontally acquired antibiotic resistance genes (acquired resistome profiling), (2) PDC designation, and (3) evaluation of the structural integrity of the P. aeruginosa porin OprD.
Horizontally acquired antibiotic resistance genes are detected with the ResFinder analysis tool and its manually curated database (v.2.4.0) (9). The resulting ResFinder .json files are further processed with a Python-based script that removes P. aeruginosa intrinsic core resistance genes, incomplete ones, and/or duplicate results. In the final PaREx reports, all acquired resistance genes along with their percentage of identity are listed and classified into four groups: beta-lactamases, aminoglycoside resistance genes contributing to tobramycin and/or amikacin resistance, fluoroquinolone resistance determinants, and other acquired resistance genes.
For PDC designation, de novo assemblies are processed with a Python-based script that uses the Basic Local Alignment Search Tool (BLAST+, v2.9.0-2) to compare assembled contigs against a custom-built database (https://arpbigidisba.com/pseudomonas-aeruginosa-derived-cephalosporinase-pdc-database/) in which all the PDC allele variants from the NCBI’s Pathogen Detection Reference Gene Catalog (last accession date: 03/06/2025) have been included. The detected PDC allele variant and any amino acid variations encountered relative to the PAO1 reference cephalosporinase sequence (PDC-1), which can be useful for understanding some resistance phenotypes, are included in the final reports.
Finally, to characterize the mutational resistome, PaREx employs a reference-guided assembly approach. For the identification of single-nucleotide variants (SNVs) and insertions/deletions (InDels) between the P. aeruginosa PAO1 reference genome sequence (NC_002516.2) and isolates’ quality-filtered reads, PaREx uses the Snippy tool applying default options (https://github.com/tseemann/snippy), and, then, the resulting snps.vcf file is parsed by a Python-based script.
In recent years, our understanding of mutation-driven antibiotic resistance mechanisms in P. aeruginosa has significantly advanced, primarily due to the screening of comprehensive mutant libraries and whole-genome sequencing data obtained from in vitro antibiotic resistance evolution assays, in vivo monitoring of antimicrobial resistance development, longitudinal analysis of sequential cystic fibrosis isolates, and from the characterization of epidemic high-risk clones. With this information, we have previously defined a panel of 164 chromosomal genes based on the PAO1 reference genome to analyze the mutational resistome of P. aeruginosa (15, 26) and for which we have recently defined the naturally occurring polymorphisms (20). For the PaREx pipeline, we have extended this panel and included genes that have been linked to cefiderocol and/or ceftolozane/tazobactam and/or ceftazidime/avibactam resistance (27–31), as well as some additional genes related to the recycling and modification of P. aeruginosa peptidoglycans (Data S5).
From the snps.vcf file, PaREx extracts this new panel that comprises a total of 221 chromosomal genes related to mutation-driven antibiotic resistance in P. aeruginosa and filters the naturally occurring polymorphisms from missense and nonsense mutations by using a custom-built database that includes about 2,875 natural polymorphisms (Data S5).
Finally, given the existence of different sequence variants of the gene encoding OprD (32) and its relevant role in carbapenem resistance, a specific module for assessing its structural integrity has been developed. In PaREx, a custom Python-based script that uses the Basic Local Alignment Search Tool (BLAST+, v2.9.0-2) and a custom-built database including the OprD reference sequences of P. aeruginosa strains PAO1, LESB58, UCBP-PA14, MTB-1, FRD1, and F23197 is used for the evaluation of the OprD porin. This script first selects the most suitable reference sequence and then explores the integrity of the porin, thereby minimizing errors derived from the use of an inappropriate reference sequence.
PaREx final reports and outputs
As the final reports, PaREx provides a csv file that integrates all generated results from all the isolates analyzed in the same batch along with individual summary reports for each of the isolates in pdf format. The csv file contains four different sheets corresponding to the basic and the extended resistomes, each provided in both raw and clean formats. The basic resistome includes the core set of chromosomic genes considered the main drivers of clinical resistance to classical antipseudomonal antibiotics, whereas the extended resistome includes additional genes that either have been associated with resistance to classical antipseudomonals but their effect is lower and/or has not been sufficiently demonstrated or are genes linked to resistance to newer β-lactam/β-lactamase inhibitor combinations (ceftolozane/tazobactam, ceftazidime/avibactam) and/or cefiderocol. Both clean resistome sheets contain only mutations with a documented or predicted impact on resistance as naturally occurring polymorphisms have been removed. In addition, during PaREx execution, different output folders are generated containing intermediate files, such as the de novo assemblies or the output folder of Snippy, that are conserved and can be used by users for other purposes not covered in the pipeline.
PaREx running options
The PaREx pipeline has been developed with flexibility in mind, allowing users to (1) skip some steps/modules, (2) adjust the running parameters for each step, and/or (3) run the analyses on single or multiple isolates. Detailed usage instructions are provided in the README file (https://github.com/ARPBIGIDISBA/PaREx).
PaREx lists and databases
Core resistance gene list
The manually curated ResFinder database includes genes that have been documented in the literature to be horizontally transferred among bacterial species, some of which are P. aeruginosa chromosomal genes that belong to its intrinsic core resistome (9). To identify the P. aeruginosa intrinsic core resistance genes included in the ResFinder database, we analyzed, along with PAO1 and PA14 reference genomes, the draft genomes from a collection of 461 P. aeruginosa clinical isolates that we have previously sequenced and which includes a set of 304 isolates from a multicenter study performed in 2017, which includes isolates exhibiting different antibiotic susceptibility profiles (13; European Nucleotide Archive Project Numbers: PRJEB40140 and PRJEB31047) and a set of 157 MDR and XDR isolates obtained from another multicenter study performed in 2022 (17; European Nucleotide Archive Project Number: PRJEB61879). This collection of 461 P. aeruginosa genomes was representative of all 17 Spanish regions and exhibited a high level of genetic diversity, with up to 192 different STs identified (Shannon diversity index, H’ = 4.47). All genes detected with ResFinder in at least 95% of the draft genomes were included in the PaREx core resistance genes database. In addition, all detected blaOXA allele variants were individually investigated, and those classified as blaOXA-50 (PoxB) derivatives were also included in the database. Finally, and based on the current evidence, we excluded the crpP phosphotransferase as it does not appear to play any role in P. aeruginosa resistance to fluoroquinolones (33).
Natural polymorphisms database
For defining P. aeruginosa naturally occurring polymorphisms in the 221 chromosomal antibiotic resistance genes included in the mutational resistome analysis of the PaREx pipeline, the same approach, consisting in the analysis of the genomes of 100 wild-type P. aeruginosa strains, that we used previously for defining the natural polymorphism in a subset of these genes was applied (13; European Nucleotide Archive project number PRJEB40140). Identified natural polymorphisms are listed in Data S5.
Hardware and software setup
The PaREx pipeline has been developed and executed on Ubuntu 20.04.6 LTS using Python v3.10.12 (packaged by conda-forge and compiled with GCC 12.3.0). Core third-party tools are installed using a dedicated Bash installer script that manages dependencies and sets up symbolic links for ease of use. Installation instructions are provided in the project’s GitHub repository and are compatible with any UNIX-based environment.
A system with at least 8 CPU cores, 16 GB RAM, and 512 GB SSD storage is recommended for optimal performance. The pipeline can also be executed on Windows systems using the Windows Subsystem for Linux 2 (WSL2).
The PDC analyzer web tool
The PDC analyzer web tool is based on the Python-based script and custom-built database of the PDC designation module included in the PaREx pipeline. For the list of AmpC mutations associated or that may contribute to resistance to the novel antipseudomonal cephalosporins, we performed a comprehensive and exhaustive literature review on PubMed using the following search terms: “ceftolozane tazobactam resistance OR ceftazidime avibactam resistance OR cefiderocol resistance AND pseudomonas AND mut*” (accession date: 29/05/2025).
P. aeruginosa collections used for the validation of the functionalities of PaREx and the PDC analyzer web tool
Analysis of a P. aeruginosa clinical isolate collection
For the validation of PaREx and its functionalities, we sequenced and analyzed a set of 260 P. aeruginosa clinical isolates exhibiting different antibiotic susceptibility profiles (sequencing files have been deposited in the European Nucleotide Archive under study accession number PRJEB94805). For this purpose, four clinical isolates were randomly selected from each of the 66 hospitals (covering all 17 Spanish regions) participating in a national survey in 2022 (17). Minimum inhibitory concentrations of piperacillin/tazobactam (4/4–256/4 mg/L), ceftazidime (1–64 mg/L), cefepime (1–64 mg/L), ceftolozane/tazobactam (0.5/4–32/4 mg/L), ceftazidime/avibactam (0.5/4–32/4 mg/L), aztreonam (2–128 mg/L), imipenem (0.5–64 mg/L), meropenem (0.5–64 mg/L), ciprofloxacin (0.12–16 mg/L), tobramycin (0.25–32 mg/L), amikacin (2–128 mg/L), and colistin (0.5–16 mg/L) have been previously determined by broth microdilution using SensiTitr panels (Plate Code:FRCNRP2, Thermo Fisher Diagnostics, S.LU) and using EUCAST 2025 (v15.0), and clinical breakpoints were used for interpretation of SIR categories. The modR profile was defined as resistance to at least 1 agent in 1 or 2 of 7 antibiotic classes including antipseudomonal penicillins + β-lactamase inhibitor combinations (piperacillin/tazobactam), antipseudomonal cephalosporins (ceftazidime and cefepime), monobactams (aztreonam), antipseudomonal carbapenems (imipenem and meropenem), fluoroquinolones (ciprofloxacin), aminoglycosides (tobramycin and amikacin), and polymyxins (colistin); the MDR profile is defined as resistance to at least 1 agent in at least 3 of 7 antibiotic classes, and the XDR profile is defined as resistance to at least 1 agent in all but 1 or 2 antibiotic classes (34).
Analysis of the NCBI Microbial Browser for Identification of Genetic and Genomic Elements (MicroBIGG-E) database
Recently, Mack and collaborators (22) explored the presence of β-lactamase alleles in more than 30,000 P. aeruginosa isolates collected worldwide and deposited in the NCBI Pathogen Detection databases to gain insights into the P. aeruginosa β-lactamases. In this work, we used the PDC analyzer web tool to analyze this global collection and to determine the global prevalence of mutations linked to resistance to the novel antipseudomonal combinations ceftolozane/tazobactam, ceftazidime/avibactam, and/or cefiderocol.
PaREx availability and requirements
Project name: P**seudomonas a**eruginosa Resistome Explorer
Project home page: https://github.com/ARPBIGIDISBA/PaREx.
Operating systems: Unix (Ubuntu 20.04.6 LTS) and Windows (Windows Subsystem for Linux 2)
Programming language: Python
Other requirements: Python 3.8.10 or higher
License: Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0)
Any restrictions to use by non-academics: Commercial use is prohibited without prior permission. Attribution and the same license are required for modifications.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Horcajada JP, Montero M, Oliver A, Sorlí L, Luque S, Gómez-Zorrilla S, Benito N, Grau S. 2019. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin Microbiol Rev 32:e 00031-19. doi:10.1128/CMR.00031-1931462403 PMC 6730496 · doi ↗ · pubmed ↗
- 2Ciofu O, Tolker-Nielsen T. 2019. Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents-how P. aeruginosa can escape antibiotics. Front Microbiol 10:913. doi:10.3389/fmicb.2019.0091331130925 PMC 6509751 · doi ↗ · pubmed ↗
- 3López-Causapé C, Rojo-Molinero E, Macià MD, Oliver A. 2015. The problems of antibiotic resistance in cystic fibrosis and solutions. Expert Rev Respir Med 9:73–88. doi:10.1586/17476348.2015.99564025541089 · doi ↗ · pubmed ↗
- 4Oliver A, Rojo-Molinero E, Arca-Suarez J, Beşli Y, Bogaerts P, Cantón R, Cimen C, Croughs PD, Denis O, Giske CG, et al.. 2024. Pseudomonas aeruginosa antimicrobial susceptibility profiles, resistance mechanisms and international clonal lineages: update from ESGARS-ESCMID/ISARPAE group. Clin Microbiol Infect 30:469–480. doi:10.1016/j.cmi.2023.12.02638160753 · doi ↗ · pubmed ↗
- 5World Health Organization. 2024. WHO bacterial priority pathogens list, 2024: bacterial pathogens of public health importance to guide research, development and strategies to prevent and control antimicrobial resistance. Available from: https://www.who.int/publications/b/64088
- 6Petit RA III, Read TD. 2020. Bactopia: a flexible pipeline for complete analysis of bacterial genomes. m Systems 5:e 00190–20. doi:10.1128/m Systems.00190-2032753501 PMC 7406220 · doi ↗ · pubmed ↗
- 7Xavier BB, Mysara M, Bolzan M, Ribeiro-Gonçalves B, Alako BTF, Harrison P, Lammens C, Kumar-Singh S, Goossens H, Carriço JA, Cochrane G, Malhotra-Kumar S. 2020. Bac Pipe: a rapid, user-friendly whole-genome sequencing pipeline for clinical diagnostic bacteriology. i Science 23:100769. doi:10.1016/j.isci.2019.10076931887656 PMC 6941874 · doi ↗ · pubmed ↗
- 8Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, Philippon A, Allesoe RL, Rebelo AR, Florensa AF, et al.. 2020. Res Finder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 75:3491–3500. doi:10.1093/jac/dkaa 34532780112 PMC 7662176 · doi ↗ · pubmed ↗