Mesenchymal stromal cells modulate survival and regeneration of human hematopoietic stem cells via PGE2/cAMP signaling
Siva Sai Naga Anurag Muddineni, Chen Katz-Even, Adi Zipin-Roitman, Diana Rasoulouniriana, Debanjan Singha Roy, Rabeaa Aborgies, Katia Beider, Yael Raz, Neta Solomon, Gal Hershkovitz, Yael Shulman, Rony Chen, Eviatar Weizman, Claudia Waskow, Arnon Nagler, Michael Milyavsky

TL;DR
Mesenchymal stromal cells protect human blood stem cells from radiation damage through a signaling pathway involving PGE2 and cAMP.
Contribution
The study identifies PGE2/cAMP signaling as a novel mechanism by which mesenchymal stromal cells protect hematopoietic stem cells from genotoxic stress.
Findings
MSC-secreted PGE2 activates cAMP/CREB signaling to protect HSPCs from irradiation-induced apoptosis.
Pharmacological activation of the cAMP pathway with Forskolin/IBMX preserves HSPC function and self-renewal.
cAMP agonists reduce pro-apoptotic proteins and stabilize anti-apoptotic proteins in HSPCs.
Abstract
Ionizing radiation and chemotherapy significantly impair hematopoietic stem and progenitor cell (HSPC) function, increasing the risk of bone marrow failure and secondary malignancies. Mesenchymal stromal cells (MSCs), critical regulators within the hematopoietic niche, maintain HSPC quiescence, self-renewal, survival, and differentiation. However, the specific pro-regenerative signaling pathways activated by MSCs in human HSPCs remain incompletely defined. Here, we show that bone marrow-derived MSCs effectively suppress irradiation-induced apoptosis and preserve the in vivo repopulation capacity of human HSPCs. Transcriptomic analysis of HSPCs revealed a pronounced upregulation of CREB target genes following MSC co-culture, consistent with increased activation of the cAMP/CREB signaling pathway. Mechanistically, MSC-secreted prostaglandin E2 (PGE2) emerged as a key mediator of cAMP…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
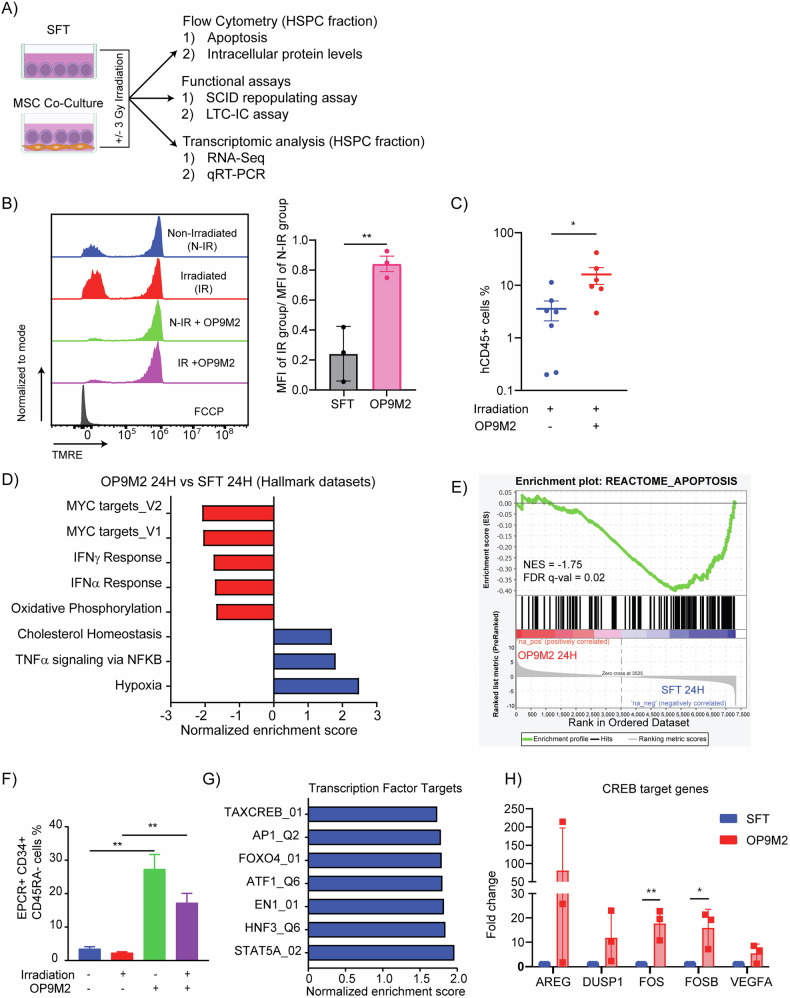 Figure 1
Figure 1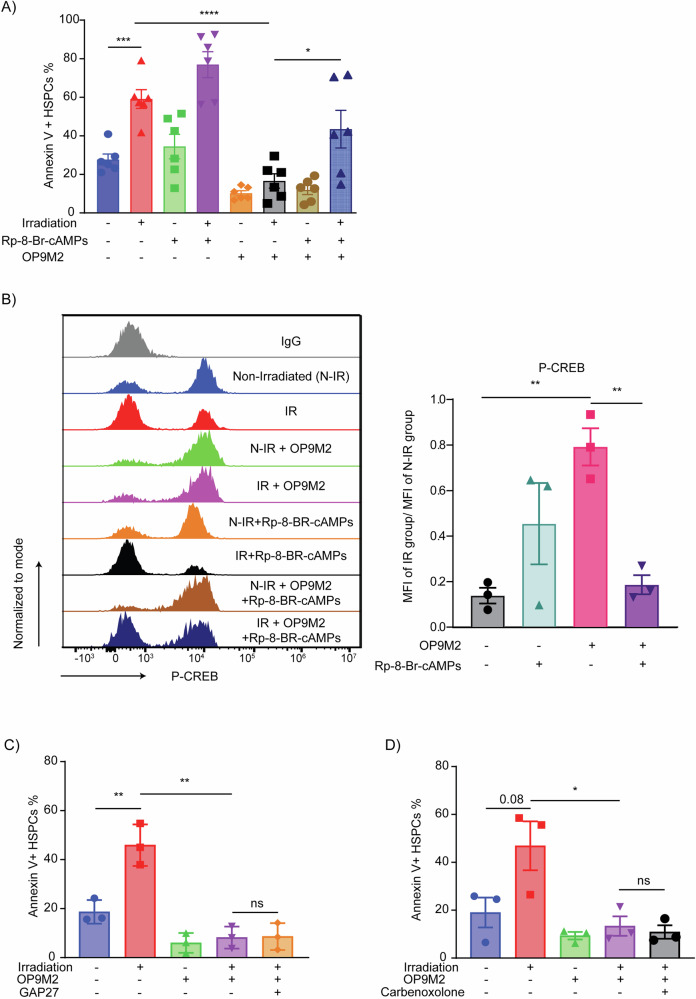 Figure 2
Figure 2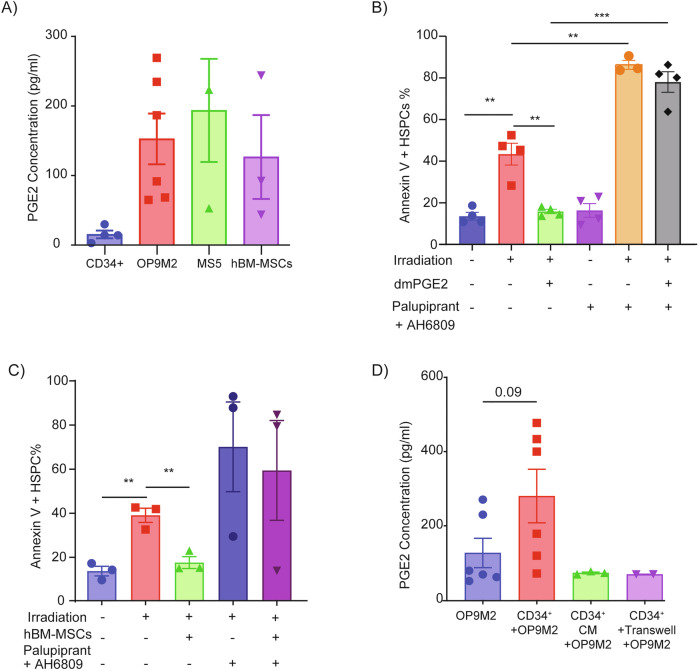 Figure 3
Figure 3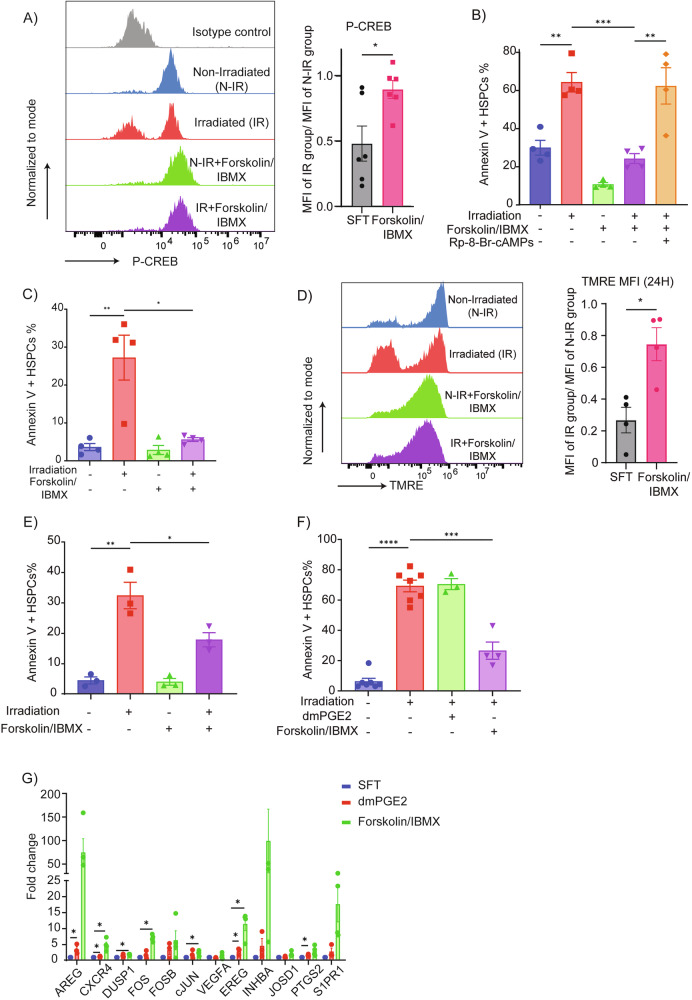 Figure 4
Figure 4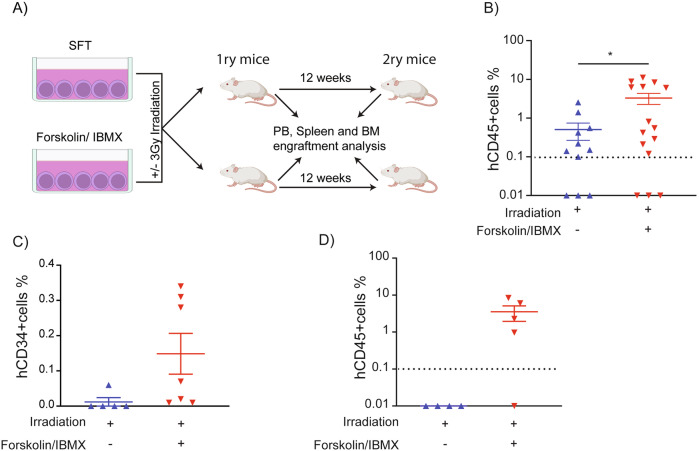 Figure 5
Figure 5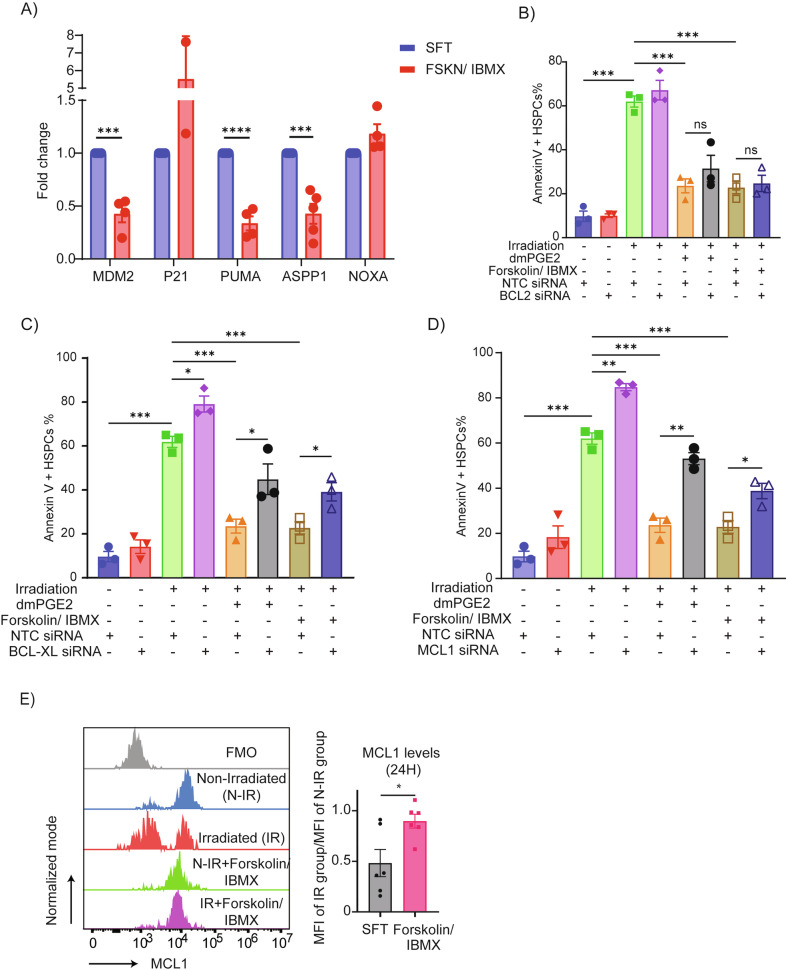 Figure 6
Figure 6 Figure 7
Figure 7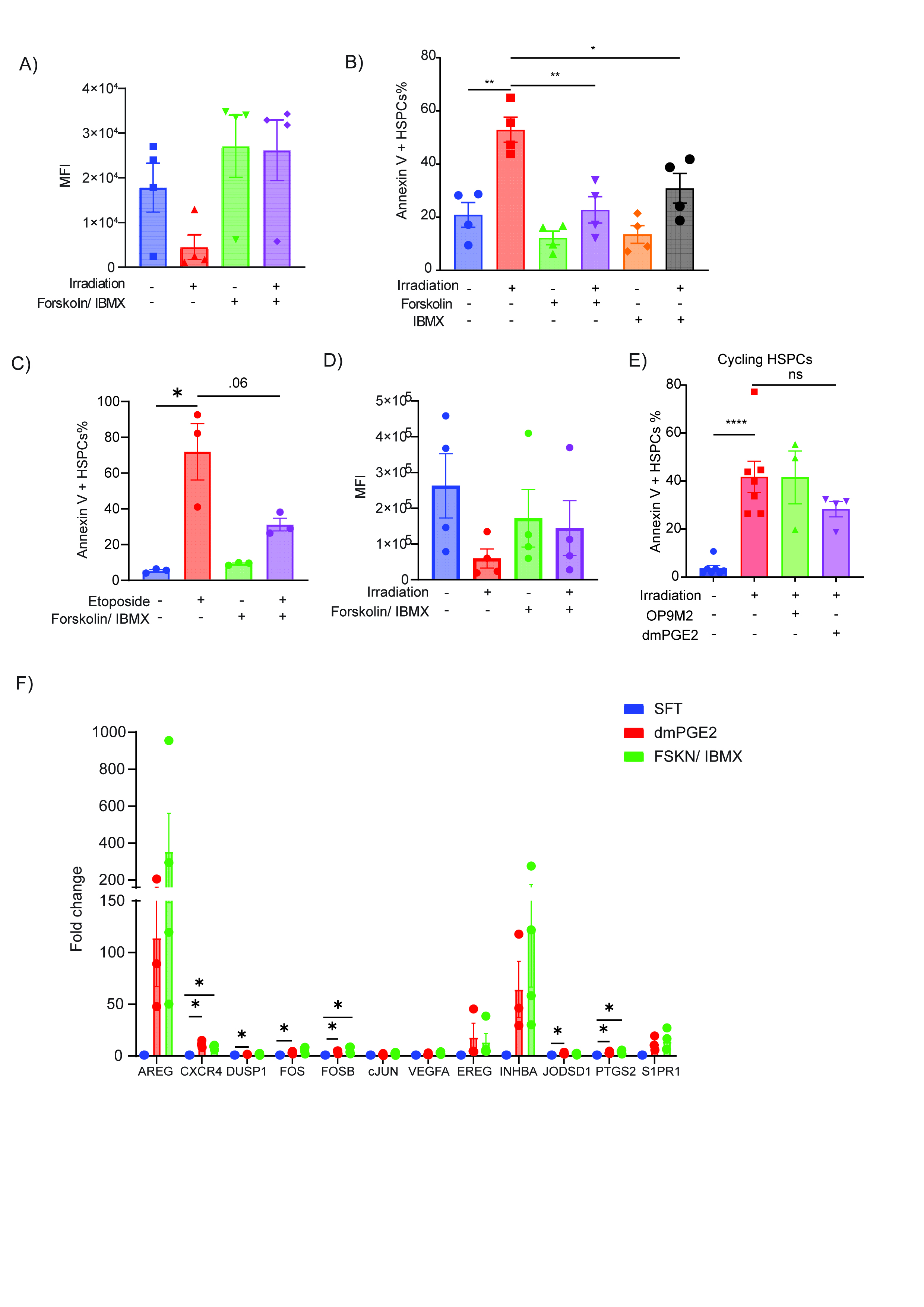 Figure 8
Figure 8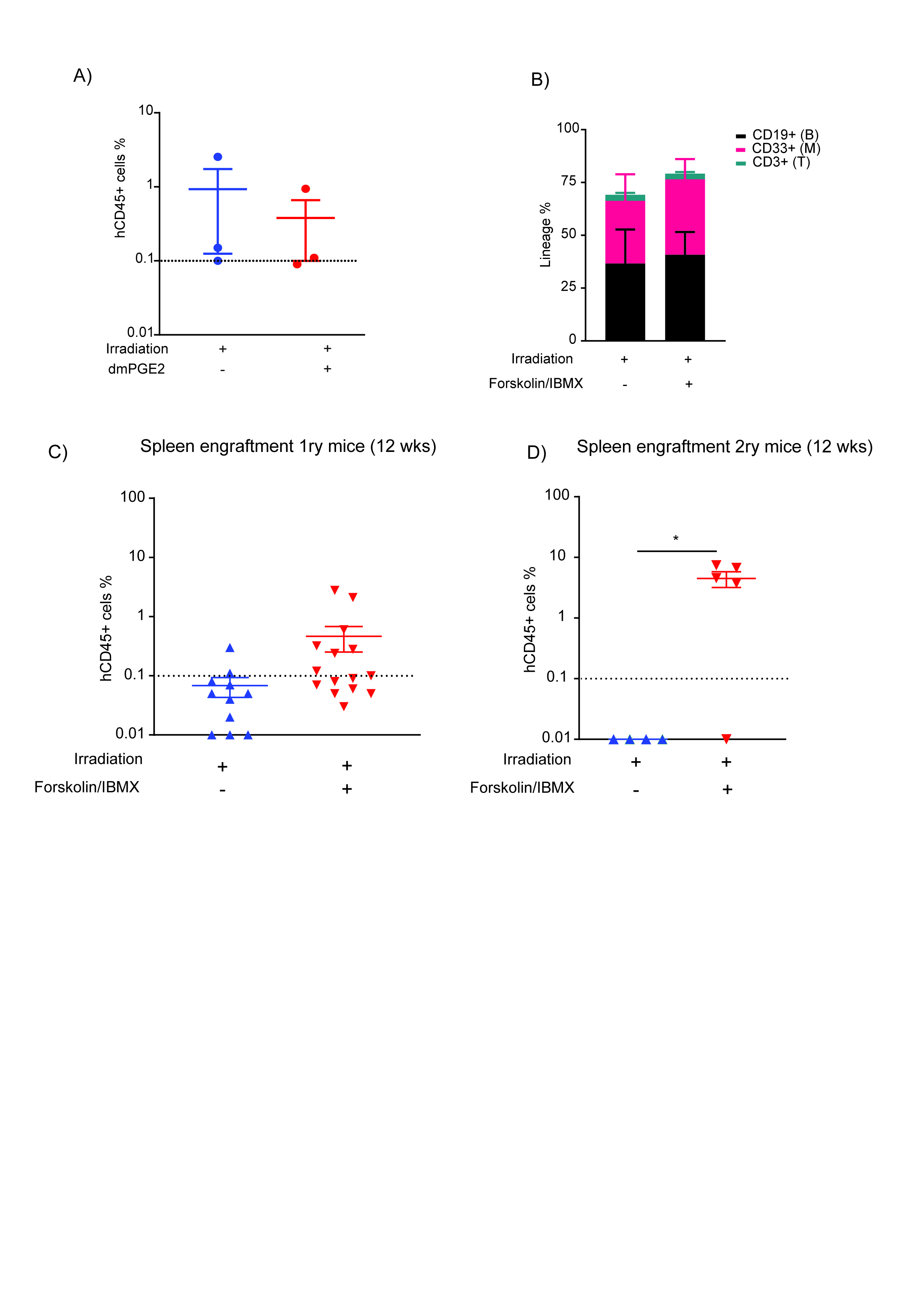 Figure 9
Figure 9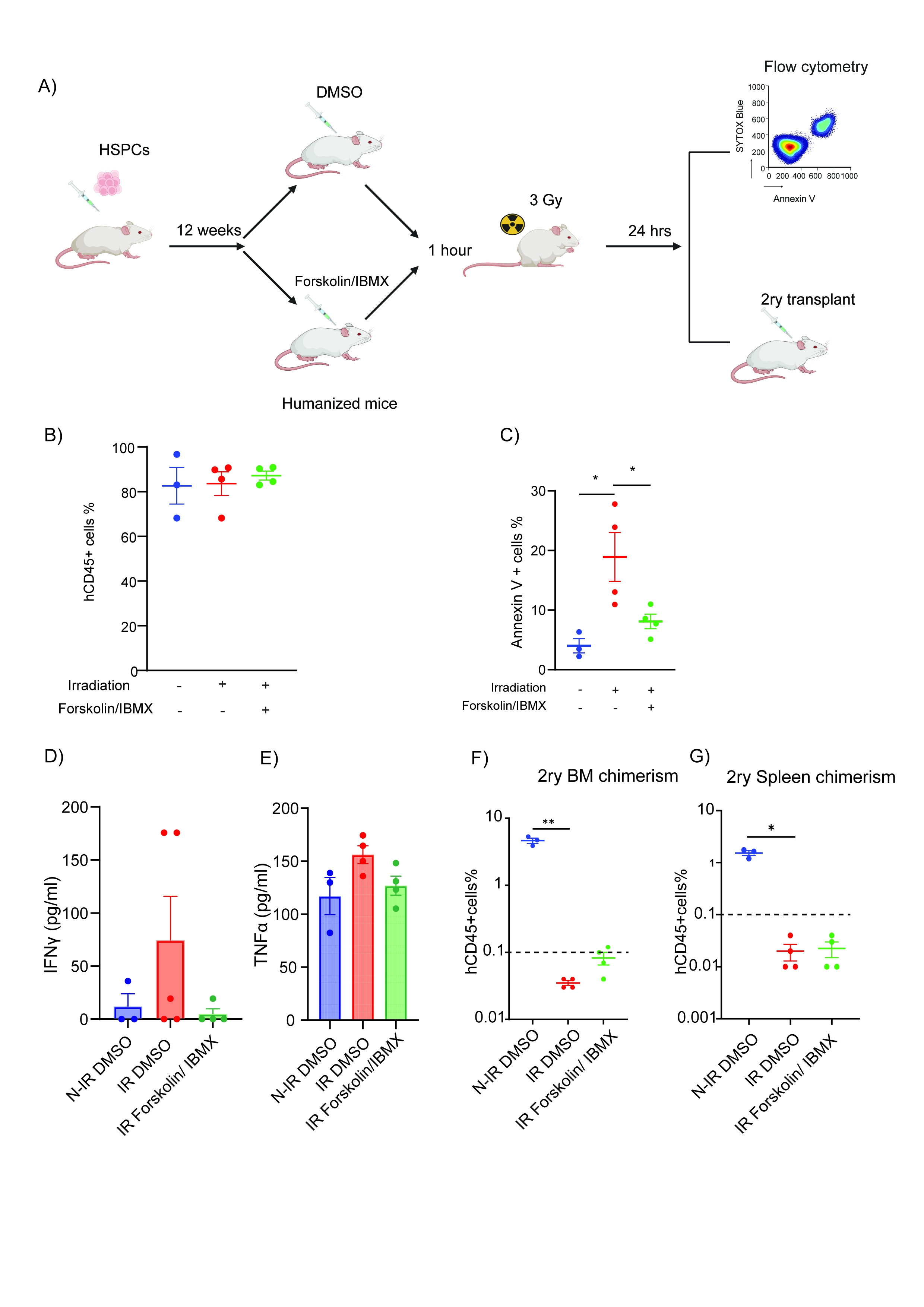 Figure 10
Figure 10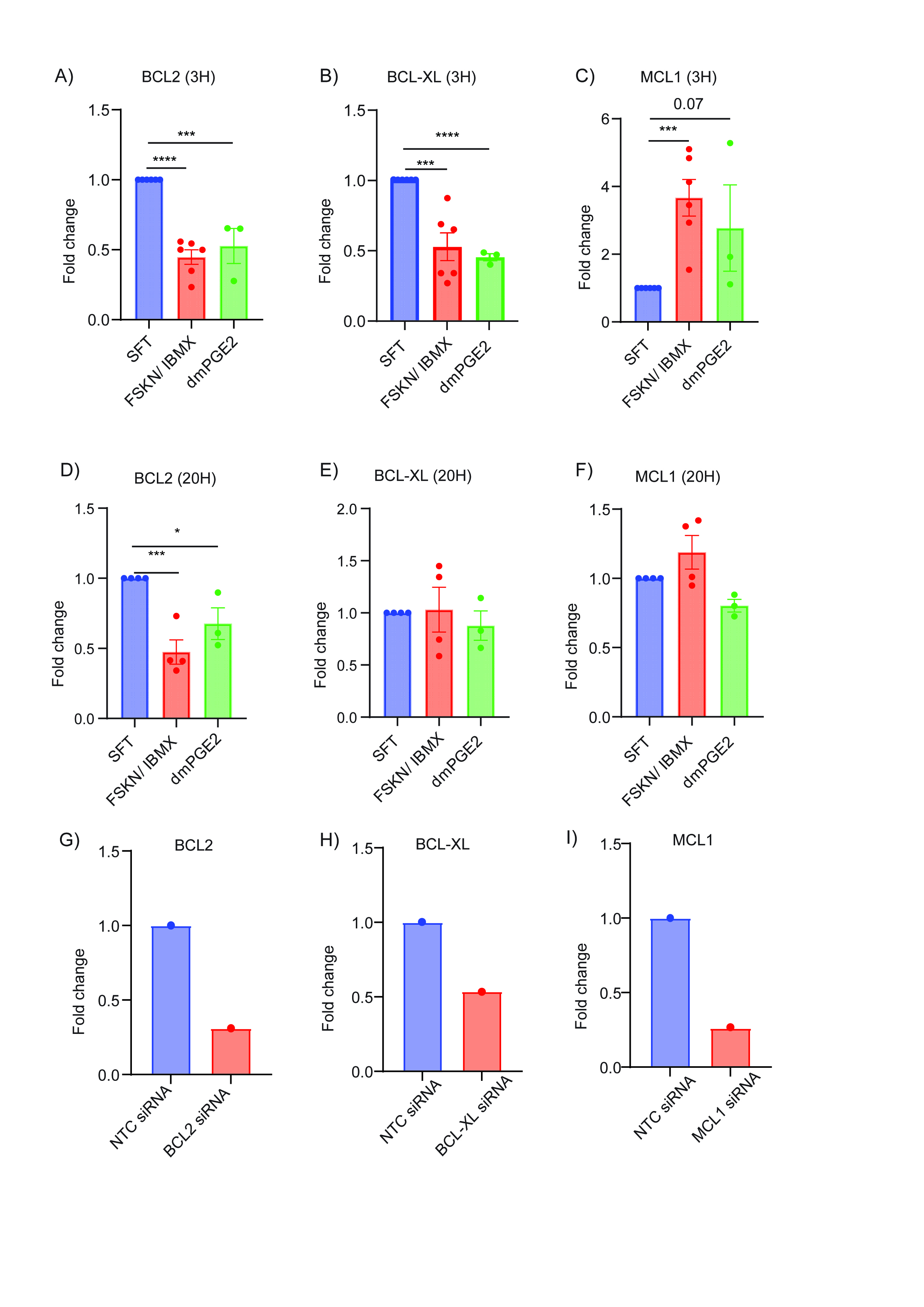 Figure 11
Figure 11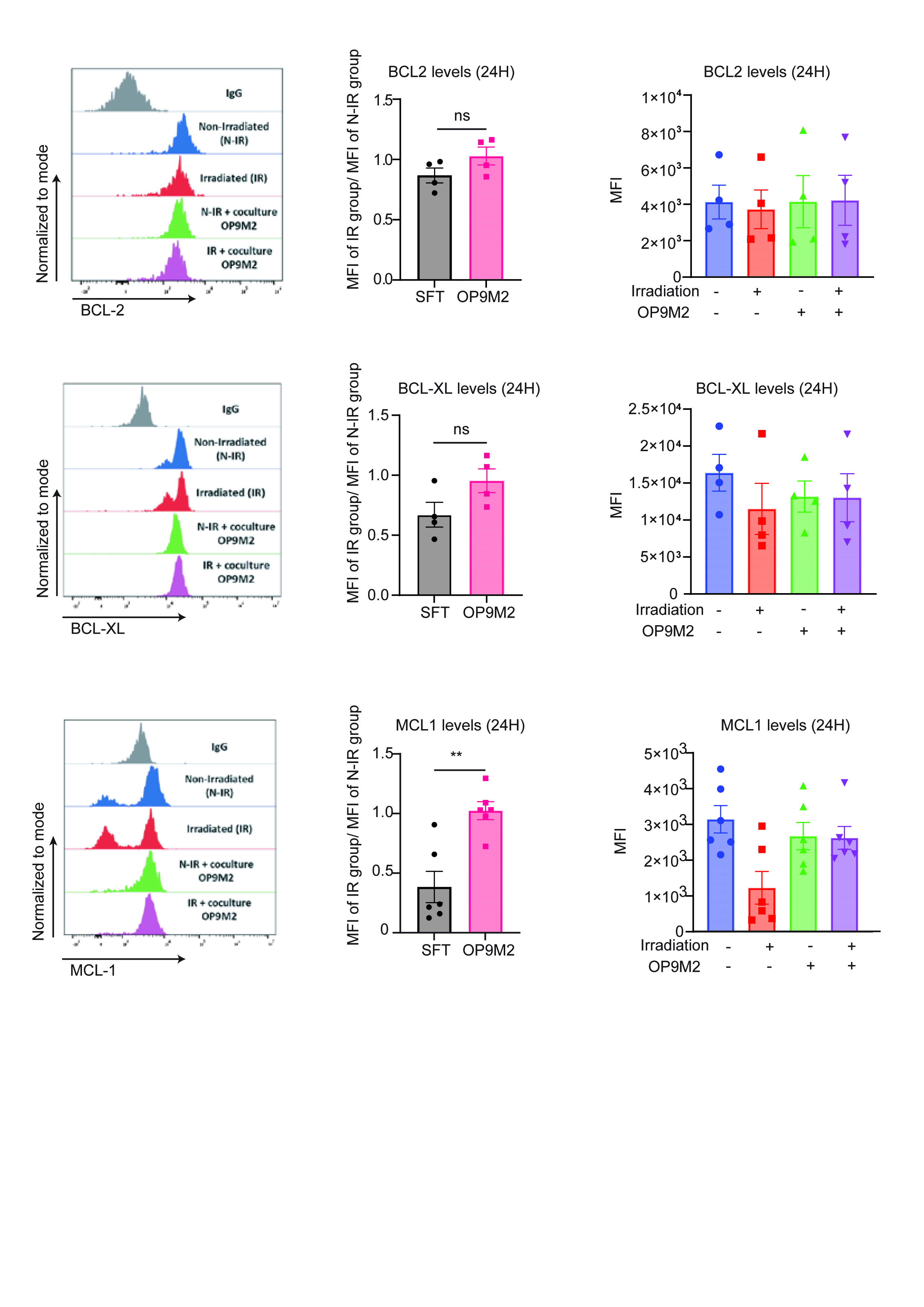 Figure 12
Figure 12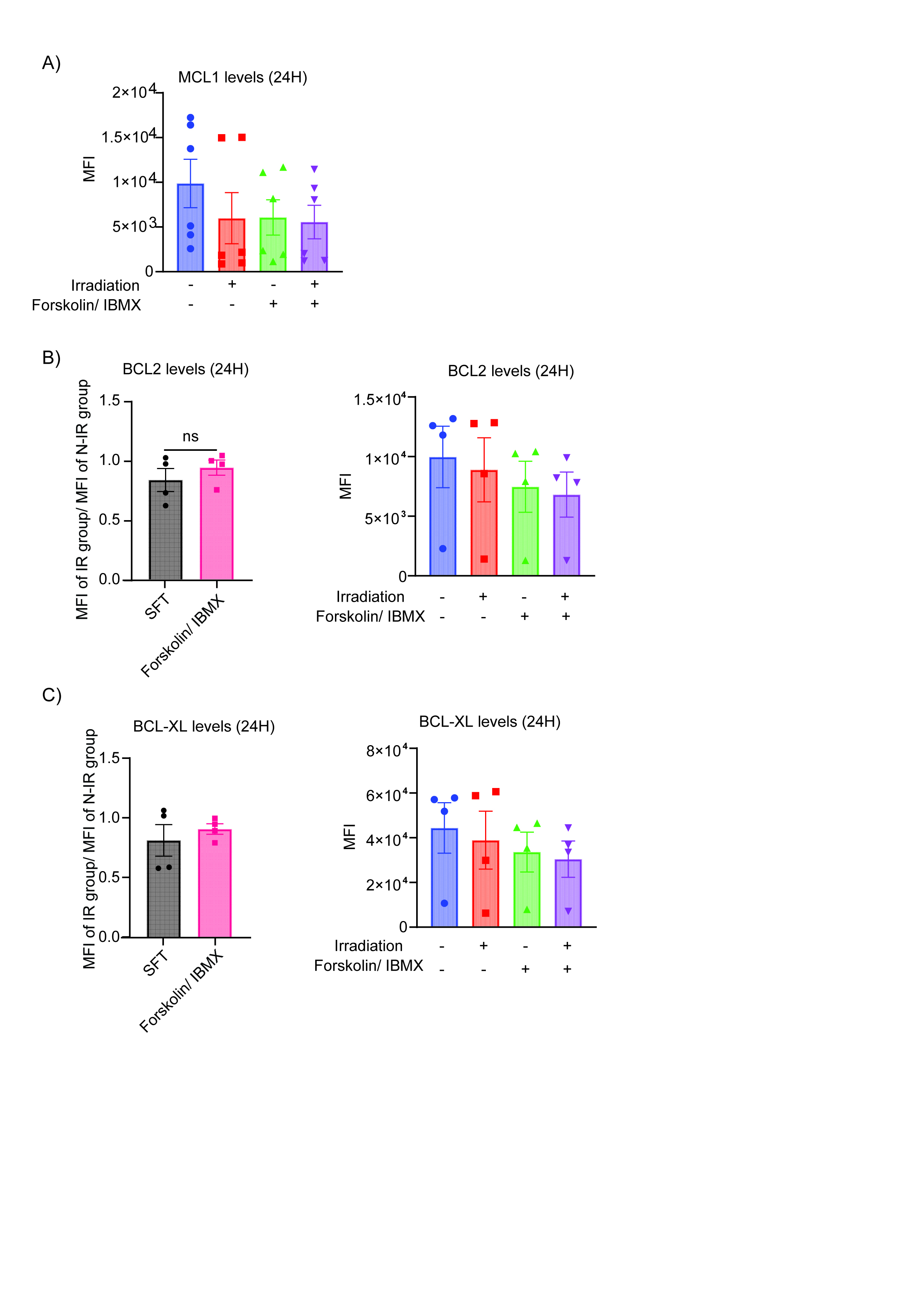 Figure 13
Figure 13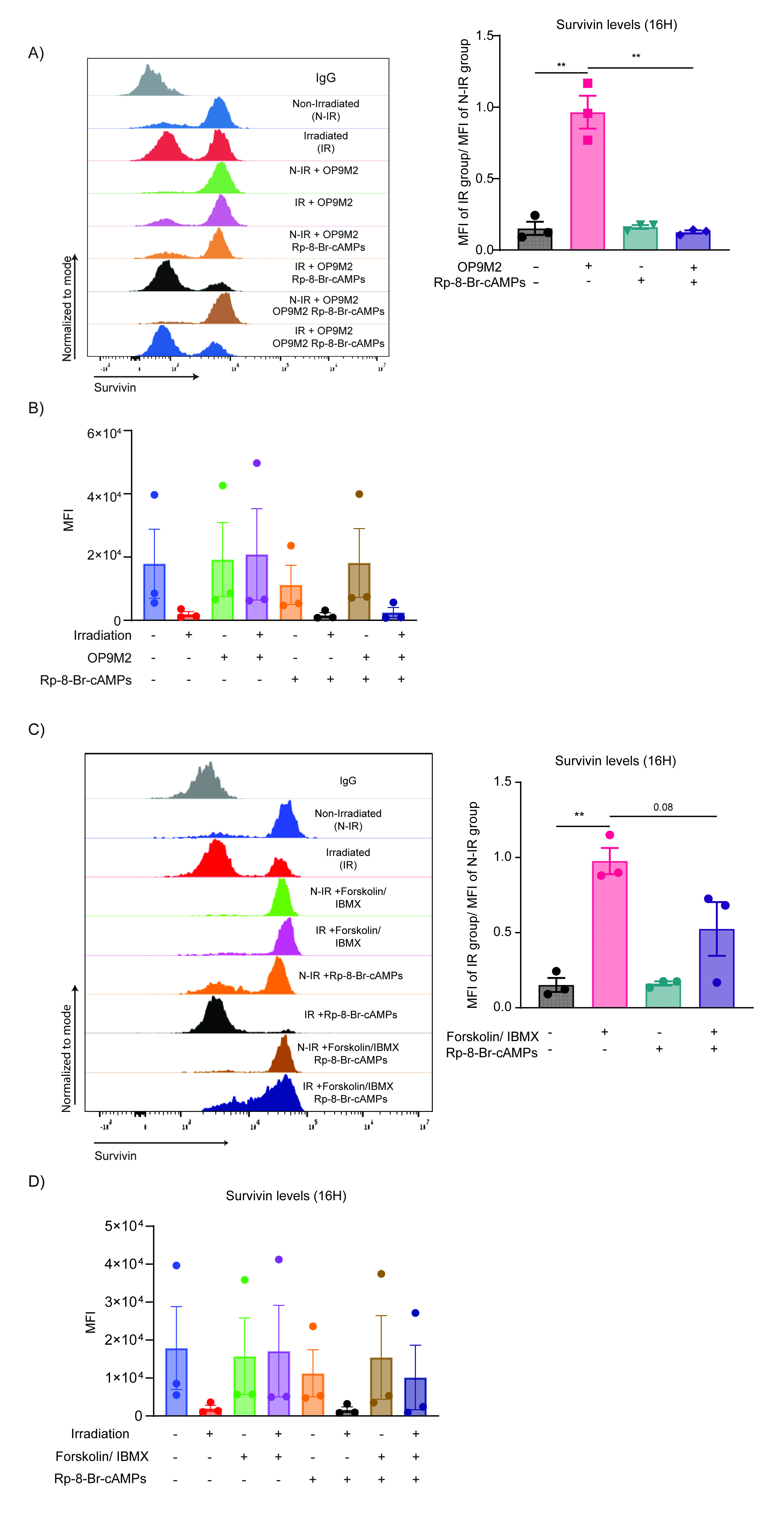 Figure 14
Figure 14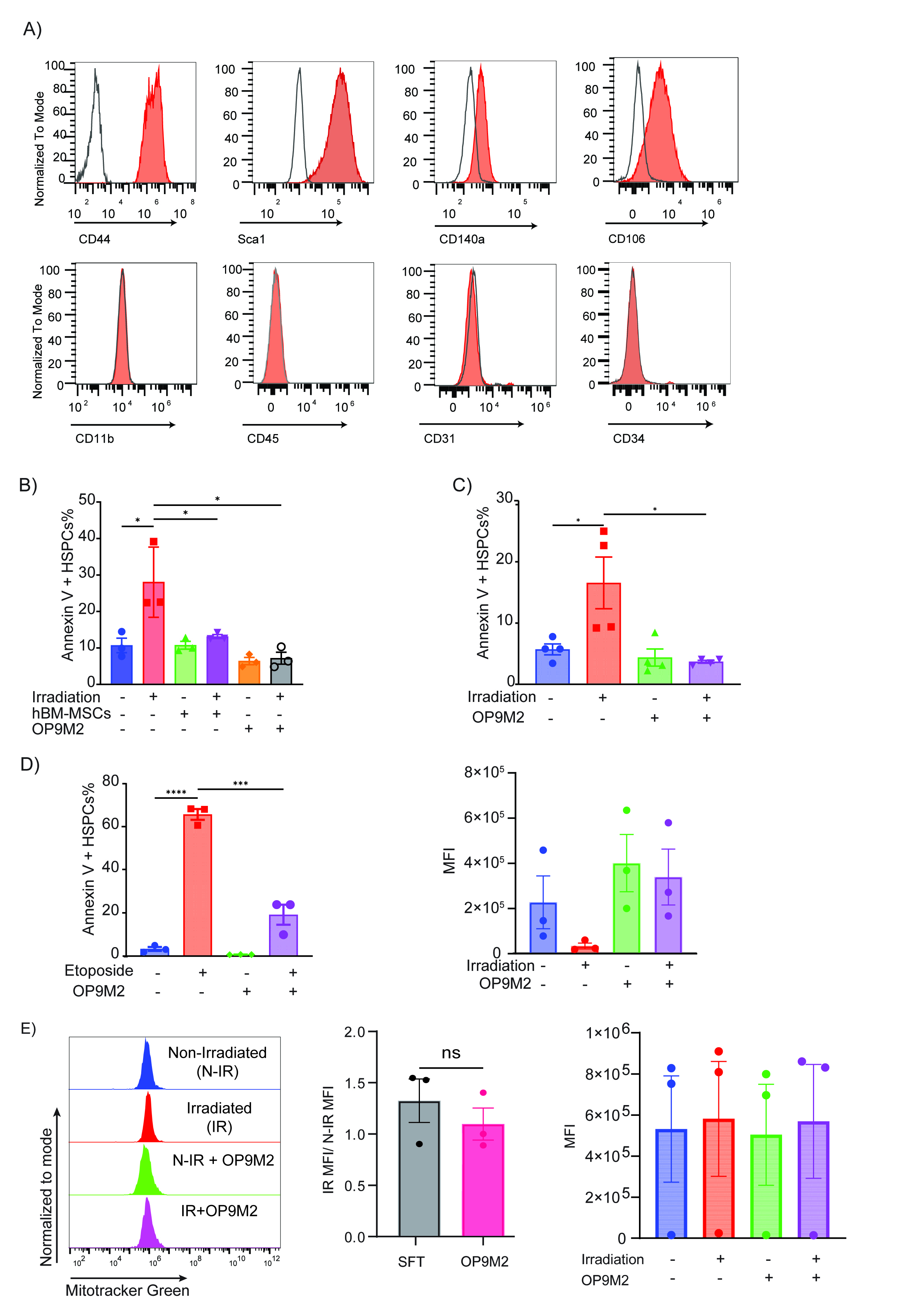 Figure 15
Figure 15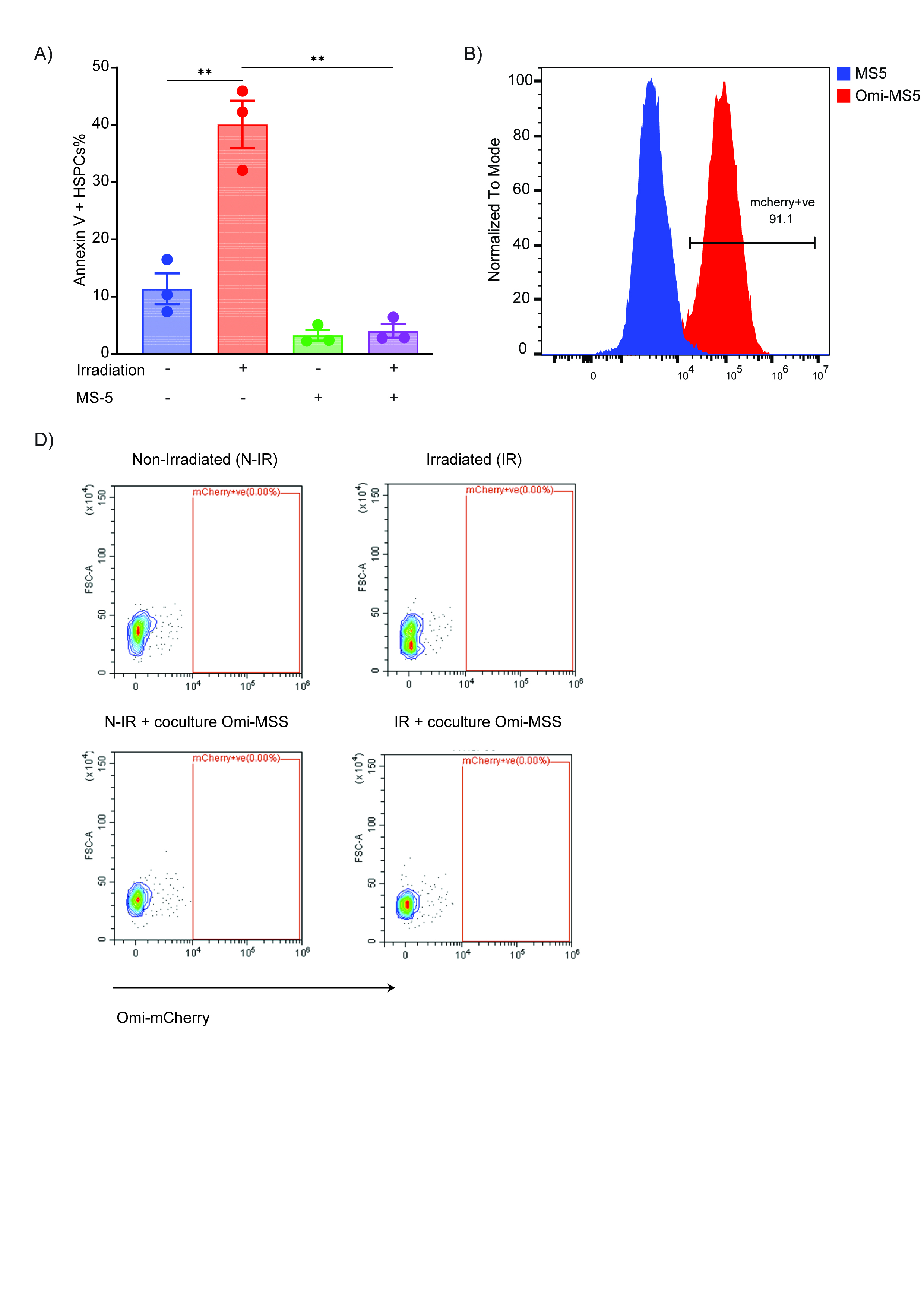 Figure 16
Figure 16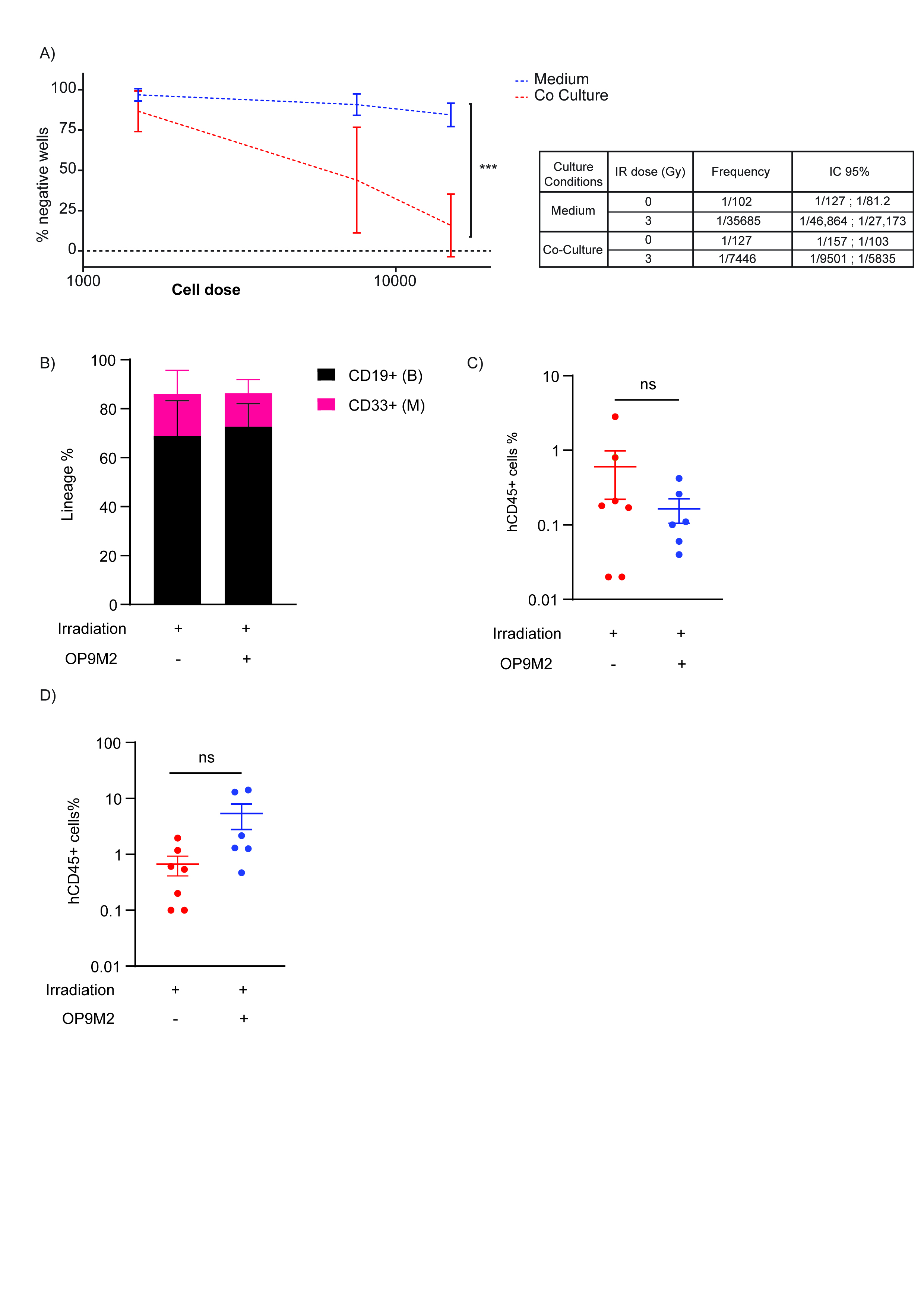 Figure 17
Figure 17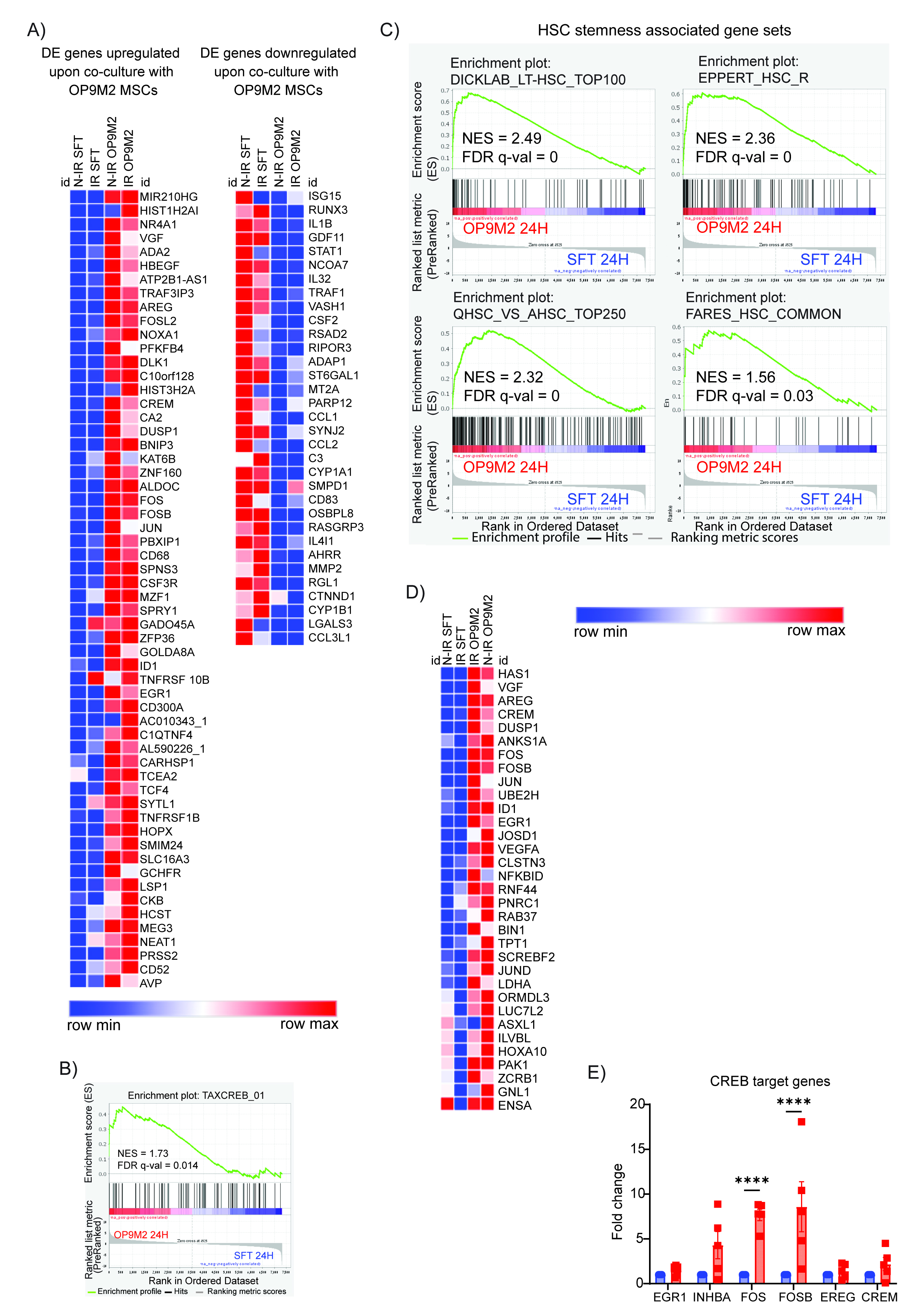 Figure 18
Figure 18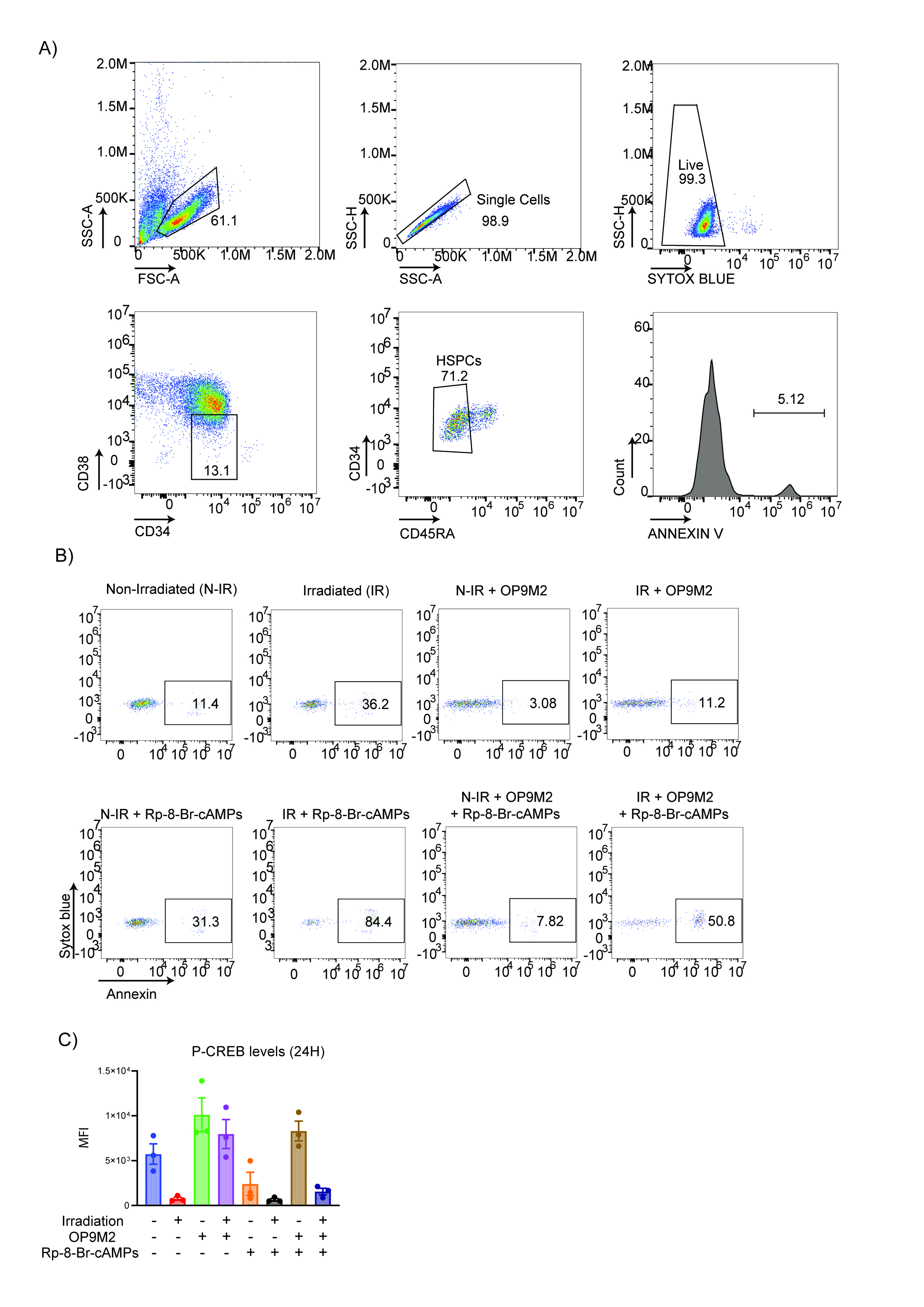 Figure 19
Figure 19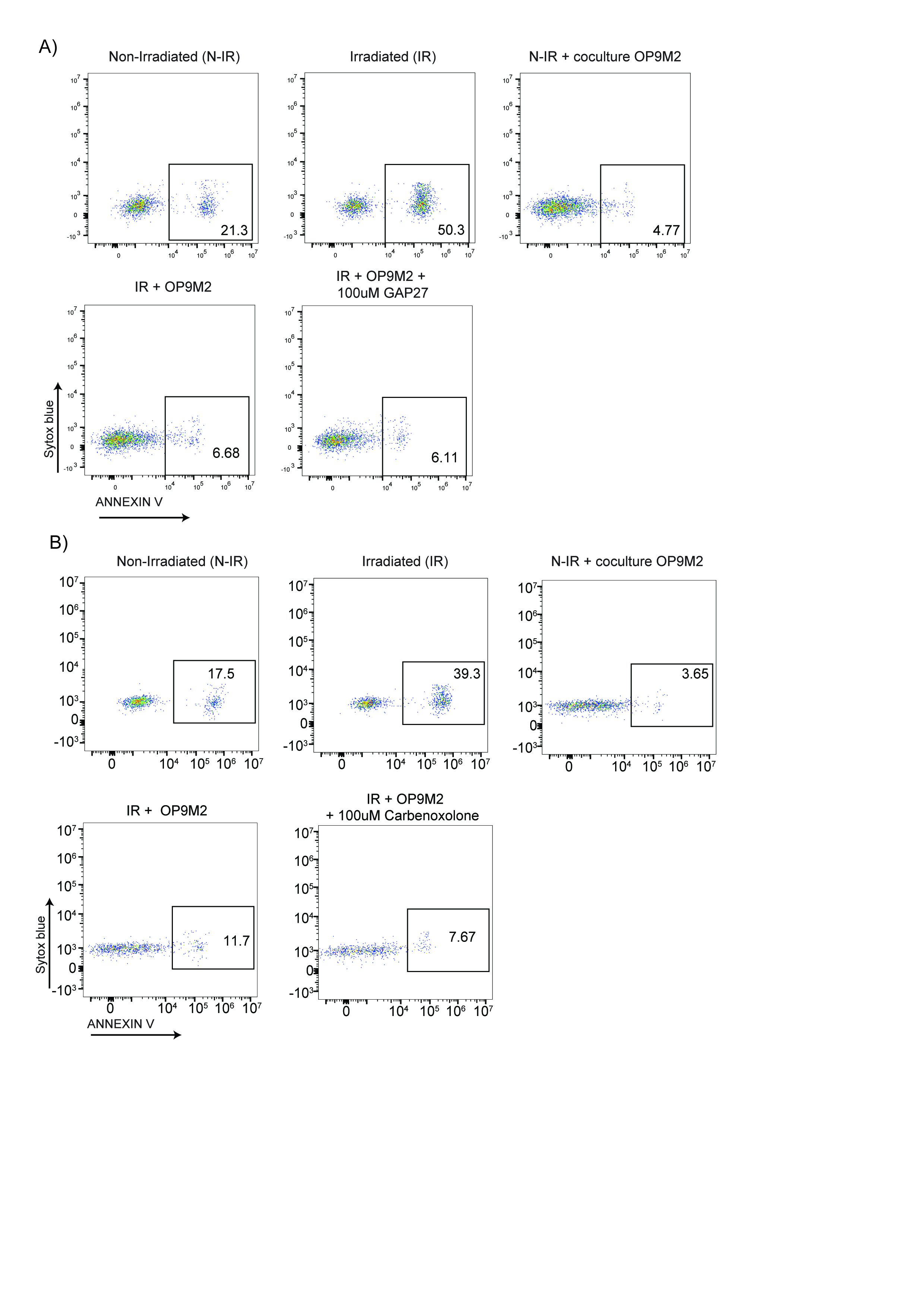 Figure 20
Figure 20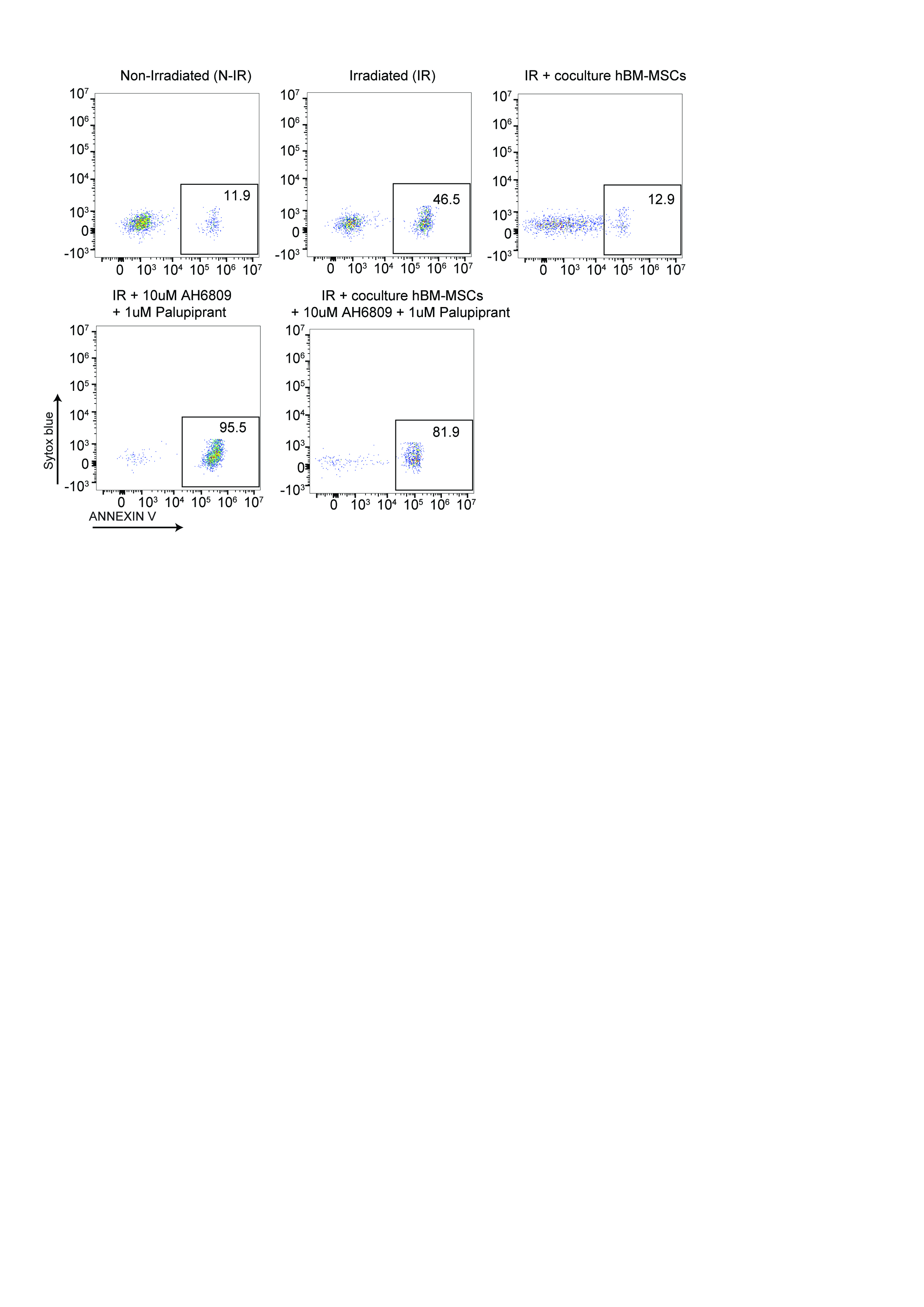 Figure 21
Figure 21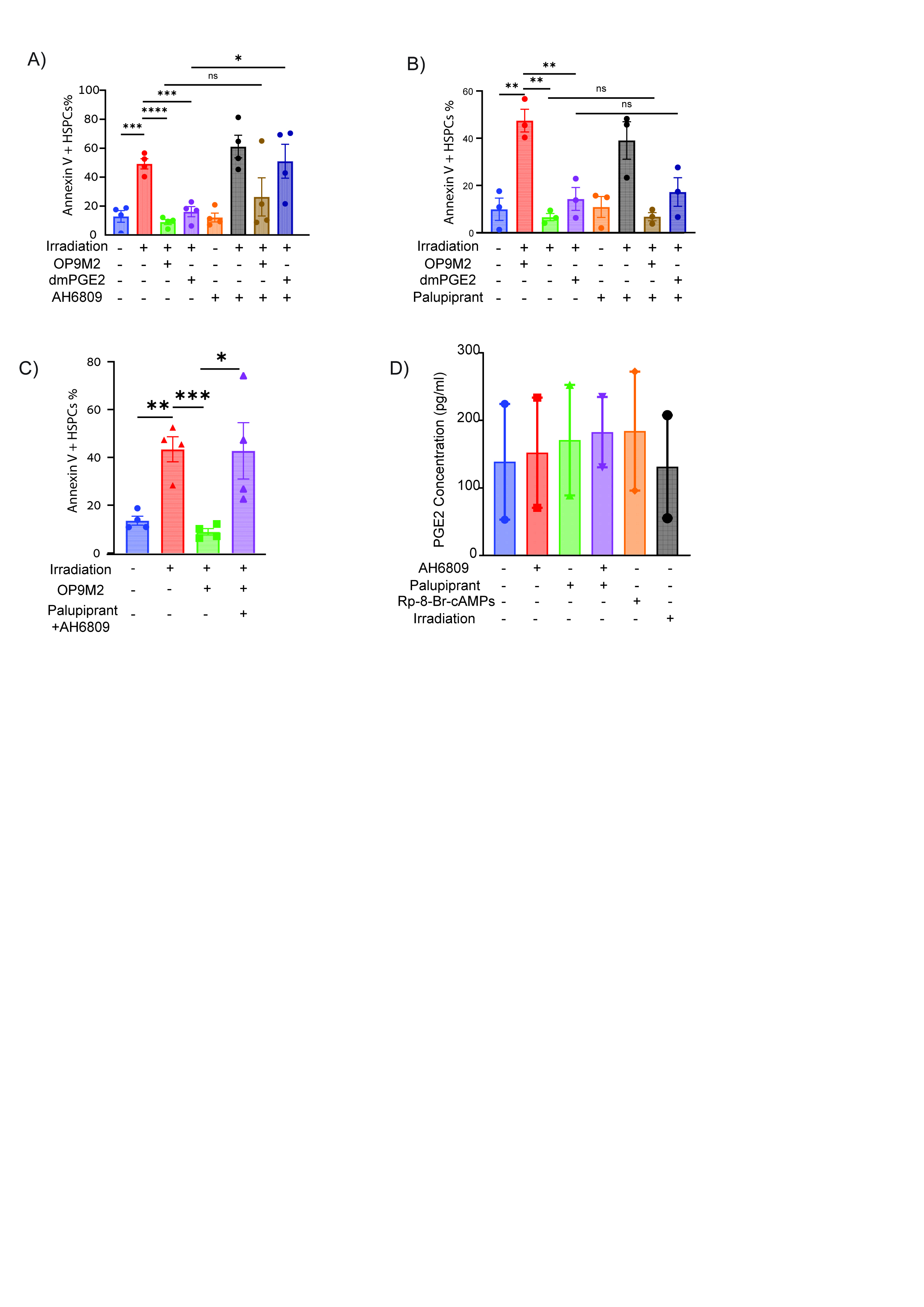 Figure 22
Figure 22- —https://doi.org/10.13039/501100011634TAU | Varda and Boaz Dotan Research Center for Hemato-Oncology Research, Tel Aviv University (Dotan Research Center in Hemato-Oncology, Tel Aviv University)
- —https://doi.org/10.13039/501100003977Israel Science Foundation (ISF)
- —https://doi.org/10.13039/501100011600TAU | Cancer Biology Research Center, Tel Aviv University (CBRC, TAU)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMesenchymal stem cell research · Oral health in cancer treatment · Hematopoietic Stem Cell Transplantation
Introduction
Hematopoietic Stem and Progenitor Cells (HSPCs) are rare bone marrow (BM) cells positioned at the apex of the hematopoietic hierarchy. They possess the unique ability to self-renew and differentiate into multiple blood cell lineages. These regenerative properties are crucial for lifelong blood production, the success of autologous or allogeneic HSPC transplantation, and the regeneration of hematopoiesis following ionizing radiation (IR) or chemotherapy [1, 2].
The hematopoietic system is among the most radiosensitive tissues in our body. DNA damage to HSPCs critically limits hematopoietic regeneration by inducing apoptosis, impairing self-renewal, and causing aberrant differentiation. Patients with inherited defects in DNA damage response genes, as well as murine models of these diseases, frequently exhibit marked genomic instability, progressive BM failure due to impaired HSC function, and an increased incidence of hematopoietic malignancies [3]. A comprehensive understanding of the molecular pathways that regulate HSPC survival, maintenance, and regeneration under genotoxic stress is critical for developing therapeutic strategies to mitigate these adverse outcomes.
Prior work, including our own, have established that irradiated human HSPCs undergo rapid apoptosis and exhibit a pronounced loss of long-term repopulation capacity [4–7]. Genetic approaches have implicated key regulators such as p53, ASPP1, BCL-2, and MCL1 in the HSPC response to genotoxic injury [4, 8–10].
The heightened susceptibility of hematopoietic cells to genotoxic stress has driven pioneering studies in murine models to identify factors that promote HSPC regeneration. These studies implicated numerous BM niche-secreted factors in mitigating DNA damage-induced HSPC injury, including SCF [11, 12]. EGF [13, 14]. VEGF [15]. Angiopoietin [16]. Pleiotrophin [17–19]. Dickkopf-1 [20]. Angiogenin [21]. FGF [22, 23]. and PGE2 [24]. However, when tested on human HSPCs, key factors such as SCF, FLT3L, TPO, and EGF failed to inhibit DNA damage-induced apoptosis in vitro or protect human hematopoiesis in immunodeficient xenograft models [25]. These discrepancies underscore species-specific differences in irradiation susceptibility [26]. which can stem from variations in transcriptional responses [27]. cytokine requirements, telomere/telomerase biology, and the inbred nature of murine strains, complicating the direct extrapolation of rodent findings to humans [28–30].
The BM microenvironment consists of a variety of non-hematopoietic stromal cells that engage in dynamic crosstalk with HSPCs within specialized niche structures [31, 32]. Mesenchymal stromal cells (MSCs), a critical niche component, support HSPC long-term regenerative capacity in both in vivo and ex vivo settings [33–36]. Substantial research has explored MSCs therapeutic potential in protecting the hematopoietic system from IR-induced injury [35–43]. While these studies have demonstrated increased survival of irradiated hematopoietic cells in co-culture, the identity of MSC-derived pro-survival factors, the molecular pathways engaged in human HSPCs, and their functional impact in vivo remains largely unknown.
Prostaglandin E2 (PGE2) is a bioactive lipid that regulates vertebrate HSPCs via EP2 (PTGER2) and EP4 (PTGER4) Gs-protein-coupled receptors, activating the cyclic AMP (cAMP) signaling pathway [44–46]. The cAMP-responsive element-binding protein (CREB), a master transcription factor activated by elevated cAMP levels, mediates PGE2-dependent transcriptional responses [47]. Short-term (2 h) PGE2 exposure enhanced HSPC migration, survival under cytokine deprivation, homing ability, and human chimerism in xenograft models [48–50]. but failed to improve neutrophil recovery in a clinical trial [49]. Likewise, prolonged (24–48 h) ex vivo PGE2 stimulation before xenotransplantation did not enhance human chimerism [51–53]. While, systemic PGE2 administration pre-IR rescued murine HSPC function [54]. It showed no protective effect in humanized mice [25]. Thus, whether physiological PGE2 mediates MSC-mediated pro-survival signaling and promotes human HSPC regeneration after DNA damage remains unresolved.
Here we demonstrate that MSC-secreted PGE2 activates cAMP signaling via EP2 and EP4 receptors in HSPCs, preventing IR-induced apoptosis by rapidly modulating ASPP1, p21, and PUMA expression. Notably, pharmacological cAMP activation with small-molecule agonists replicated MSC-mediated protection, and markedly enhanced HSPC functionality ex vivo and in vivo.
Materials and methods
Experimental procedures are described in detail in Supplemental Materials.
Results
MSCs promote stemness and anti-inflammatory transcriptional program in human HSPCs
To decipher the molecular mechanisms underlying MSC-mediated protection against genotoxic stress, we established a niche mimicking human CD34^+^ -MSC co-culture system (Fig. 1A). As expected, upon IR the more immature fraction defined here as CD34 + CD38-CD45RA- HSPCs showed a significant increase in Annexin^+^ apoptotic cells, whereas co-culture with human BM-MSCs completely prevented apoptosis (Supplemental Fig. 1B). Due to the limited availability of primary human BM-MSCs, we used the well-characterized murine OP9M2 MSC cell line to support and extend results obtained with human MSCs [55, 56]. (Supplemental Fig. 1A). Co-culturing HSPCs with OP9M2 cells prevented apoptosis induced by IR or Etoposide treatments (Supplemental Fig. 1B, D).Fig. 1MSCs activate a CREB-associated stemness and anti-apoptotic transcriptional program to protect irradiated human HSPCs.A Schematic of the experimental design to evaluate HSPC survival and functionality following co-culture with MSCs post-irradiation. B Analysis of mitochondrial membrane potential in HSPCs under the indicated conditions. Left: representative TMRE histograms; right: quantification expressed as the ratio of mean fluorescence intensity (MFI) between irradiated and non-irradiated cells (n = 3). C In vivo assessment of HSPC functionality. CB CD34+ HSPCs cultured for 24 h under the indicated conditions were transplanted retro-orbitally into NSGW41 mice. Percentage of human CD45+ cells in murine bone marrow 15 weeks post-transplant. Each symbol represents an individual recipient; horizontal bars denote the mean (n = 2). D GSEA showing significant enrichment of hallmark gene sets in HSPCs co-cultured with OP9M2 MSCs compared to cytokine-only culture. E Gene Set Enrichment Analysis (GSEA) plot illustrating the enrichment of apoptosis pathway genes in HSPCs co-cultured with OP9M2 MSCs versus those cultured in serum-free medium with cytokines. F Flow cytometric quantification of EPCR expression on CD34 + CD45RA- HSPCs cultured under the indicated conditions for up to 96 h (n = 3). G GSEA demonstrating significantly enriched transcription factor target gene sets in HSPCs co-cultured with OP9M2 MSCs relative to cytokine-only conditions. H qRT-PCR analysis validating upregulation of CREB target genes in HSPCs co-cultured with OP9M2 MSCs compared to controls (n = 3). Data are mean ± SEM. Statistical significance was assessed by unpaired two-tailed Student’s t-test.
Given the key role of mitochondria in cell functionality, we assessed mitochondrial mass and integrity in HSPCs cultured with cytokines or in contact with OP9M2. In the frame of 24 h, we detected no changes in mitochondrial mass or mitochondrial membrane potential between HSPCs cultured alone or in presence of MSCs. In contrast, while IR did not affect mitochondrial mass, it induced mitochondrial depolarization in HSPCs cultured alone, correlating with increased apoptosis. Co-irradiated MSCs preserved mitochondrial polarization in HSPCs, consistent with reduced rate of apoptosis (Fig. 1B; Supplemental Fig. 1E, 1F). We observed no mitochondrial transfer from MSCs to HSPCs, ruling out this possibility from MSC mediated protection (Supplemental Fig. 2A–C).
To functionally evaluate the impact of MSC-mediated protection on HSPC regenerative potential, we performed in vitro and in vivo assays. In long-term culture-initiating cell (LTC-IC) assays, irradiated HSPCs in monoculture exhibited significantly reduced colony-forming potential. In contrast, MSC co-cultures enhanced LTC-IC frequency of irradiated CD34^+^ cells by nearly five-fold, indicating maintained clonogenicity (Supplemental Fig. 3A). We next evaluated the in vivo engraftment ability of irradiated HSPCs using immunodeficient mouse models. HSPCs cultured without MSCs post-IR engrafted poorly (~3% human CD45+ cells), whereas MSC co-culture substantially improved engraftment to ~16% (Fig. 1C). Notably, multilineage differentiation capacity and splenic homing of HSPCs remained unaltered, suggesting that MSC interaction preserved the fundamental properties of hematopoietic stem cells, including lineage potential and trafficking competency (Supplemental Fig. 3B, D).
To elucidate transcriptional programs underlying MSC-mediated protection, we performed RNA-seq on CD34 + CD38-CD45RA- HSPCs cultured alone or in direct contact with MSCs for 24 h. MSC co-culture led to a robust transcriptional shift characterized by significant upregulation of AP-1 transcription factors (FOS, FOSB, FOSL2, JUN) and metabolic genes involved in glycolysis (ALDOC, PFKFB4). Concurrently, transcripts encoding pro-inflammatory cytokines and chemokines, including IL1B, STAT1, CCL1, and CCL2, were downregulated (Supplemental Fig. 4A). Gene set enrichment analysis (GSEA) revealed suppression of stress-associated signatures, including apoptosis, unfolded protein response, and oxidative phosphorylation, alongside repression of MYC target genes and both type I and II interferon response pathways (Fig. 1D, E). In parallel, MSC-exposed HSPCs showed enrichment of stemness-associated programs, including HSC quiescence, glycolytic metabolism, and transcriptional signatures of regenerative EPCR+ HSPCs (Supplemental Fig. 4C). Flow cytometry confirmed a marked increase in EPCR surface expression in MSC co-cultured HSPCs, consistent with expansion of functionally superior regenerative subsets (Fig. 1F).
MSCs activate cAMP/ CREB signaling pathway to protect human HSPCs
To identify upstream regulators responsible for the observed transcriptional shifts, we performed GSEA using the Transcription Factor Targets (TFT) gene set collection. We found strong predicted activation of bZIP family transcription factors, notably CREB1 and ATF1, along with the AP-1 complex (Fig. 1G; Supplemental Fig. 4B, 4D). Consistent with this, targeted HOMER analysis of promoter regions showed modest enrichment of the canonical CRE motif (TGACGTCA) and a CRE-like/AP-1 motif (TGACTCA) in upregulated genes relative to background (Table 1). CREB1 is a well-established downstream effector of cAMP signaling, known to regulate stress response genes and survival in a cell type specific manner. We validated upregulation of canonical CREB target genes in both OP9M2 and hBM-MSC co-cultured HSPCs via qRT-PCR, consistent with transcriptome-level predictions (Fig. 1H; Supplemental Fig. 4E). Importantly, treatment with the PKA inhibitor Rp-8-Br-cAMPs blocked MSC-mediated protection and reduced CREB phosphorylation in HSPCs, indicating that MSC-derived cues induce CREB activation through a cAMP-dependent mechanism (Fig. 2A, B; Supplemental Fig. 5A–5C).Fig. 2MSCs activate the cAMP/CREB signaling pathway to protect human HSPCs.A CD34+ cells in mono- or MSC co-culture were pre-incubated with Rp-8-Br-cAMPS followed by IR (3 Gy). Fraction of Annexin V + CD34 + CD38-CD45RA- HSPCs was determined 24 h after IR (n = 6). B Flow cytometric analysis of phospho-CREB (Ser133) levels in CB CD34 + CD38-CD45RA- HSPCs after 24 h under the indicated conditions (n = 3). C, D CB CD34+ cells cultured for 24 h in the presence of gap junction inhibitors (Carbenoxolone and GAP27), fraction of Annexin V + CD34 + CD38-CD45RA- HSPCs was determined 24 h after IR (n = 3). Data are mean ± SEM. Statistical significance was assessed by unpaired two-tailed Student’s t-test.Table 1HOMER motif enrichment for the canonical CRE motif (TGACGTCA) and the CRE-like/AP-1 motif (TGACTCA) in promoters of genes upregulated in HSPCs upon OP9M2 MSC co-culture.MotifsP valueTarget (%)Background (%)AP11e-312.055.29CREB11e-24.22****1.13
To test whether cAMP is transferred from MSCs to HSPCs through direct cell-cell contact, we disrupted gap junctions using Carbenoxolone and GAP27 inhibitors. Neither agent affected MSC-mediated protection, suggesting that intracellular cAMP is not shuttled through connexin-based channels (Fig. 2C, D; Supplemental Figs. 6A, B).
PGE2 secreted by BM-MSCs promote HSPCs survival via EP2 and EP4 receptors
We next aimed to identify factors secreted by MSCs that trigger can cAMP/CREB activation in HSPCs. Based on literature mining and receptor expression analysis, we compiled a list of Gs-coupled ligands known to be secreted by MSCs and examined their cognate receptor expression on human HSPCs. Among all candidates, PGE2 emerged as the most likely mediator. EP2 and EP4, two Gs-coupled PGE2 receptors, were robustly expressed on HSPCs, while other Gs-coupled receptors exhibited negligible expression (Table 2). PGE2 levels measured in culture supernatants confirmed that PGE2 was abundantly secreted by different MSCs but not by CD34+ HSPCs themselves (Fig. 3A). Importantly, pharmacological blockade of EP receptors using AH6809 (EP1-3 antagonist) or Palupiprant (EP4-specific antagonist) abrogated the cytoprotective effects of both MSCs and exogenously added dmPGE2 (Fig. 3B, C; Supplemental Fig. 7A, 8A-C). AH6809 and Palupiprant treatment did not alter PGE2 secretion by MSCs (Supplemental Fig. 8D), further supporting a direct signaling mechanism.Fig. 3BM-MSCs secrete PGE2 to protect human HSPCs from IR-induced apoptosis.A ELISA-based quantification of PGE2 in conditioned media collected 24 h after growing different cell types in SFEM SFT medium (n = 4). B, C Annexin V analysis of apoptosis in CB CD34 + CD38-CD45RA- HSPCs cultured for 24 h under the indicated conditions, with or without dmPGE2/ hBM-MSCs and selective EP receptor antagonists (AH6809 for EP1–3 and Palupiprant for EP4) (n = 4). D Comparison of PGE2 secretion by OP9M2 MSCs when co-cultured with CD34+ cells either in direct contact or separated by a 0.4μm transwell (n = 3).Table 2. Potential cAMP activating ligands and their receptors in the context of MSC-HSPC crosstalk.BM-MSC-secreted molecules reported to activate Gs coupled GPCRsCognate Gs coupled receptorsReceptor expression on CD34 + CD38-CD45RA- HSPCsProstaglandin E2 (PGE₂) [74].PTGER2LowPTGER4YesAdenosine [75].ADORA2ANot expressedADORA2BLowParathyroid hormone-related protein (PTHrP) [76].PTH1RNot expressedOnly ligands capable of stimulating Gs coupled receptors and produced by MSCs were considered.
Interestingly, co-culture with HSPCs enhanced PGE2 secretion from OP9M2 MSCs, revealing a positive feedback loop and highlighting the reciprocal nature of MSC-HSPC communication (Fig. 3D). Thus, PGE2 emerges as the dominant MSC-secreted factor that signals through EP2 and EP4 to activate cAMP/CREB and safeguard human HSPCs.
Pharmacological activation of cAMP/CREB signaling pathway protects human HSPCs from IR induced apoptosis
To directly activate cAMP signaling and assess its cytoprotective potential, we treated HSPCs with Forskolin (an adenylyl cyclase activator) and IBMX (a broad phosphodiesterase inhibitor). This combination synergistically restored P-CREB levels in irradiated HSPCs (Fig. 4A; Supplemental Fig. 10A). Importantly, Forskolin/IBMX significantly reduced IR-induced apoptosis in neonatal and adult BM HSPCs, an effect that was reversed by PKA inhibition (Fig. 4B, C; Supplemental Fig. 9B, 9D). Forskolin/IBMX treatment of HSPCs also preserved mitochondrial membrane potential following IR (Fig. 4D; Supplemental Fig. 10D).Fig. 4. Pharmacological activation of the cAMP/CREB signaling pathway protects human HSPCs from IR-induced apoptosis.A Flow cytometric quantification of phospho-CREB (Ser133) levels in CB CD34 + CD38-CD45RA- HSPCs after 24 h of treatment with Forskolin/IBMX, demonstrating sustained CREB activation post-irradiation (n = 4). B Annexin V staining showing that Forskolin/IBMX pre-treatment significantly reduces IR-induced apoptosis in CB CD34 + CD38-CD45RA- HSPCs; this protective effect is abrogated by the cAMP antagonist Rp-8-Br-cAMPs (n = 4). C Apoptosis analysis in human BM-derived CD34 + CD38-CD45RA- HSPCs cultured under the indicated conditions for 24 h (n = 3). D TMRE staining demonstrating preserved mitochondrial membrane potential in Forskolin/IBMX-treated CD34 + CD38-CD45RA- HSPCs: representative histograms (left) and MFI quantification (right) (n = 3). E Annexin V analysis of apoptosis in cycling CB HSPCs (pre-cultured for 72 h) after 24 h exposure to the indicated treatments (n = 3). F Quantification of apoptosis in CB CD34+ cells cultured for 72 h post-irradiation under the indicated conditions (n = 3). G qRT-PCR analysis of CREB target gene expression in CD34+ cells treated with either dmPGE2 or Forskolin/IBMX for 20 h (n = 4). Data are mean ± SEM. Statistical significance was assessed by unpaired two-tailed Student’s t-test.
Interestingly, we observed that both OP9M2 and dmPGE2 were unable to rescue cycling HSPCs from IR-induced apoptosis (Supplemental Fig. 10E). Unlike dmPGE2, Forskolin/IBMX conferred protection to both quiescent and actively cycling HSPCs over 72 hours, indicating a broader therapeutic window and sustained efficacy (Fig. 4E, F). Furthermore, induction of CREB-responsive genes was more sustained with Forskolin/IBMX than with dmPGE2. (Fig. 4G; Supplemental Fig. 10F).
Pharmacological activation of the cAMP/CREB pathway enhances the function of irradiated human HSPCs
To specifically evaluate the intrinsic effects of cAMP-elevating drugs on the function of irradiated human HSPCs - independent of systemic influences that may arise in vivo - we pre-treated CD34+ cells with Forskolin/IBMX ex vivo, irradiated them and transplanted them into NSGW41 mice (Fig. 5A). Compared to vehicle-treated controls, Forskolin/IBMX-treated cells showed significantly higher levels of human chimerism in the bone marrow (6.6-fold increase) and successful engraftment in a higher proportion of recipients (12/15 vs. 4/11) (Fig. 5B, C; Supplemental Fig. 11B, C). In contrast, dmPGE2-treated HSPCs showed no significant benefit in engraftment capacity (Supplemental Fig. 11A). In secondary transplants, only Forskolin/IBMX-pretreated HSPCs retained multilineage reconstitution potential, confirming preserved stemness and self-renewal (Fig. 5D; Supplemental Fig. 11D).Fig. 5. Pharmacological activation of the cAMP/CREB signaling pathway enhances engraftment, self-renewal, and protects engrafted human hematopoietic cells from IR-induced apoptosis in vivo.A Schematic representation of the experimental design to assess HSPC functionality following irradiation and Forskolin/IBMX treatment ex vivo. B Human chimerism in the bone marrow of NSGW41 mice 12 weeks post-injection of control or Forskolin/IBMX-treated and irradiated (3 Gy) hCD34+ cells (each symbol represents an individual recipient; horizontal bars denote the mean; n = 2). C Quantification of immature (hCD34 + ) cell frequency in the bone marrow of mice transplanted with control versus Forskolin/IBMX-treated CD34⁺ cells (n = 2). D Human chimerism in the bone marrow of secondary recipient mice following transplantation of total bone marrow from primary recipients (each symbol represents an individual recipient; horizontal bars denote the mean). Data are mean ± SEM. P values determined by one-way ANOVA with Tukey’s multiple comparisons test.
To test whether systemic pharmacological activation of cAMP signaling could protect human hematopoietic cells, we injected human CD34+ cells into NSG mice. Twelve weeks post-engraftment, mice were stratified into three groups based on peripheral blood chimerism and treated with either 1) DMSO (non-irradiated), 2) DMSO followed by total body irradiation (TBI, 3 Gy) or 3) Forskolin/IBMX followed by TBI (3 Gy), one hour later (Supplemental Fig. 12A). Twenty-four hours post IR, flow cytometric analysis of bone marrow revealed no significant difference in total human CD45+ chimerism across groups, but TBI markedly increased apoptosis in human CD45+ hematopoietic cells (Supplemental Fig. 12B, C). Notably, Forskolin/IBMX pre-treatment significantly reduced this irradiation-induced apoptosis compared to vehicle-treated controls, indicating that transient in vivo activation of the cAMP pathway can attenuate acute genotoxic injury to human hematopoietic cells within the bone marrow niche (Supplemental Fig. 12C).
TBI is known to induce pro-inflammatory cytokines that can also trigger inflammatory cell death pathways in bone marrow cells in addition to apoptosis [57]. Thus, we next examined the inflammatory milieu after irradiation. TBI caused only a modest, rise in human IFNγ and no change in TNFα (Supplemental Fig. 12D, E), and Forskolin/IBMX did not further elevate IFNγ, indicating that systemic cAMP activation does not alter IR-induced cytokine release under radiation regiment employed. Despite reducing Annexin+ cell fraction after IR, pharmacological cAMP activation failed to preserve human long-term BM and splenic repopulating capacity in secondary recipients (Supplemental Fig. 12F, G), highlighting the complexity of the in vivo pathways ultimately regulating HSPC survival. Together, these long-term in vivo and in vitro data establish that pharmacological activation of cAMP signaling pathway can mimic pro-survival phenomenon of MSC co-culture and offers significant radioprotection on ex vivo irradiated human HSPCs.
Pro-survival effects of cAMP agonists are dependent on functional anti-apoptotic MCL1/BCL-XL proteins and altered P53/ASPP1 program
To identify downstream effector proteins mediating cAMP-induced radioprotection, we analyzed transcript and protein levels of pro- and anti-apoptotic BCL-2 family members. At early time points (3 hours post-treatment), MCL1 mRNA was strongly induced by both Forskolin/IBMX and dmPGE2, while BCL2 and BCL-XL transcripts were transiently suppressed (Supplemental Figs. 13A, C). By 20 hours, mRNA expression of MCL1 and BCL-XL returned to baseline, while BCL2 remained low (Supplemental Figs. 13D–F). In addition, we revealed that cAMP agonist treatment attenuated expression of selected p53 targets (e.g. MDM2, PUMA), and co-factors (e.g. ASPP1), while inducing cell cycle regulator p21 (Fig. 6A), collectively supporting an anti-apoptotic shift in transcriptional programming.Fig. 6. Pro-survival effects of cAMP/CREB signaling depend on functional MCL1 and BCL-XL proteins.A qRT-PCR analysis of selected p53 pathway-related genes in CD34+ cells treated with Forskolin/IBMX for 2 h (n = 4). CB CD34+ cells were nucleofected with non-targeting control (NTC) or siRNA pools targeting BCL2 (B), BCL-XL (C), or MCL1 (D) and allowed to recover for 24 h. Nucleofected cells were then exposed to the indicated treatments followed by apoptosis analysis in CD34 + CD38-CD45RA- HSPCs after 24 h (n = 3). E Flow cytometric analysis of MCL1 protein levels in CD34 + CD38-CD45RA- HSPCs cultured with or without Forskolin/IBMX for 24 h. Left: representative histograms; right: quantitative MCL1 expression (n = 6).
To test functional relevance, we used siRNA to knock down BCL2, BCL-XL, or MCL1 in HSPCs (Supplemental Figs. 13G–I). While BCL2 depletion had no significant effect, knockdown of either MCL1 or BCL-XL abolished the protective effects of Forskolin/IBMX and dmPGE2 (Fig. 6B–D), indicating their non-redundant roles. Intracellular flow cytometry analysis confirmed that Forskolin/IBMX and MSC co-culture stabilize MCL1 protein levels in irradiated HSPCs, whereas BCL2 and BCL-XL proteins remained unchanged (Fig. 6E; Supplemental Figs. 14A–C, 15A–C).
Finally, we assessed expression of Survivin (BIRC5), a known regulator of apoptosis previously implicated in PGE2 signaling [58]. Survivin transcript and protein levels were not significantly altered by dmPGE2 or Forskolin/IBMX treatment under steady-state conditions. However, IR-induced downregulation was reversed in OP9M2 MSC coculture and dmPGE2 conditions, suggesting a context-dependent stabilization (Supplemental Figs. 16A–D).
In summary, our integrative analysis demonstrates that dmPGE2 and Forskolin/IBMX conferred radioprotection depends on the stabilization of MCL1 and BCL-XL proteins in human HSPCs. This dual requirement underscores the complexity of IR induced apoptosis regulation in HSPCs and highlights the importance of a coordinated anti apoptotic signaling by cAMP elevation.
Discussion
Our results establish that activation of cAMP signaling pathway potently inhibits DNA damage-induced apoptosis and enhances the long-term regenerative potential of human HSPCs. In an ex vivo niche-mimicking model, MSCs secrete PGE2 upon contact with HSPCs, engaging EP2 and EP4 receptors to suppress IR-induced apoptosis. Pharmacological modeling revealed that both PGE2 and the Forskolin/IBMX (Forskolin/IBMX) combination mitigate early apoptosis, but only Forskolin/IBMX enhanced the repopulation capacity of irradiated HSPCs in both primary and secondary transplantation assays. Mechanistically, cAMP agonists downregulated pro-apoptotic ASPP1 and PUMA, and upregulated MCL1 and p21, aligning with reduced apoptosis and enhanced survival. These findings demonstrate that physiological or pharmacological activation of cAMP signaling ameliorates IR-induced functional loss of human HSPCs.
Dissection of the MSCs impact on the transcriptome of irradiated human HSPCs is unique to this study and extends prior knowledge obtained from murine HSPCs isolated after whole body IR [9, 54]. Our transcriptomic analyses revealed that MSCs induces a distinct gene expression program in HSPCs, characterized by upregulation of stemness-associated transcription factors (e.g., FOS, JUN, EGR1 and CREB) and a concurrent suppression of pro-inflammatory, stress-related and proliferative pathways (e.g. IFNa, IFNg and MYC). Abrupt transcriptional induction of multiple stress pathways in human HSPCs ex vivo was recently proposed to negatively impact expansion and functionality [56, 59]. On the other hand, our transcriptomic analysis reveals that MSCs can stabilize human HSPCs ex vivo in the state reminiscent of their freshly isolated counterparts. Interestingly, MSC-induced transcriptional signature in HSPCs was not significantly affected by IR. Our transcriptomic results underscore niche signaling complexity and argue that MSCs can prime HSPCs for enhanced resilience to IR and other genotoxic insults.
While dissecting the possible mechanisms by which MSCs can elevate cAMP levels in human HSPCs, we excluded the involvement of mitochondrial transfer or gap junction mediated exchange [60, 61]. Instead, we found that both human and murine MSCs in contact with MSCs increased secretion of physiological cAMP agonist PGE2, which blocked IR-induced apoptosis and promoted regeneration. The hematopoietic supportive activity of MSCs is primarily attributed to the secretion of proteins such as CXCL12, SCF, ANGPT1 and Osteopontin amongst others. Our findings indicate that PGE2, a bioactive lipid as the predominant signaling mediator in the MSC secretome that protects HSPCs from IR induced apoptosis. Indeed, blocking the PGE2 receptors on HSPCs underscores the importance of BM-MSC derived PGE2 in regulating HSPC survival. Our data expands on the previous observations that PGE2 and its synthetic analogue dmPGE2 regulate HSPC survival [44, 58]. A deeper understanding of the mechanisms regulating PGE2 production and receptor expression on HSPCs will be essential for advancing MSC- or PGE2- based strategies to enhance HSPCs recovery.
Although the detailed mechanisms that account for the cell type specific control of DNA damage induced apoptosis by cAMP are still elusive, our results reveal that cAMP agonists can repress ASPP1 - the endogenous stimulator of p53-dependent apoptosis in cells, including HSPCs [4, 9]. In agreement with the established role of ASPP1 in modulation of p53 transcriptional response we observed coordinated decline in the expression of pro-apoptotic (PUMA) and upregulation of pro-survival genes (e.g. MCL1 and p21) upon HSPC treatment with cAMP agonists. This shift in p53 transcriptional response also provides plausible molecular mechanism for the observed block in IR-induced apoptosis (via decreased PUMA) and early cycling (via elevated p21) of mouse HSPCs upon injection of PGE2 [54]. Although cAMP agonists resulted only in partial blocking of PUMA induction, murine models posits that even 50% reduction in PUMA expression suffices to significantly protect from the lethal IR-hematotoxicity [6, 62]. Elevated activity of CRE-binding factors (e.g., CREB, ATF1 and AP1 complex) in HSPCs from MSCs co-culture or after treatment with cAMP agonists (this study) or of SIRT1 [63]. can additionally contribute to the diminished IR-induced apoptosis in HSPCs. Indeed, efficient transactivation of p53, as well as of CREB and NF-KB target genes, are dependent on their binding to p300 and CBP histone acetyltransferases [64]. Given limited cellular amounts of p300/CBP, higher levels of active CREB, as indicated by Ser133 phosphorylation in HSPCs, might destabilize pro-apoptosis gene regulatory programs [65]. and tilt HSPCs cell fate towards survival.
Our mechanistic studies further underscore the differential importance of anti-apoptotic BCL-2 family proteins in this process. Although early transcriptional responses to cAMP elevated treatments included a transient downregulation of BCL2 and BCL-XL, functional assays demonstrated that the radioprotective benefits of both PGE2 and Forskolin/IBMX depend critically on the stabilization of MCL1 and the activity of BCL-XL. Notably, while BCL-XL protein levels did not decrease immediately following irradiation, it is likely that BCL-XL undergoes rapid deamidation as an immediate response to IR stress [66, 67]. —a modification that may impair its function—and that its protein levels may decline at later time points. siRNA-mediated knockdowns confirmed that disruption of either MCL1 or BCL-XL, but not BCL-2, abrogated the protective effects of PGE2 and Forskolin/IBMX, emphasizing the need for a coordinated anti-apoptotic response in mitigating IR-induced damage. In addition to mediating cAMP pro-survival effects as uncovered here, elevated dependency of HSPCs on both BCL-XL and MCL-1 for their survival after DNA damage, as uncovered here, and by Erlacher group [8, 68]. represents a human specific phenomenon, as mouse counterparts dependent only on MCL-1 [69]. and would predict severe hematotoxicity upon clinical targeting with selective inhibitors.
Importantly, despite efficient IR-induced apoptosis abrogation in HSPCs by cAMP agonists ex vivo, total human engraftment (hCD45+ cells) remained markedly reduced, indicating in vivo reactivation of DNA damage induced mechanisms that limit HSPC proliferation. Remarkably, our re-transplantation experiments demonstrated that even few Forskolin/IBMX pre-treated irradiated HSPCs preserved significant self-renewal ability that was lost completely in the cytokine only cultured irradiated HSPCs. Moreover, they chart a critical time window in which modulation of DNA damage induced apoptosis can play a critical role for preservation of human HSPC functionality. These findings also independently support a concept obtained in transgenic animals with switchable p53 [70]. or deleted PUMA [71, 72]. suggesting that while transient inactivation of p53-dependent apoptosis can rescue myelosuppression, its delayed functional restoration suffices to suppress IR-induced and compensatory proliferation driven carcinogenesis.
While agents like dmPGE2 have shown promise in murine models [44, 54, 63]. our data indicates that direct modulation of the cAMP/CREB axis—via compounds such as Forskolin/IBMX—may provide a more robust and sustained protective effect in human HSPCs. Our in vivo data indicate that transient cAMP activation can attenuate the immediate apoptotic response of human hematopoietic cells to irradiation, independent of measurable changes in donor-derived IFNγ or TNFα. This points to a direct effect on intrinsic apoptosis pathways, although the impact on the host microenvironment requires further investigation. Indeed, the lack of improvement in secondary repopulation suggests that IR-induced persistent DNA damage signaling, oxidative stress, and radiation-induced bystander effects - still undermines long-term HSC function [36, 73]. These broader lesions likely lie beyond the protective reach of short-term cAMP elevation alone. Incorporating additional agents that mitigate oxidative damage or preserve the stromal microenvironment may therefore be required to achieve durable radioprotection in vivo.
In conclusion, our integrated molecular and functional analyses establish that activation of cAMP/CREB signaling pathway strongly potentiates human HSPCs regenerative abilities after DNA damage via decreasing ASPP1/p53 transcriptional activity and preserving vital MCL1 and BCL-XL protein levels. We predict that focusing on the pharmacological modulation of ASPP1/p53 axis in primary human HSPCs may be more directly relevant for therapeutic strategies aimed to mitigate DNA damage associated myelosuppression and genome editing efficacy without promoting transformation.
Supplementary information
Supplemental methods and figure legends Supplemental Figure 1 Supplemental Figure 2 Supplemental Figure 3 Supplemental Figure 4 Supplemental Figure 5 Supplemental Figure 6 Supplemental Figure 7 Supplemental Figure 8 Supplemental Figure 9 Supplemental Figure 10 Supplemental Figure 11 Supplemental Figure 12 Supplemental Figure 13 Supplemental Figure 14 Supplemental Figure 15 Supplemental Figure 16
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rangarajan A, Hong SJ, Gifford A, Weinberg RA, Parekh C, Crooks GM. Erratum to Species- and Cell Type-Specific Requirements for Cellular Transformation. [Cancer Cell. 2004;6:171–183. J Clin Immunol. 2013;24:394–8.10.1016/j.ccr.2004.07.00915324700 · doi ↗ · pubmed ↗
- 2Sporrij A, Choudhuri A, Prasad M, Muhire B, Fast EM, Manning ME, et al. PGE 2 alters chromatin through H 2A.Z-variant enhancer nucleosome modification to promote hematopoietic stem cell fate. Proc Natl Acad Sci USA. 2023;120.10.1073/pnas.2220613120 PMC 1017584237126722 · doi ↗ · pubmed ↗
- 3Magnusson M, Sierra MI, Sasidharan R, Prashad SL, Romero M, Saarikoski P, et al. Expansion on Stromal Cells Preserves the Undifferentiated State of Human Hematopoietic Stem Cells Despite Compromised Reconstitution Ability. P Lo S One. 2013;8.10.1371/journal.pone.0053912 PMC 354705023342037 · doi ↗ · pubmed ↗
- 4Shu M, Zhang J, Peng Y, Li Z, Shu X, Wang J, et al. Programmed cell death regulates hematopoietic cell homeostasis under radiation conditions. Stem Cell Res Ther [Internet]. [cited 2025 Dec 2];16:390. Available from: (2025) https://pmc.ncbi.nlm.nih.gov/articles/PMC 12281764/.10.1186/s 13287-025-04502-3PMC 1228176440691803 · doi ↗ · pubmed ↗
- 5de Oliveira Bravo M, Carvalho JL, Saldanha-Araujo F. Adenosine production: a common path for mesenchymal stem-cell and regulatory T-cell-mediated immunosuppression. Vol. 12, Purinergic Signalling. Springer Netherlands; 2016. p. 595–609.10.1007/s 11302-016-9529-0PMC 512400627557887 · doi ↗ · pubmed ↗