Novel splice-site variants in TMPRSS3 impair hearing via exon skipping and abrogated protease activity
Xiang Wang, Wei-Qian Wang, Sha-Sha Huang, Xi Wang, Li-Min Hu, Xing Liu, Qiu-Quan Wang, Zhen-Dong Wang, Jin-Cao Xu, Yong-Yi Yuan, Xue Gao

TL;DR
This study identifies new genetic variants in the TMPRSS3 gene that cause hearing loss by disrupting protein function and splicing, offering insights into how different mutations lead to varying severity of hearing impairment.
Contribution
The study expands the known spectrum of TMPRSS3 variants and demonstrates that splice-site variants cause more severe protease activity loss than missense variants.
Findings
Three Chinese families with compound heterozygous TMPRSS3 variants were identified, including novel splice-site and missense mutations.
Splice-site variants caused exon skipping and significantly reduced protease activity compared to missense variants.
The findings explain genotype-phenotype heterogeneity in TMPRSS3-associated hearing loss.
Abstract
Hearing loss (HL) is genetically heterozygous, making its genetic diagnosis challenging. Identification of novel HL-associated genes and variants will enhance our understanding of the molecular mechanisms and improve genetic diagnosis. TMPRSS3, encoding a transmembrane serine protease, is implicated in autosomal recessive nonsyndromic hearing loss (ARNSHL), designated as DFNB8 (postlingual onset) or DFNB10 (prelingual onset). Although over 100 pathogenic TMPRSS3 variants have been reported, only seven splice-site variants have been documented to date. This study investigates the molecular etiology of ARNSHL in three Chinese family and functionally characterizes five novel TMPRSS3 variants, including one non-canonical splice-site variant, two canonical splice-site variants, and two missense variants. Whole-exome sequencing and gene panel sequencing identified candidate variants,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
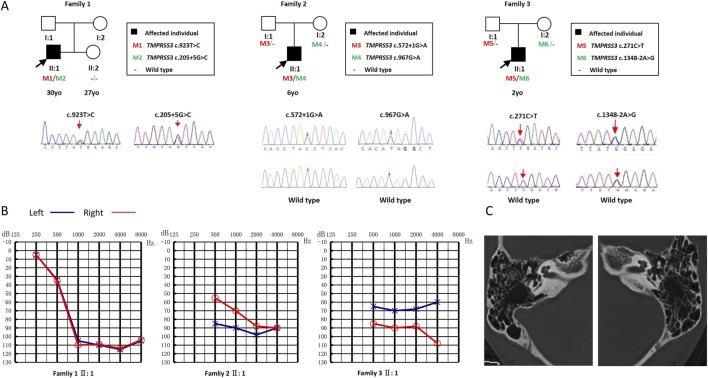 FIGURE 1
FIGURE 1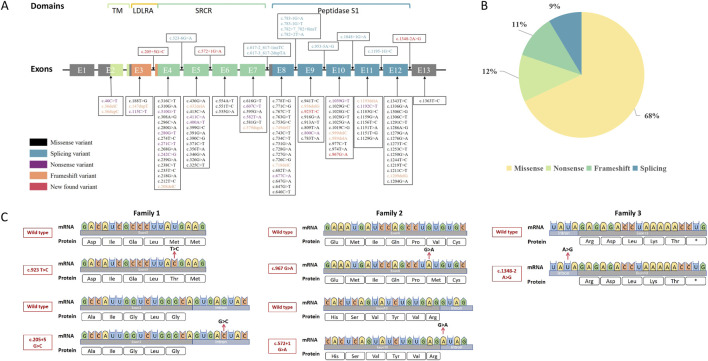 FIGURE 2
FIGURE 2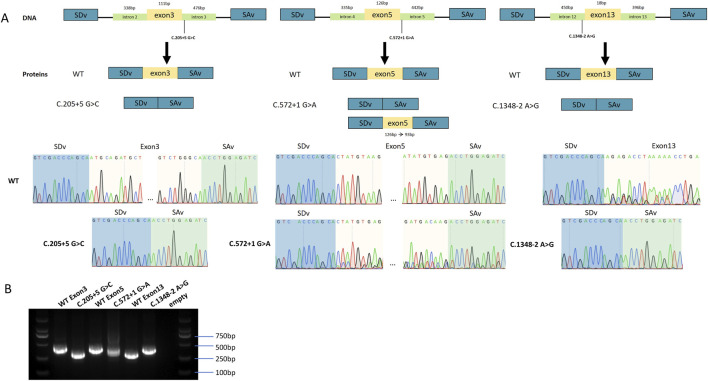 FIGURE 3
FIGURE 3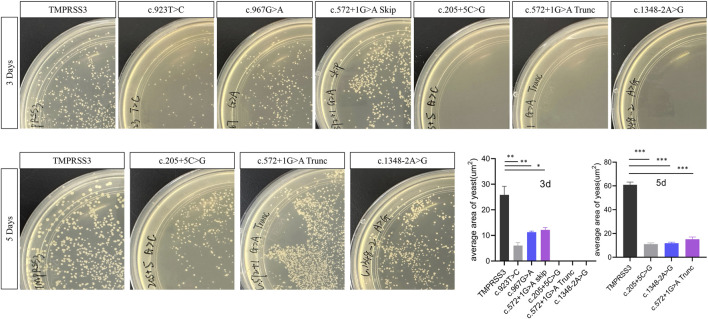 FIGURE 4
FIGURE 4- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHearing, Cochlea, Tinnitus, Genetics · Genomics and Rare Diseases · Vestibular and auditory disorders
Introduction
1
Hearing loss (HL) is among the most prevalent sensory disorders worldwide, with genetic factors contributing to over 50%–80% of congenital HL cases (Shearer et al., 1993; Choe et al., 2023). The extreme genetic heterogeneity of HL, involving hundreds of genes, poses significant challenges for molecular diagnosis and genetic counseling. Autosomal recessive nonsyndromic hearing loss (ARNSHL) accounts for approximately 70%–80% of hereditary HL cases, with 88 associated genes identified to date (Hereditary Hearing Loss Homepage, accessed, 10/19/25).
TMPRSS3 (MIM *605511), which encodes a transmembrane serine protease, is an important causative gene associated with both prelingual (DFNB10, MIM #605316) and postlingual (DFNB8, MIM #601072) forms of ARNSHL. TMPRSS3-related hearing loss is highly heterogeneous in severity, ranging from mild to profound, and typically follows a progressive course. Pathogenic variants in this gene are among the top ten genetic cause of HL (Sang et al., 2019). The TMPRSS3 protein is expressed in key cochlear cell types, including hair cells, supporting cells, the stria vascularis, and the spiral ganglion neurons (Chen et al., 2022; Guipponi et al., 2002; Arora et al., 2025; Tang et al., 2019).
To date, over 100 pathogenic TMPRSS3 variants have been reported, including missense, nonsense, frameshift, and splice-site alterations (Nisenbaum et al., 2023). Among these, splice-site variants remain exceptionally rare, with only seven documented cases (Gao et al., 2017a; Lechowicz et al., 2017; Imtiaz et al., 2011; Richard et al., 2019). The phenotypic severity of HL is influenced by both the type and genomic location of the variant, with splice-site and missense variants often exerting distinct functional effects (Guipponi et al., 2008).
Despite advances in genetic testing, molecular diagnosis of HL remains challenging due to the large number of candidate genes and the presence of variants of uncertain significance (VUS). Functional validations are therefore essential, particularly for variants suspected to affect splicing or enzymatic activity. Although a few TMPRSS3 splice-site variants have been reported, their precise molecular mechanisms and genotype-phenotype correlations are not fully understood (Boudewyns et al., 2018; Kim et al., 2017).
In this study, we investigated three Chinese families with ARNSHL and identified five novel compound heterozygous TMPRSS3 variants. The variants include one non-canonical splice-site variant (c.205 + 5G>C), two canonical splice-site variants (c.572 + 1G>A, c.1348-2A>G), and two missense variants (c.923T>C, p. Met308Thr, c.967G>A, p. Val323Met). We performed functional analyses—minigene splicing assays and yeast-based protease activity assays—to assess their pathogenicity. Our findings expand the mutational spectrum of TMPRSS3 and provide new mechanistic insights into the genotype-phenotype correlations underlying DFNB8/10-related HL.
Materials and methods
2
Study subjects
2.1
This retrospective study utilized in-house databases of genetic hearing loss from the College of Otolaryngology Head and Neck Surgery at the Chinese PLA General Hospital. The clinical assessment for HL included medical history collection (via questionnaire), otoscopy, pure tone audiometry, acoustic immittance, and stapedius reflexes. HL severity was classified according to the 2021 WHO guidelines, based on the average pure tone audiometry (PTA) threshold at 0.5, 1, 2, and 4 kHz (mild: 26–40 dB HL; moderate: 41–60 dB HL; severe: 61–80 dB HL; profound: ≥81 dB HL). Frequency-specific HL was defined as low (125–500 Hz), mid (1–2 kHz), and high (4–8 kHz). Prelingual (onset before age 3) and postlingual (onset after age 3) HL were distinguished. Vestibular function was evaluated using tandem gait, Romberg, and caloric test, while temporal bone CT were also assessed.
Whole exome sequencing (WES) and variant analysis
2.2
Whole-exome sequencing (WES) and targeted gene panel sequencing were performed for each proband. Sequence reads were aligned to the GRCh38 reference genome, followed by variant calling and annotation. Variant filtering was prioritized on known HL-related genes. Candidate variants were classified based on American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines for genetic hearing loss and validated by Sanger sequencing (Richards et al., 2015).
Construction of TMPRSS3 minigene plasmids
2.3
Three TMPRSS3 splice-site variants (c.205 + 5G>C, c.572 + 1G>A, and c.1348-2A>G) were functionally tested using the pSPL3 exon-trapping vector (Miaolinbio, Wuhan, China) (Westin et al., 2021). Genomic DNA from HEK293T cells served as the template for generating mutant fragments by PCR using gene-specific primers (Supplementary Table S1, TsingkeBio, Beijing, China). For the c.205 + 5G>C mutant, exon 3 along with 338 bp of the upstream intron and 476 bp of the downstream intron were amplified. The first PCR (primers 205-F1 and 205-R1) amplified exon 3 with a 338 bp segment of intron 2, while the second PCR (primers 205-F2 and 205-R2) amplified exon 3 with a 476 bp segment of intron 3. These two fragments were then fused by overlap-extension PCR using primers 205-F1 and 205-R2 to generate the full-length mutant construct. The same strategy was applied for constructing the c.572 + 1G>A mutant fragment (exon 5 with 335 bp of upstream intron and 442 bp of downstream intron) and the c.1348-2A>G mutant fragment (exon 13 with 450 bp of upstream intron and 396 bp of downstream intron).
For cloning, the pSPL3 plasmid was digested with EcoRI and NotI (NEB, US). The ligation ratio was determined using the online tool provided by Vazyme (https://crm.vazyme.com/cetool/restructure.html). The ligation products were transformed into competent E. coli DH5α cells (Takara, Japan) and plated onto ampicillin-containing agar plates, followed by overnight incubation at 37 °C. Single colonies were picked and inoculated into LB medium supplemented with ampicillin, then cultured for 3 h. Colony PCR and Sanger sequencing were performed using primers SD-6 and SA2 to confirm correct insertions. Colonies with correct constructs were expanded in 8 mL of ampicillin-containing LB medium overnight at 37 °C with shaking. Plasmid DNA was subsequently extracted using the EndoFree Plasmid Midi Kit (CWbio, Beijing, China).
Cell transfection and splicing assay
2.4
HEK293T cells were cultured in DMEM (Thermo, US) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (Beyotime, Shanghai, China) and seeded into 12-well plates. After 24 h, the cells were transfected in triplicate with either wild-type or mutant pSPL3 plasmids (c.205 + 5G>C, c.572 + 1G>A, and c.1348-2A>G) using the jetPRIME® transfection system (Polyplus, Shanghai, China), with 0.8 μg DNA, 1.6 μL jetPRIME® reagent, and 75 μL jetPRIME® buffer per well. Total RNA was extracted 48 h post-transfection using a kit (ABclonal, Beijing, China) and reverse-transcribed into cDNA (ABclonal, Beijing, China).
PCR amplification was performed in a 40 μL reaction volume. The cycling conditions were as follows: 95 °C for 12 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s; followed by a final extension at 72 °C for 5 min. The PCR products were separated on a 2% agarose gel at 160 V for 15 min and visualized under UV light. The remaining products were subjected to Sanger sequencing to confirm the splicing alterations resulting from the variants.
Molecular cloning and construction of wild-type (WT) and mutant TMPRSS3 expression plasmids
2.5
After confirming that splice-site variants alter mRNA expression, wild-type TMPRSS3 cDNA was obtained from HeLa cell (which express TMPRSS3 protein according to the Human Protein Atlas, www.proteinatlas.org). Mutant TMPRSS3 constructs were subsequently generated by overlap extension PCR using gene-specific primers (see Supplementary Table S1). For functional assays in yeast, wild-type and mutant TMPRSS3 sequences were cloned into the pGBKT7 vector (carrying a TRP1 auxotrophic marker), and the TMPRSS3 substrate epithelial sodium channels (ENaC, Coolaber, Beijing, China) was cloned into the pGADT7 vector (carrying a LEU2 auxotrophic marker). Following transformation and selection, yeast strains of each genotype were plated onto YPD medium, incubated, and colony numbers were quantified using the ImageJ software. The experiments were performed in triplicate, and the collected data are presented as mean ± SD. Statistical significance was analyzed using one-way ANOVA followed by Tukey’s post hoc test.
Results
3
Clinical findings
3.1
Family 1: the proband (II:1, a 30-year-old Han Chinese male, Figure 1A) presented with progressive, postlingual, severe sensorineural HL with onset at age 10 (Figure 1B). Audiograms revealed mid-to-high-frequency HL with absent stapedius reflexes. Tinnitus was reported. Vestibular function tests (tandem gait, Romberg, caloric tests) were unremarkable. There were no structural abnormalities of the outer, middle or inner ear were observed on temporal bone CT (Figure 1C). The patient currently uses bilateral hearing aids for auditory rehabilitation. No family history of HL was reported.
Pedigree, audiograms, temporal bone CT of families with TMPRSS3 causatives. (A) Family 1: WES identified compound heterozygous TMPRSS3 variants c.205 + 5G>C and c.923T>C (p.Met308Thr); the spouse tested negative for common deafness genes. Family 2: A hearing-loss gene panel detected c.572 + 1G>A and c.967G>A (p.Val323Met). Family 3: WES revealed c.1348-2A>G and c.271C>T. (B) Audiological findings. Family 1 II:1: Hearing loss noted at age 10; audiogram obtained at 24 years. Family 2 II:1: First audiological assessment at 1 year 2 months. Family 3 II:1: Hearing impairment observed after age 1; preoperative thresholds recorded at 2 years 10 months before right cochlear implantation. (C) Temporal-bone CT of the Family 1 proband. CT demonstrated patent external auditory canals, normal tympanic membranes, well-aerated middle ears, and symmetrical internal auditory canals without stenosis or dilation.
Family 2: the proband (II:1, a 6-year-old Han Chinese male) presented with prelingual, severe sensorineural HL. The patient underwent left cochlear implantation at 1 year and 2 months of age, and the audiogram represents his preoperative hearing status (Figure 1B). Following implantation, his auditory and speech-language development demonstrated significant improvement. No family history of HL was reported.
Family 3: the proband (II:1, a 10-year-old Han Chinese male) presented with progressive, prelingual, severe sensorineural HL with onset at age 1. The patient underwent right cochlear implantation at 2 years and 10 months of age (Figure 1B). The presented audiogram, obtained preoperatively, documents the baseline hearing level prior to surgery. His postoperative listening and language abilities have shown marked improvement. No family history of HL was reported.
Identification of TMPRSS3 variants
3.2
Previously reported TMPRSS3 variants were compiled from the Human Gene Mutation Database (HGMD) and cross-referenced with the PubMed literature. These data were then integrated to construct a comprehensive map of known TMPRSS3 variants (Figure 2A). The retrieved variants, detailed in Supplementary Table S2, revealed that splice-site variants are considerably less frequent than missense variants. Among all documented variants, missense variants constituted the majority (68%), followed by nonsense (12%), frameshift (11%), and splice-site (9%) variants (Figure 2B). In this study, we identified five novel variants, comprising three splice-site variants and two missense variants (Figure 2C). WES identified three novel TMPRSS3 variants c.205 + 5G>C, c.923T>C (p.Phe308Leu), and c.1348-2A>G with robust sequencing quality (median depth >100× for c.205 + 5G>C and c.923T>C, >200× for c.1348-2A>G; >98–99% of targeted regions covered at ≥30×). Based on ACMG/AMP guidelines, c.205 + 5G>C was classified as a variant of uncertain significance (PM2, PP3, PP4), c.923T>C as likely pathogenic (PM2, PM3, PP3, PP4), and c.1348-2A>G as pathogenic (PVS1, PM2, PM3, PP3, PP4, PP5). Panel sequencing revealed two additional novel variants with adequate coverage: c.572 + 1G>A (splicing), classified as pathogenic (PVS1, PM2, PM3, PP4), and c.967G>A, classified as a variant of uncertain significance (PM2, PM3, PP4).
Summary of reported and novel TMPRSS3 variants. (A) Distribution of known and novel TMPRSS3 variants. Previously reported variants were compiled from HGMD and PubMed and mapped across the gene (see Supplementary Table S2). (B) Proportion of variant types. Incorporating five novel variants from this study, the overall distribution was: missense 68%, nonsense 12%, frameshift 11%, and splice-site 9%. (C) Novel variants identified in this study. Comparative mRNA and protein sequence diagrams illustrate the local molecular changes for each family.
The two missense variants are located in close proximity to each other, the c.923T>C variant results in a substitution of methionine by threonine at position 308 of the protein, and the c.967G>A variant leads to a substitution of valine by methionine at position 323. These variants alter a highly conserved residue within the catalytic serine protease domain. Located proximal to the most common pathogenic variant, c.916G>A, these variants are likely to exert similar deleterious effects (Gao et al., 2017a; Weegerink et al., 2011).
Splicing analysis
3.3
In vitro splicing assays were performed using pSPL3 plasmids carrying the splice site variants c.205 + 5G>C, c.572 + 1G>A, and c.1348-2A>G. To analyze the effect of each variant on splicing, we constructed minigenes containing either the wild-type or mutant sequences, which included the exons flanking the splice sites and portions of the adjacent introns. These constructs were transfected into HEK293T cells to evaluate splicing outcomes.
Sequencing confirmed that the wild-type TMPRSS3 fragments were correctly expressed, with proper splicing of exons 3, 5, and 13 (Figures 3A,B).
Analysis of the effects of splice-site variants using minigene assays. (A) Sequencing chromatograms of wild-type and mutant minigene constructs. SAv and SDv correspond to the upstream and downstream exons surrounding the inserted target gene. For c.205 + 5G>C, the minigene (338 bp upstream and 476 bp downstream of exon 3) showed normal splicing in the wild type and exon 3 skipping in the mutant. For c.572 + 1G>A, the construct (335 bp upstream and 442 bp downstream of exon 5) produced mixed transcripts in the mutant, including a 33 bp truncation and complete exon 5 skipping. For c.1348-2A>G, located downstream of the stop codon, the wild-type construct showed no SAv expression, whereas the mutant induced exon 13 skipping and restored SAv expression. (B) PCR results of wild-type and mutant minigenes. Gel electrophoresis showed paired bands for each exon: wild-type and mutant products for exon 3, exon 5, and exon 13, respectively.
The c.205 + 5G>C is a non-canonical donor site variant in intron 2. It is predicted to cause skipping of exon 3, resulting in a 111 bp truncation of the transcript (Figure 3A). This event is predicted to disrupt the LDL receptor class A (LDLRA) domain, affecting protein stability.
The c.572 + 1G>A is a canonical donor site variant in intron 5 within the scavenger receptor cysteine-rich (SRCR) domain. This variant results in two splicing outcomes: complete skipping of the exon 5, similar to c.205 + 5G>C, resulting in a 126 bp truncation of the transcript; or a 33 bp truncation within exon 5 (Figure 3A). Both outcomes are likely to induce misfolding of the highly conserved SRCR domain, leading to loss of enzymatic activity or impaired substrate recognition, thereby inhibiting ENaC activation.
The c.1348-2A>G is a canonical acceptor site variant upstream of exon 13 within the serine protease domain. It led to exon 13 skipping (Figure 3A). Since exon 13 contains the stop codon, its exclusion results in a frameshift and incorporation of downstream intronic sequences, leading to the formation of an aberrant serine protease (SP) domain.
Minigene assays confirmed that the splice-site variants led to aberrant splicing. While the wild-type construct produced the expected normal transcript, the mutant constructs resulted in abnormal splicing patterns. These findings were consistently replicated in three independent experiments in HEK293T cells. Sequencing all bands confirmed break points and splicing events.
Mutant TMPRSS3 proteins showed aberrant protease activity
3.4
Previous studies have demonstrated that TMPRSS3 cleaves ENaC, and the cleavage efficiency can serve as an indicator of TMPRSS3 enzymatic activity (Guipponi et al., 2002). Cleavage efficiency correlates with colony growth on selective medium. Yeast successfully co-transformed with the TMPRSS3 and its substrate plasmids carried the LEU and TRP markers, respectively, and were selected on minimal media containing 2% glucose lacking leucine and tryptophan (Leu^−^/Trp^−^). Subsequently, the Leu^+^/Trp^+^ transformants were then replica-plated onto YPD medium supplemented with 2% sucrose and 0.5 mg/mL antimycin A. Quantification of colony number and size was performed using the ImageJ software, serve as a proxy for protease activity (Kim et al., 2017; Wong et al., 2020).
To assess the pathogenic potential of the identified TMPRSS3 variants, we evaluated the proteolytic activity of the mutant alleles in comparison to the wild-type protein. The results demonstrated a marked reduction in enzymatic activity across all mutant constructs. When colonies larger than 0.005 mm^2^ were quantified, those carrying the variants exhibited a significantly slower growth rate than the wild-type. By day 3, colonies harboring splice-site variants were scarcely visible, while those with missense variants were detectable but had a significantly smaller average area. Mutant colonies that remained undetectable by day 3 were further incubated alongside wild-type controls until day 5, at which point colonies corresponding to all three splice-site variants became visible, confirming their viability and excluding the possibility of complete growth failure. Collectively, these functional findings support the pathogenic potential of all five novel variants identified in this study (Figure 4).
*Functional assessment of TMPRSS3 variants using a yeast protease assay. Colony growth was quantified using standard morphological criteria. After 3 days at 30 °C (top row), the assay shows the wild-type control followed by the missense variants c.923T>C and c.967G>A, and the splice-site variants c.572 + 1G>A, c.205 + 5G>C, and c.1348-2A>G. After 5 days (bottom row), the wild-type control and the three exon-skipping mutants (c.205 + 5G>C, c.572 + 1G>A, c.1348-2A>G) are shown. Colony numbers at both time points are presented in the accompanying graphs, demonstrating deleterious effects for all variants, with splice-site variants causing more severe growth impairment than missense variants. All experiments were performed with three independent biological replicates, and data are presented as mean ± SD. Statistical significance was analyzed using one-way ANOVA followed by Tukey’s post hoc test, with p < 0.05 considered statistically significant. *p < 0.05, **p < 0.01, **p < 0.001.
While observing a decrease in protein activity, we also predicted the protein structural changes caused by each variant. Point variants did not cause obvious changes in the protein backbone, whereas splice-site alterations had a greater impact. However, predictions for the intramembrane and transmembrane regions differed markedly, and the confidence of the predictions was low (Supplement figure).
Interestingly, the colony growth defect associated with the non-canonical splice-site variant (c.205 + 5G>C) appeared less severe than that caused by the two canonical splice-site variants (c.572 + 1G>A and c.1348-2A>G) (Figure 4). This observation suggests a potential gradation in pathogenicity based on splice-site type, although further evidence is required to support this conclusion.
Discussion
4
In this study, we identified five novel compound heterozygous TMPRSS3 variants (c.205 + 5G>C, c.923T>C, c.967G>A, c.572 + 1G>A, and c.1348-2A>G) in three Chinese individuals with prelingual autosomal recessive non-syndromic hearing loss (ARNSHL). The splice-site variants are particularly noteworthy, as they not only expand the mutational spectrum of TMPRSS3 but also exhibit more severe pathogenic effects than the missense variants.
While over 100 TMPRSS3 variants have been reported, splice-site variants remain exceptionally rare, with only seven had been clearly documented prior to this report. Our identification of three additional splice-site variants contributes significantly to this limited dataset. Functional characterization through minigene splicing assays confirmed that these variants disrupt normal mRNA processing, and protease activity assays demonstrated a marked reduction in enzymatic function. These findings extend beyond prior studies, which have predominantly focused on missense variants that impair protease activity, and provide further mechanistic support for establishing the genotype-phenotype correlations in TMPRSS3-related HL.
Yeast-based protease activity assays have served as an important functional tool for evaluating TMPRSS3 since 2003. Studies have consistently shown that pathogenic variants such as Asp103Gly, Arg109Trp, Cys194Phe, Trp251Cys, Pro404L, Cys407Arg, and Ala426Thr abolish yeast growth on sucrose plates, indicating a severe loss of proteolytic activity, whereas benign polymorphisms like Gly111Ser and Ile253Val do not impair function (Lee et al., 2003). Subsequent studies has corroborated this pattern, with variants like Val116Met and Val291Leu being highly deleterious, and others such as Arg80His, Leu184Ser, Ala418Val, and Trp251Cys also demonstrated severe functional impairment (Kim et al., 2017; Wong et al., 2020).
Previous work has shown that loss-of-function variants in any of the three major domains—LDLRA, SRCR domain, and serine protease—can result in HL (Guipponi et al., 2002; Masmoudi et al., 2001). In line with this, the variants identified in our study, including two missense and three splice-site alterations, all resulted in impaired protease function. Minigene assays specifically demonstrated that the splice-site variants c.205 + 5G>C, c.572 + 1G>A, and c.1348-2A>G caused the skipping of exons 3, 5, and 13, respectively. Notably, these splicing defects were associated with the most severe loss of enzymatic in our functional assays.
To date, 10 TMPRSS3 splice-site variants have been associated with HL of varying severity. A pattern emerges where canonical splice-site variants (e.g., c.1195-1G>C and c.783-1G>A) generally cause prelingual, profound HL, likely due to complete exon skipping and protein truncation. In contrast, non-canonical variants such as c.323-6G>A and c.205 + 5G>C tend to produce postlingual or progressive phenotypes (DFNB8). This clinical distinction may be explained by partial splicing defects that preserve residual protein function, a hypothesis consistent with the possibility of incomplete exon skipping induced by non-canonical variants.
The pathogenic severity of TMPRSS3 variants vary widely, and compound heterozygosity further complicates clinical interpretation. While missense variants have been more extensively studied, splice-site variants remain relatively under-investigated. Our study addressed this gap by functionally validation three novel splice-site variants, confirming they cause aberrant splicing and exon skipping, and demonstrating that all five novel variants impair proteolytic function. This provides direct molecular evidence of their pathogenicity.
The molecular pathogenesis underlying TMPRSS3-related HL have been elucidated through cellular and animal studies. Mice carrying a nonsense variant (Tmprss3 Y206X) develop postnatal degeneration of cochlear hair cells beginning at postnatal day 12 (P12), demonstrating the protein’s essential role in hair cell survival and cochlear homeostasis (Shearer et al., 2025). Mechanistically, TMPRSS3 is known to activate the ENaC, maintains potassium homeostasis, and support normal outward K^+^ currents in inner hair cells. Taken together, these findings reinforce the critical functional role of TMPRSS3 in the auditory system (Molina et al., 2013).
Globally, TMPRSS3 variants account for approximately 0.36%–12% of genetic HL cases (Walsh et al., 2006; Wattenhofer et al., 2005; Damrongchietanon et al., 2025; Bharadwaj et al., 2025; Li et al., 2024), with notable differences among populations (e.g., only 0.36% in a large Japanese cohort) (Nisenbaum et al., 2023; Colbert et al., 2024; Moon et al., 2021; Gao et al., 2017b). Although not the most common ARNSHL gene, TMPRSS3 remains clinically important due to: (i) its association with both prelingual (DFNB10) and postlingual (DFNB8) HL forms; (ii) phenotypic variability that correlates with variant type and domain; and (iii) its potential as a target for emerging gene therapies.
Recent advances in TMPRSS3-targeted gene therapy have been encouraging. Adeno-associated virus (AAV)-mediated gene replacement has successfully restored hearing in mouse models, paving the way for potential clinical translation (Du et al., 2023; Richard and Delprat, 2024). Our findings demonstrate that splice-site variants lead to adjacent exon skipping and that splice-site and missense variants differentially affect TMPRSS3 function, suggest that TMPRSS3-related HL may be amenable to tailored therapeutic strategies. Specifically, antisense oligonucleotide-based approaches designed to correct splicing defects could represent a viable treatment avenue for patients carrying splice-site variants.
Conclusion
5
In conclusion, our findings advance the understanding of TMPRSS3-related HL by functionally characterizing novel splice-site and missense variants and delineating their distinct impact on protein function. The more severe enzymatic impairment caused by splice-site variants provides a mechanistic explanation for the phenotypic heterogeneity observed in patients. This work underscores the necessity of precision medicine approaches in HL treatment, where therapeutic strategies may need to be tailored based on the specific molecular consequences of each patient’s variants. As gene therapies for HL advance toward clinical application, functional studies like this will be crucial for accurately matching patients with the appropriate therapeutic interventions. Integrating genetic diagnosis with detailed functional validation is a critical step toward achieving personalized medicine for hereditary hearing loss.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arora R. PifkováL. Deutschmann A. U. Reisinger E. (2025). Tmprss 3 is expressed in several cell types of the inner ear including type II but hardly in type I spiral ganglion neurons. Front. Cellular Neuroscience 19, 1690227. 10.3389/fncel.2025.1690227 41312438 PMC 12647049 · doi ↗ · pubmed ↗
- 2Bharadwaj T. Acharya A. Manyisa N. R. Aboagye E. T. Peigou Wonkam R. Xhakaza L. (2025). The diverse genetic landscape of hearing impairment in South African families. Clin. Genetics 108 (5), 511–520. 10.1111/cge.14765 40371963 PMC 12501749 · doi ↗ · pubmed ↗
- 3Boudewyns A. van den Ende J. Sommen M. Wuyts W. Peeters N. Van de Heyning P. (2018). Role of targeted next generation sequencing in the etiological Work-Up of congenitally deaf children. Otology and Neurotology Official Publication Am. Otological Soc. Am. Neurotol. Soc. Eur. Acad. Otology Neurotol. 39 (6), 732–738. 10.1097/mao.0000000000001847 29889784 · doi ↗ · pubmed ↗
- 4Chen Y. S. Cabrera E. Tucker B. J. Shin T. J. Moawad J. V. Totten D. J. (2022). TMPRSS 3 expression is limited in spiral ganglion neurons: implication for successful cochlear implantation. J. Medical Genetics 59 (12), 1219–1226. 10.1136/jmg-2022-108654 35961784 PMC 10245338 · doi ↗ · pubmed ↗
- 5Choe G. Park S. K. Kim B. J. (2023). Hearing loss in neonates and infants. Clin. Experimental Pediatrics 66 (9), 369–376. 10.3345/cep.2022.01011 36634668 PMC 10475863 · doi ↗ · pubmed ↗
- 6Colbert B. M. Lanting C. Smeal M. Blanton S. Dykxhoorn D. M. Tang P. C. (2024). The natural history and genotype-phenotype correlations of TMPRSS 3 hearing loss: an international, multi-center, cohort analysis. Hum. Genetics 143 (5), 721–734. 10.1007/s 00439-024-02648-3 38691166 PMC 11098735 · doi ↗ · pubmed ↗
- 7Damrongchietanon T. Wattanasirichaigoon D. Khongkraparn A. Noojarern S. Tiravanitchakul R. Kasemkosin N. (2025). Diagnostic yield of whole exome sequencing with targeted gene analysis in prelingual sensorineural hearing loss in Thailand. Sci. Reports 15 (1), 32784. 10.1038/s 41598-025-18038-2 40998904 PMC 12464309 · doi ↗ · pubmed ↗
- 8Du W. Ergin V. Loeb C. Huang M. Silver S. Armstrong A. M. (2023). Rescue of auditory function by a single administration of AAV-TMPRSS 3 gene therapy in aged mice of human recessive deafness DFNB 8. Mol. Therapy The Journal Am. Soc. Gene Ther. 31 (9), 2796–2810. 10.1016/j.ymthe.2023.05.005 37244253 PMC 10491991 · doi ↗ · pubmed ↗