Targeting Plasmodium falciparum with purine antimetabolites as a therapeutic strategy
Worlanyo Tashie, Harry P. de Koning, Nancy O. Duah-Quashie, Neils B. Quashie

TL;DR
This paper explores using purine antimetabolites to target Plasmodium falciparum, a malaria-causing parasite, by exploiting its reliance on purine salvage from the host.
Contribution
The paper highlights Pf ENTs as a novel delivery route for purine-based antimalarial drugs.
Findings
P. falciparum's purine salvage pathway is a viable drug target due to its lack of de novo purine synthesis.
Purine analogs can enter infected erythrocytes via Pf ENTs and disrupt parasite metabolism.
Targeting Pf ENTs offers a framework for developing new antimalarial therapies.
Abstract
Plasmodium falciparum lacks the de novo purine biosynthesis pathway and relies exclusively on salvaging free purines from the host to meet its metabolic requirements. This absolute dependence on the purine salvage pathway provides a compelling opportunity for antimalarial drug development, particularly in the face of rising resistance to current therapies. Although the purine salvage system has been extensively studied as a potential drug target in P. falciparum, no purine-based antimalarial drug has yet reached clinical use. In this review, we summarize the potential of targeting the purine salvage pathway in antimalarial drug development, with a focus on strategies that leverage P. falciparum Equilibrative Nucleoside Transporters (Pf ENTs) as conduits for therapeutic agents. Purine analogs that efficiently enter P. falciparum-infected erythrocytes, reach Pf ENTs, and undergo selective…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
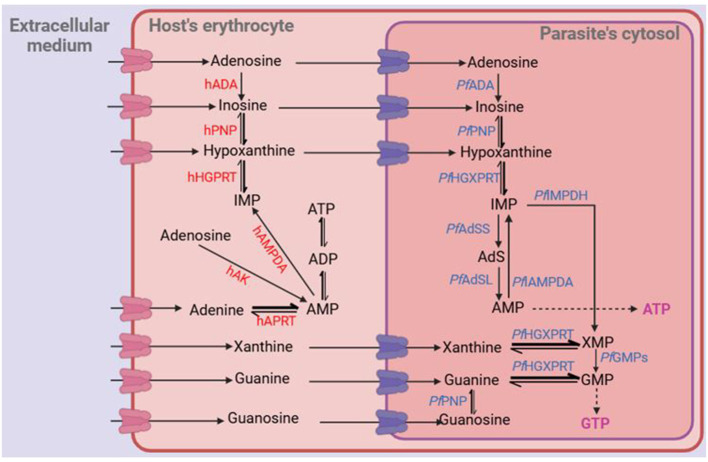 Figure 1
Figure 1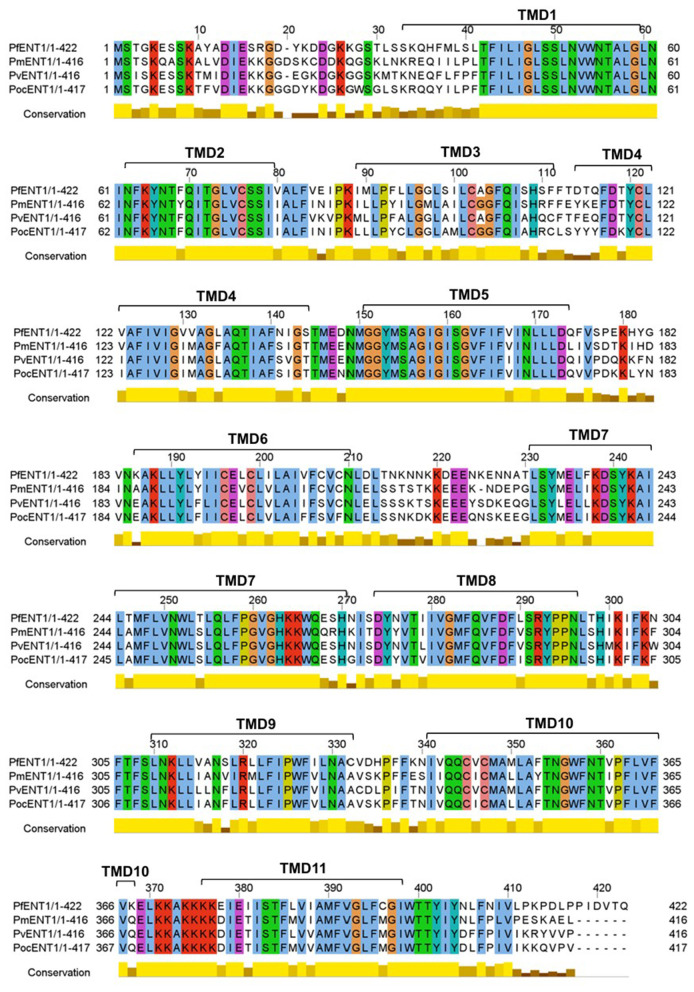 Figure 2
Figure 2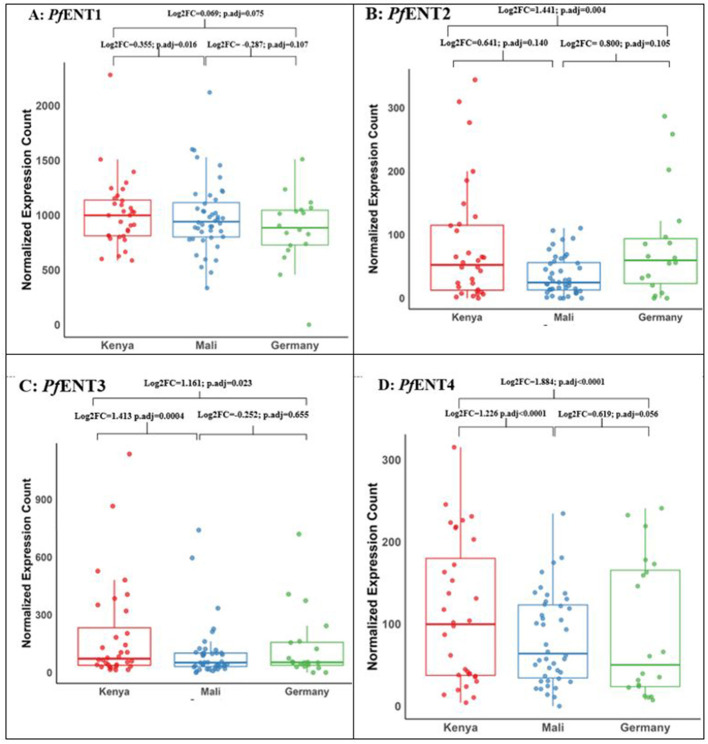 Figure 3
Figure 3|
| |||||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
| |
| Hypoxanthine | 410 | 0.34 | |||||
| Adenosine | 13.2 | 320 | 1,860 | 1,450 | 2.0 | ||
| Inosine | 253 | ||||||
| Adenine | 320 | 820 | 1,200 | ||||
| Thymidine | 1,330 | 1,110 | |||||
| Uridine | 3,500 | ||||||
| Hypoxanthine | ## | 0.751 | |||||
| Guanine | – | ### | 0.112 | ||||
| Adenosine | ## | 4.02 | |||||
| Inosine | # | 2.01 | |||||
| Adenine | – | ## | 2401 | ||||
| Guanosine | ++ | # | 11.62 | ||||
| Thymidine | + | # | |||||
| Uridine | +/– | # | |||||
|
|
|
| ||
|---|---|---|---|---|
| Adenine | 13 ± 1 | 12 ± 1 | 16 ± 4.5 | |
| Guanine | 37 ± 2 | 50 ± 3 | 28 ± 2 | |
| Hypoxanthine | 180 ± 12 | 200 ± 170 | 290 ± 21 | |
| Xanthine | >150 | |||
| 6-thioguanine | 95 ± 8 | |||
| 7-deazaguanine | 15 ± 5 | |||
| 9-methylguanine | 7.4 ± 2 | |||
| 9-deazaguanine | 8.0 ± 2 | |||
| Allopurinol | 150 ± 9 | |||
| 2-thioaminopurinol | 20 ± 6 | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHIV/AIDS drug development and treatment · Malaria Research and Control · Biochemical and Molecular Research
Introduction
1
Malaria continues to pose a major public health challenge, with sub-Saharan Africa bearing the highest burden (Phillips et al., 2017; WHO, 2025c). In 2024, an estimated 282 million cases of malaria were reported worldwide, resulting in 579,000 deaths. Africa is the continent most affected and accounts for approximately 94% of all malaria cases (WHO, 2025c). Among the five Plasmodium species that infect humans, Plasmodium falciparum is the most virulent and is responsible for the majority of malaria-related deaths (Daily and Parikh, 2025; WHO, 2025c).
Chemotherapy remains a cornerstone of malaria control. However, the increasing resistance of P. falciparum to existing antimalarial drugs poses a significant threat to global efforts to control and eradicate the disease (Theodoridis and Carvalho, 2025; WHO, 2025c). In response to the growing problem of resistance, the World Health Organizations (WHO) recommended artemisinin-based combination therapies (ACTs) as the first-line treatment for uncomplicated malaria, which have since become the cornerstone of malaria case management (Nosten and White, 2007; WHO, 2025b). The success of ACTs lies in their pharmacological synergy: a rapidly acting artemisinin derivative which has a short plasma half-life is combined with a longer acting partner drug. This strategy limits the parasite's exposure time to the artemisinin, thereby reducing the likelihood of developing resistance (Kavishe et al., 2017). Nevertheless, the efficacy of ACTs is increasingly threatened by the emergence and spread of artemisinin resistance, first reported in Western Cambodia in the late 2000s (Dondorp et al., 2009; Noedl et al., 2008), but now increasingly reported from a much wider geographical area (Ward et al., 2022). Artemisinin partial resistance is characterized by delayed clearance of P. falciparum from the bloodstream following treatment with artemisinin-based therapies. Accumulating evidence demonstrates that mutations in the PfKelch13 propeller domain (PfK13) are associated with this delayed parasite clearance both in vitro and in vivo after artemisinin treatment (Rosenthal et al., 2024; WHO, 2025a). In Africa, artemisinin partial resistance has been confirmed in multiple countries, including Rwanda (Straimer et al., 2022; Uwimana et al., 2021), Uganda (Balikagala et al., 2021; Conrad et al., 2023; Ogwang et al., 2024), Eritrea (Mihreteab et al., 2025), and Tanzania (Juliano et al., 2024). These artemisinin partial resistant parasites have emerged independently and have not spread from South-East Asia (WHO, 2025a). Although ACTs remain highly effective in most African settings (Assefa et al., 2024), the recent emergence of partial resistance to artemisinin-based therapies, including combinations, in parts of Africa underscores the urgent need for novel therapeutic antimalarial strategies.
One promising strategy lies in targeting the parasite's absolute dependency on host-derived purines for survival. Purines and pyrimidines are essential for the synthesis of DNA, RNA, and are critical metabolites in all living organisms (el Kouni, 2017; Krungkrai and Krungkrai, 2016). Unlike humans, who can synthesize purine nucleotides both de novo and through salvage pathways, Plasmodium species lack the de novo purine biosynthesis pathway (de Koning et al., 2005; Webster et al., 1984) but do synthesize pyrimidines de novo (Cassera et al., 2011a; de Koning et al., 2005). Consequently, P. falciparum relies entirely on salvaging purines precursors from its host to meet metabolic demands (Campagnaro and de Koning, 2020; Gero and O'Sullivan, 1990; Webster et al., 1984). This unique dependence represents a critical vulnerability in the parasite's biology and offers a valuable target for antimalarial drug development.
The uptake of purines into the parasite's cytosol, mediated by Equilibrative Nucleoside Transporters (Pf ENTs), is a critical step in the purine salvage pathway (Campagnaro and de Koning, 2020; Carter et al., 2003; Riegelhaupt et al., 2010). Compromising this pathway could effectively deprive the parasite of its essential purine supply and subsequently lead to its death. A promising strategy is to exploit the Pf ENTs as conduits for delivering drugs into the parasite. This approach would allow for the inhibition of essential enzymes or the disruption of nucleic acid synthesis, leading to chain termination or lethal mutagenesis. Pf ENT1 is a high affinity, high capacity transporter and most likely energy-dependent, and therefore directs effective, monodirectional and concentrative uptake of purines and their analogs into the parasite. Its broad specificity for nucleosides and bases doubtlessly aids in the design of antimalarial purine antimetabolites.
While the purine salvage system has been extensively studied as potential drug targets in P. falciparum (Cheviet et al., 2019; Frame et al., 2015a; Quashie et al., 2008), no purine-based antimalarial drug has yet reached clinical application or even, to the best of our knowledge, clinical development. This is in large part because efforts have centered more on the development of inhibitors of key enzymes of P. falciparum purine metabolism, including transition-state inhibitors of P. falciparum purine nucleoside phosphorylase (Pf PNP) such as DADMe-Immucillin-G (Cassera et al., 2011b) and 5′-methylthio-immucillin-H (Ting et al., 2005), as well as inhibitors targeting P. falciparum hypoxanthine-guanine-xanthine-phosphoribosyl transferase (Pf HGXPRT), rather than on the challenges of delivering these analogs into the infected human erythrocyte and thence into the parasite.
This review focuses on the untapped potential of Pf ENTs as therapeutic targets for inhibitors and as conduits for purine antimetabolites, emphasizing their critical role in the parasite's survival and the opportunities they offer for the development of purine-based antimalarial strategies. With the escalating threat of drug-resistant malaria, and the implementation of a truly effective vaccine always fading into the future (Sibomana et al., 2025), innovative approaches such as these are urgently needed to strengthen global efforts to combat this deadly disease.
Purine transport and metabolism in P. falciparum
2
Transport of purines across biological membranes
2.1
Purine nucleobases and nucleosides are hydrophilic and hence cannot freely cross the lipid bilayer of plasma membranes (Campagnaro and de Koning, 2020; Carter et al., 2003). This makes the internalization of purine nucleobases and nucleosides dependent on specialized transport systems to facilitate their import into, or export out of, prokaryotic and eukaryotic cells. The type of purine transporter present in cellular membranes determines the purine nucleobases and nucleosides that are transported across it (Campagnaro and de Koning, 2020; Rehan et al., 2019).
In protozoa, these can be specific for one or more nucleosides (Al-Salabi et al., 2007; Carter et al., 2000b), for nucleobases (Aldfer et al., 2024; Burchmore et al., 2003), or both (de Koning and Jarvis, 1999). Moreover, protozoan transporters can be purine-specific, pyrimidine specific, or mixed purine/pyrimidine transporters (Aldfer et al., 2022a; Alzahrani et al., 2017; de Koning and Jarvis, 1999; Elati et al., 2023). Many studies have implicated protozoan ENT transporters in the uptake of purine antimetabolites (Aldfer et al., 2022a; Al-Salabi and de Koning, 2005; Hulpia et al., 2019a; Ungogo et al., 2023; Vasudevan et al., 1998) and/or other drugs, including the diamidine and melaminophenyl arsenical trypanocides (Carter et al., 1999; de Koning et al., 2000a; Stewart et al., 2010). This has also been demonstrated for P. falciparum (Cassera et al., 2011b; Sosa et al., 2019; Tashie et al., 2025b; Ting et al., 2005) and will be discussed in section 3.2, below.
The expression levels of protozoan ENT transporters are regulated by various factors such as substrate availability, cell cycle phase and life cycle stage (de Koning et al., 2000b; Hall et al., 1996; Martin et al., 2014; Menezes et al., 2025). Where this has been studied, protozoan nucleoside and nucleobase transporters have been found to be proton symporters (de Koning and Jarvis, 1997, 1998; de Koning et al., 1998; Landfear et al., 2004; Ortiz et al., 2009), which enables them to efficiently transport substrate at low concentrations and against a concentration gradient. These are ideal characteristics for an antimetabolite transporter.
Purine salvage in P. falciparum
2.2
The genome of P. falciparum is rich in adenine (80.6% A-T content), and replication thus requires a very efficient supply of adenine nucleotides (Cheviet et al., 2019; Gardner et al., 2002), especially given the very high rate of replication in the human host. In the intraerythrocytic stages of P. falciparum, only one of the ten enzymes required for de novo synthesis of purine, namely adenylosuccinate lyase, has been identified (Krungkrai and Krungkrai, 2016) and this is used in the conversion of inosine monophosphate to AMP. Consequently, the parasite, being unable to synthesize purines de novo, must import purines into its cytosol at a very high rate to meet its purine requirements. The low concentration of free purine nucleosides and nucleobases in the erythrocyte cytosol necessitates high substrate affinity and the required quantity necessitates a high rate of uptake (Vmax); nucleotides such as ADP, ATP, and GTP are not taken up across the plasma membrane. The combination of high affinity and high capacity in turn necessitates a form of active transport, likely in the form of proton symport as shown for other protozoan purine transporters (see above—section 2.1).
The import of purine nucleobases (adenine, guanine, hypoxanthine, and xanthine) and nucleosides (adenosine, guanosine, and inosine) into the parasite's cytosol is mediated by the Pf ENTs. Genome analysis reveals four Pf ENTs: Pf ENT1, Pf ENT2, Pf ENT3, and Pf ENT4 (Fox and Bzik, 2020; Frame et al., 2015a; Ghérardi and Sarciron, 2007). Pf ENT1 and Pf ENT4 are localized in the parasite's plasma membrane (Frame et al., 2012, 2015a), while Pf ENT2 localizes to the endoplasmic reticulum (Frame et al., 2015a); the localization of Pf ENT3 has not yet been reported. All Pf ENTs are expressed during the intraerythrocytic stages of the parasite's life cycle (Frame et al., 2012, 2015a; Ghérardi and Sarciron, 2007; Quashie et al., 2008).
Following the import of purine nucleobases and nucleosides into the parasite's cytosol, their incorporation into the nucleotide pool is mediated by a set of well-characterized purine metabolism enzymes. Pf HGXPRT converts salvaged hypoxanthine, guanine and xanthine into inosine monophosphate (IMP), guanosine monophosphate (GMP) and xanthosine monophosphate (XMP), respectively. Pf PNP convert nucleosides into their corresponding nucleobases and sugar counterparts, thereby funneling inosine, xanthosine, and guanosine as nucleobases through Pf HGXPRT, yielding the corresponding mononucleotides. Unlike many other protozoa (Vodnala et al., 2008) including the apicomplexan Toxoplasma gondii (Kim et al., 2008), P. falciparum cannot directly convert adenosine to AMP, because it lacks the gene for adenosine kinase, and must undergo a two-step conversion to hypoxanthine instead: P. falciparum adenosine deaminase (Pf ADA) catalyzes the deamination of adenosine into inosine, followed by the phosphorolysis of inosine to hypoxanthine by Pf PNP (Cassera et al., 2011a; Cheviet et al., 2019; Ducati et al., 2013; Krungkrai and Krungkrai, 2016). The overall purine salvage pathway in P. falciparum is depicted in Figure 1, and has been extensively reviewed by Cassera et al. (2011a), Ducati et al. (2013), and Ginsburg (2016).
Purine salvage in P. falciparum. Bold arrows on reversible steps indicate the metabolically favored direction. hADA, human adenosine deaminase; hPNP, human purine nucleoside phosphorylase; hHGPRT, human hypoxanthine-guanine phosphoribosyl transferase; hAK, human adenosine kinase; hAMPDA, human adenosine 5′-monophosphate deaminase; hAPRT, human adenine phosphoribosyl transferase; AMP, adenosine 5′-monophosphate; ADP, adenosine 5′-diphosphate; ATP, adenosine 5′-triphosphate; IMP, inosine 5′-monophosphate; XMP, xanthosine 5′-monophosphate; GMP, guanosine 5′-monophosphate; AdS, adenylosuccinate; PfADA, P. falciparum adenosine deaminase; PfPNP, P. falciparum purine nucleoside phosphorylase; Pf HGXPRT, P. falciparum hypoxanthine-guanine-xanthine phosphoribosyl transferase; Pf AMPDA, P. falciparum adenosine 5′-monophosphate deaminase; Pf IMPDH, P. falciparum inosine 5′-monophosphate dehydrogenase; Pf GMPs, P. falciparum guanosine 5′-monophosphate synthase; Pf AdSS, adenylosuccinate synthetase; Pf AdSL, adenylosuccinate lyase. Adapted from Ducati et al. (2013).
The development of purine-based antimalarial drugs must consider the unique purine metabolism in the Plasmodium parasite. As shown in Figure 1, P. falciparum only incorporates oxopurine nucleobases directly into its nucleotide pool, through a streamlined one-step phosphorylation catalyzed by Pf HGXPRT. In contrast, nucleosides, if they are to become nucleotides, must first be catabolised into their respective nucleobases by Pf PNPs, introducing an additional and potentially rate-limiting step (Cassera et al., 2011a; Cheviet et al., 2019; Ducati et al., 2013) during which any modifications to the ribose moiety are lost. In addition, adenosine analogs would first have to be deaminated to inosine analogs while antiprotozoal analogs with modifications of the purine ring [e.g., 7-deaza (Hulpia et al., 2019b) or 2-F (Vodnala et al., 2013)] may not be ADA substrates. However, if the adenosine analogs are ADA substrates, they are broken down in human serum (Rottenberg et al., 2005). Therefore, to effectively target the purine salvage pathway in antimalarial drug design, nucleobases should be prioritized over nucleosides due to their direct and efficient incorporation into the parasite's nucleotide pool. This approach was demonstrated by Tashie et al. (2025b) where the authors demonstrated that nucleobases, particularly guanine derivatives, inhibited the in vitro growth of P. falciparum parasites.
PfENTs as drug targets in P. falciparum
2.3
Pf ENTs represent a critical link in the parasite's purine salvage pathway and can therefore be exploited in the development of therapeutic agents. Among the Pf ENTs, Pf ENT1 serves as the primary route for purine uptake in P. falciparum and is essential for the parasite's intraerythrocytic survival, validating it as a promising target for antimalarial drug development El Bissati et al., (2006), (2008); Zhang et al., (2018).
Using targeted gene manipulation approaches, El Bissati et al. (2006) and El Bissati et al. (2008) provided compelling genetic evidence for the essential role of Pf ENT1 in the parasite's purine salvage pathway. Transgenic parasites lacking Pf ENT1 (ΔPf ENT1) exhibited a conditionally lethal growth phenotype under physiological purine concentrations, demonstrating an inability to complete the intraerythrocytic life cycle when hypoxanthine, inosine, adenosine, xanthine, guanine or guanosine were provided at concentrations sufficient to support wild-type parasite growth (El Bissati et al., 2006, 2008). Reintroduction of Pf ENT1 into the knock-out background fully restored purine utilization and growth across a broad range of nucleobases and nucleosides, confirming that the observed phenotype was attributable specifically to loss of Pf ENT1 rather than secondary genetic effects (El Bissati et al., 2008). Together, these findings establish Pf ENT1 as an essential and primary purine uptake route in P. falciparum, providing strong experimental validation of Pf ENT1 as a potential drug target in the parasite.
In addition to genetic validation, pharmacological evidence further supports Pf ENT1 as a viable drug target in the parasite. Studies by Frame et al. (2015b) and Sosa et al. (2019) identified small-molecule inhibitors of Pf ENT1 that inhibited the in vitro growth of P. falciparum, providing direct functional evidence for the therapeutic potential of targeting purine transport in P. falciparum.
Despite being the primary purine uptake route in the parasite, with a high rate of substrate translocation, various studies, employing different expression systems and experimental methodologies to characterize Pf ENT1, as well as slightly different primary sequence, have yielded inconsistent purine uptake kinetics, fueling ongoing scientific debate about its substrate preference (Table 1). Among the main contributors of the variability is the expression in Xenopus laevis oocytes compared to studies using live P. falciparum trophozoites freshly isolated from human erythrocytes (without codon optimisation); we have ourselves attempted the expression of Pf ENTs including Pf ENT1 in other protozoa, after optimizing the highly AT-rich sequences for the codon preferences of the heterologous expression cells, and found the results insufficiently reliable (unpublished data) although we have been highly successful with ENT and non-ENT transporters from other organisms [e.g., Aldfer et al., 2022b; Natto et al., 2021b]. More important is the difference in assay conditions. Whereas the studies by Downie et al. (2006), Downie et al. (2008), Parker et al. (2000), and Carter et al. (2000a) used high radiopermeant concentrations (10–100 μM), our earlier work (Quashie et al., 2008) utilized far lower concentrations of radiolabelled permeant (30–250 nM), allowing the detection rather than saturation of high affinity transporters. Moreover, the saturating radiolabel concentrations cause the transporter to operate at Vmax and may thereby saturate the downstream metabolic enzymes as well, causing the permeant to back up and exceed the extracellular concentration. In contrast, we utilized the lowest possible [^3^H]-permeant concentrations, which allowed the accurate determination of a sub-micromolar Km value for hypoxanthine and low micromolar Km for adenosine—reasonable values given the low concentration of free purines in freshly obtained human erythrocytes [~10 nM (Casali et al., 2016)] and the sub-micromolar Km value of Pf HGXPRT for hypoxanthine [0.46 μM (Queen et al., 1988); 0.9 μM (Keough et al., 1999)]. Proposed substrate Km values in the 100s of μM for Pf ENT1 make no physiological sense as a transporter with a Km of 250 μM would operate at just 0.04% of Vmax at a substrate concentration of 0.1 μM, or 0.4% at a concentration of 1 μM. Moreover, all other protozoan nucleoside and nucleobase transporters yet identified are of the ENT family and display sub-micromolar to low micromolar Km values for their primary substrates (Aldfer et al., 2022a),b; Campagnaro and de Koning, 2020; de Koning et al., 2005; Elati et al., 2023; Landfear et al., 2004).
Nevertheless, converging evidence suggests that P. falciparum relies primarily on high-affinity oxopurine nucleobase transport, with hypoxanthine as its major purine source. This inference is supported by the observation that depletion of hypoxanthine from the culture medium using the enzyme xanthine oxidase prevents parasite growth (Berman et al., 1991); the standard supplementation of P. falciparum culture media with hypoxanthine (Basco, 2023; Tewari et al., 2019); and early biochemical studies demonstrating efficient incorporation of hypoxanthine into the parasite's nucleic acids (Büngener and Nielsen, 1968; Gutteridge and Trigg, 1970). Collectively, these findings reinforce the parasite's dependence on nucleobase transport for survival and underscore the relevance of Pf ENTs, and especially Pf ENT1, as a drug target. Findings from these studies further suggest that Pf ENT1 is a broad-specificity purine transporter, and its broad substrate selectivity/permissibility supports the delivery of purine antimetabolites through this transporter.
Purine transport in uninfected and P. falciparum-infected human erythrocytes
2.4
The mature human erythrocyte, which provides the host environment for P. falciparum growth and multiplication during the intraerythrocytic stages, lacks the enzymatic machinery necessary to synthesize purine rings de novo. Consequently, erythrocytes rely on salvaging preformed purine nucleobases and nucleosides from the plasma or by degrading purine nucleotides and nucleosides within the erythrocytes to meet their purine requirements (Dudzinska et al., 2006).
The human plasma contains purines at micromolar concentrations, with hypoxanthine (1–5 μM) and inosine (~1 μM) being the predominant forms. Adenosine is also found in the human plasma and ranges from nanomolar values up to 1–5 μM. The total concentration of purines in the plasma is below 10 μM (Campagnaro and de Koning, 2020; Frame et al., 2015a). Hypoxanthine, inosine, adenosine and other purines found in the erythrocytes originate from uptake by purine transporters and by the metabolism of erythrocyte ATP, which is present at approximately 2 mM (Frame et al., 2015a).
The steady-state purine concentration in human erythrocytes is not sufficient to support the multiple rounds of P. falciparum DNA replication that occur during the 48-h intraerythrocytic stage of the parasite's life cycle, giving rise to 16–32 merozoites (Josling and Llinás, 2015). It has been estimated that the erythrocyte's complement of ATP would be sufficient for only 1%−2% of the parasite's purine requirements for nucleic acid synthesis alone. Thus, large quantities of purine nucleobases and nucleosides must be imported into the P. falciparum-infected erythrocytes to supply enough purines to the developing intracellular parasite. The import of purines into the erythrocyte's cytosol is mediated through two main pathways: nucleosides enter through human ENT1 (hENT1) while nucleobases primarily enter via human facilitated nucleobase transporter (hFNT1; Baldwin et al., 2007; Berman et al., 1991; Domin et al., 1988; Frame et al., 2015a; Wallace et al., 2002).
Because the P. falciparum parasite is far more metabolically active than its host erythrocyte, the rate at which some key nutrients are consumed exceeds the rate at which they can be taken up by erythrocyte transporters (Counihan et al., 2021). To accommodate this increased metabolic demand, P. falciparum alters the permeability of the host erythrocyte membrane either by up-regulating existing host transport systems and/or by inducing parasite-encoded “new permeation pathways” that are inserted into the host cell plasma membrane (Counihan et al., 2021; Ginsburg et al., 1985; Thomas and Egée, 2005). These new permeation pathways are induced approximately 10–20 h after parasite invasion and allow for entry of broad range of nutrients necessary for parasite growth and survival (Ginsburg et al., 1985; Lingelbach et al., 2004). However, despite their substantial contribution to nutrient uptake in infected erythrocytes, a study by Quashie et al. (2010) demonstrated that the host erythrocyte transporters hENT1 and hFNT1, rather than the new permeation pathways, serve as the primary route for entry of adenosine, adenine and hypoxanthine into P. falciparum-infected human erythrocytes. Both human transporters were upregulated in the infected erythrocytes, leading to 2–2.5-fold higher rates of uptake of [^3^H]-adenosine and [^3^H]-hypoxanthine (Quashie et al., 2010).
Therapeutic potential of purine analogs
3
Nucleobase and nucleoside analogs as chemotherapeutic agents
3.1
Nucleobase and nucleoside analogs are mostly synthetic, chemically modified compounds designed to mimic the physiological properties of naturally occurring nucleobases and nucleosides. By mimicking these molecules, nucleobase and nucleoside analogs exploit existing cellular pathways for transport, activation, and conversion into active phosphate derivatives, which disrupt cellular processes (Hruba et al., 2023; Jordheim et al., 2013; Rehan et al., 2019).
Analogs of the naturally occurring purine nucleobases and nucleosides, modified at the nucleobase, ribose or phosphate moieties to enhance therapeutic potency and selectivity are among the most potent anticancer, antibacterial and antiviral agents. Modifications to the nucleobase often involve halogenation, azotation, N-conjugation of the base, or the introducing a different heterocyclic ring or acyclic moiety for (2′-deoxy)ribose; changes such as replacing oxygen with another atom, altering ring configurations, or substituting with acyclic fragments expand therapeutic applications. The phosphate group may be protected, substituted with another phosphorus-containing group (e.g., phosphonate) or a fragment from another compound (such as an amino acid), or entirely eliminated. These modifications have enabled the development of a wide range of analogs with therapeutic applications in clinical settings (Jordheim et al., 2013; Pastuch-Gawołek et al., 2019; Thomson and Lamont, 2019).
Due to the charged nature of nucleotides, they cannot cross the lipid bilayer of the plasma membrane. Hence, purine or pyrimidine analogs designed as therapeutic agents are administered as prodrugs, requiring sequential phosphorylation catalyzed by target cell nucleo(s/t)ide kinases to transform them into their mono-, di-, and tri-phosphorylated active forms. Once activated, the activated antimetabolites exert therapeutic effects through several mechanisms, including inhibition of key intracellular enzymes like viral DNA or RNA polymerases; interference with wider nucleotide metabolism including activated intermediates; incorporation into nucleic acid chains which eventually leads to termination of the DNA or RNA replication or altered structure; promotion of lethal mutagenesis by inducing errors in viral genomes to render them non-viable; and depletion of natural nucleotide pools to starve the cell or pathogen of essential precursors (Ali et al., 2013; Borbone et al., 2021; Galmarini et al., 2002; Kamzeeva et al., 2023; Pruijssers and Denison, 2019; Rehan et al., 2019; Thomson and Lamont, 2019).
Applications of nucleobases and nucleosides analogs as antimalarial agents
3.2
While primarily known for their use in treating cancers (Huang et al., 2022; Muggia et al., 2012) and viral infections (Feng, 2018; Geraghty et al., 2021; Pastuch-Gawołek et al., 2019), nucleobase and nucleoside analogs are also effective against parasitic diseases. Studies have highlighted their activity against Trypanosoma (Fiuza et al., 2022; Hulpia et al., 2019b; Mabille et al., 2022), Leishmania (Lin et al., 2022), Toxoplasma (Al Safarjalani et al., 2008; Elati et al., 2023), Trichomonas (Natto et al., 2021a) and Plasmodium (Sosa et al., 2019; Tashie et al., 2025b; Ting et al., 2005) parasites. These analogs interfere with parasite-specific enzymes or nucleoside transporters, thus offering a targeted approach to antiparasitic therapy (Elati et al., 2023; Fiuza et al., 2022; Hulpia et al., 2019b; Natto et al., 2021a; Sosa et al., 2019; Ting et al., 2005).
Several purine nucleobase and nucleoside analogs have shown potent activity against P. falciparum parasites. Sosa et al. (2019) identified six inhibitors of Pf ENT1 as potential therapeutic leads for antimalarial drug development. Similarly, Cassera et al. (2011b) demonstrated that the transition state nucleoside analog DADMe-Immucillin-G (BCX4945) effectively killed P. falciparum by inhibiting Pf PNP. Another transition state analog, 5′-methylthio-immucillin-H, exhibited selective inhibition of Pf PNP over the human enzyme, resulting in parasite death in vitro (Ting et al., 2005). More recently, Tashie et al. (2025b) demonstrated that the guanine derivatives 8-azaguanine, 7-deazaguanine, and 6-thioguanine significantly inhibited the in vitro growth of P. falciparum. Their findings highlight these nucleobase analogs as promising leads for purine-based antimalarial drug development and underscore the versatility of Pf ENTs in the uptake of purine antimetabolites (Tashie et al., 2025b).
Targeting PfENTs with purine analogs
4
Purine analogs targeting the Pf ENTs can act as inhibitors of purine metabolism enzymes or as “subversive substrates” that become toxic following activation by the parasite's purine salvage enzymes (Berg et al., 2010; el Kouni, 2003; Hulpia et al., 2019a). Although purine analogs could be designed to inhibit Pf ENTs directly, this approach may present with some challenges. Transporter inhibitors typically have a transient association with the target and require sustained concentrations to compete effectively, and for an extended time, with endogenous substrates—hypoxanthine and adenosine in the case of Pf ENT1. It is unlikely that depriving the parasites of purines for a short duration would do irreversible damage, and certainly this is not the case in kinetoplastids (de Koning et al., 2000b; Martin et al., 2014), although a prolonged absence of purine salvage is certainly lethal.
Ideally, novel antimalarial agents directed at Pf ENT1 should exhibit broad-spectrum activity against all human malaria parasites. Chen et al. (2025) and Deniskin et al. (2016) characterized the substrate specificity and transport kinetics of Pf ENT1 orthologs in P. vivax (PvENT1) and the rodent malaria P. berghei (PbENT1), respectively, reporting distinct substrate recognition profiles. Arora et al. (2016) expressed PbENT1 in yeast and characterized it as a nucleoside/base transporter with broad specificity and highest affinity for inosine. Two separate studies have reported characterisations of PvENT1, both reporting highest affinity for inosine followed by guanosine, albeit with an order of magnitude difference in substrate affinities between the two reports (Chen et al., 2025; Deniskin et al., 2016). Although differences in substrate affinities have been reported among Pf ENT1 orthologs, purine inhibitors shown to act on Pf ENT1 were also effective against PvENT1 (Chen et al., 2025; Deniskin et al., 2016) and PbENT1 (Arora et al., 2016). This supports the feasibility of developing Pf ENT1-targeted purine-based antimalarials with activity across multiple Plasmodium species. Nevertheless, a more comprehensive characterization of Pf ENT1 orthologs in human non-falciparum Plasmodium species would be beneficial in informing the target-based design and optimisation of such agents.
Although no functional data has yet been reported for Pf ENT1 homologs in P. malariae and P. ovale, sequence alignment analyses indicate substantial amino acid identity between Pf ENT1 and its orthologs: 74.34% for P. malariae and 75.42% for P. ovale curtisi (Figure 2). While overall amino acid conservation does not guarantee identical substrate specificity, the high level of similarity, especially across multiple transmembrane domains (TMDs), supports the possibility that purine-based antimetabolites targeting Pf ENT1 may have cross-species activity. Consistent with this, PvENT1 also shares 75% amino acid identity with Pf ENT1, and Pf ENT1-targeting antimetabolites have demonstrated inhibitory activity against PvENT1 (Chen et al., 2025; Deniskin et al., 2016). As can be seen in Figure 2, there are no major differences in amino acid sequence within the TMDs of the four Plasmodium ENT1s, such as the introduction of a polar residue in the middle of a TMD, which would be indicative of a potential change in the substrate-binding site. Indeed, it has been shown that the four residues most involved in interaction with Pf ENT1 substrate inosine are Trp53 (TMD1), Gln135 (TMD4), Asp287 and Arg291 (both TMD8; Wang et al., 2023), which are all unchanged in the various Plasmodium ENT1s (Figure 2). Together, these findings support the premise that antimetabolites targeting Pf ENT1 can achieve broad antimalarial activity across diverse Plasmodium species.
Multiple sequence alignment of ENT1 orthologs annotated with the 11 transmembrane domains (TMD1−11). Sequences included are from P. falciparum (Pf ENT1; PlasmoDB ID: PF3D7_1347200), P. malariae (PmENT1; PlasmoDB ID: PmUG01_12017700), P. vivax (PvENT1; PlasmoDB ID: PVP01_1207600) and P. ovale curtisi (PocENT1; PlasmoDB ID: PocGH01_12015900). Predicted transmembrane domains are indicated above the alignment and were defined based on Pf ENT1 and mapped across orthologs. The conservation histogram below each alignment block represents the degree of residue conservation across species, with higher bars indicating greater conservation. Pf ENT1 shares 74.34, 75.0, and 75.42% amino acid sequence identity with PmENT1, PvENT1, and PocENT1, respectively, demonstrating high conservation across human-infecting Plasmodium species.
The feasibility of exploiting the Pf ENTs as conduits for delivering drugs is supported by evidence that all four Pf ENTs are expressed in both laboratory strains (Frame et al., 2015a) and field isolates of P. falciparum (Tashie et al., 2025a; Figure 3), albeit not to the same extent. The highest expression by far was observed for Pf ENT1 in all regions from which isolates were investigated. Moreover, of the four Pf ENTs, only Pf ENT1 has been shown to be essential (El Bissati et al., 2006, 2008). Essentially is not required for effective drug targeting through a specific transporter, however. For instance, the TbAT1/P2 aminopurine transporter of T. brucei mediates the uptake of highly potent purine antimetabolites (Geiser et al., 2005; Hulpia et al., 2019b), as well as several trypanocidal drugs (Lüscher et al., 2007) but is non-essential (Matovu et al., 2003). However, a major advantage of targeting uptake through an essential transporter over a non-essential one is that transporter-related resistance is likely to develop much faster in the latter scenario (Delespaux and de Koning, 2013). In addition, the targeted transporter must be located in the plasma membrane and for P. falciparum, this is only known to be the case for Pf ENT1 (Frame et al., 2015a) and Pf ENT4 (Frame et al., 2012). Interestingly, the study by Tashie et al. (2025a) revealed that both Pf ENT1 and Pf ENT4 are highly conserved with low genetic diversity, emphasizing their importance and suitability as drug targets. Conversely, mutant alleles of TbAT1/P2 have been identified in both lab strains (Mäser et al., 1999; Stewart et al., 2010) and field isolates (Graf et al., 2013), and are associated with drug resistance (Munday et al., 2015).
Gene expression profile across distinct geographical locations. (A) Graphical representation of gene expression profile in Pf ENT1 across distinct geographical locations. (B) Graphical representation of gene expression profile in Pf ENT2 across distinct geographical locations. (C) Graphical representation of gene expression profile in Pf ENT3 across distinct geographical locations. (D) Graphical representation of gene expression profile in Pf ENT4 across distinct geographical locations. The plots above depict the normalized gene expression profiles of all four Pf ENTs across three distinct geographical locations: Kenya, Mali, and Germany. Publicly available RNA-seq datasets were used, comprising samples from individuals with uncomplicated P. falciparum mono-infections. Kenya dataset: Obtained from Kenyan children, as described by Kimenyi et al. (2024) and available under GEO accession number GSE240643; Mali dataset: Obtained from Malian children, as described by Tebben et al. (2024) and available under BioProject PRJNA962942; Germany dataset: Collected from adult travelers returning to Germany, as described by Wichers et al. (2021) and available under BioProject PRJNA679547. A total of 94 RNA-seq datasets were analyzed: 32 from Kenya, 43 from Mali, and 19 from Germany. The box plots presented here display the normalized expression count of the genes which have been adjusted to account for differences in sequencing depth and other technical variations. This normalization ensures that the expression levels are comparable across all samples. The box plot represents the interquartile range (IQR), with the middle line indicating the median. Log2FC = Log2 fold change; p. adj = adjusted p value.
Thus, the Pf ENTs, particularly Pf ENT1, offer a unique and viable pathway for developing purine-based antimalarial drugs. Rather than inhibiting these transporters, the most promising strategy is to focus on leveraging their activity to deliver effective therapeutic agents into the parasite to cause disruption in its purine metabolism and nucleic acid synthesis.
In addition to being a substrate for the Pf ENTs, any purine analog targeting the P. falciparum purine salvage pathway needs to ensure that the active compound does reach the parasite, located inside human cells—in the case of Plasmodium, erythrocytes. In turn, this means that the antimalarial purine analog must be a substrate of nucleoside transporter hENT1, nucleobase transporter hFNT1 and/or the Plasmodium-encoded new permeation pathways. The substrate binding structure-activity relationship of hENT1 and hFNT1 have already been described in considerable detail (Vickers et al., 2004; Wallace et al., 2002) and the contribution of human ENT and CNT transporters to the uptake and sensitivity of many anticancer and antiviral nucleosides has been mapped (King et al., 2006; Pastor-Anglada and Pérez-Torras, 2015; Vaskó et al., 2019).
hFNT displays highest affinity for adenine, with a Km of ~15 μM, but also good affinity for guanine with reported Km value of 37 μM and Ki of 28 μM (Table 2; Domin et al., 1988; Wallace et al., 2002)–10-fold higher than for hypoxanthine or xanthine (28 ± 2.0 μM vs. 290 ± 21 μM or >150 μM; Wallace et al., 2002). Importantly, this transporter has remarkable affinity for some therapeutic guanine analogs (Table 2), e.g., a Ki value of 7.4 μM for 9-methylguanine and 8.0 μM for 9-deazaguanine (Wallace et al., 2002). This further reinforces the notion that guanine-based antimetabolites constitute the most promising purine leads against P. falciparum, as observed by Tashie et al. (2025b). As an example, 7-deazaguanine was the second-most potent nucleobase analog against intraerythrocytic P. falciparum in the latter study (EC_50_ = 14.9 μM) and one of the highest affinity substrates of hFNT1 (K_i_ = 15 ± 4.8 μM; Table 2).
It thus follows that an effective purine antimetabolite strategy against Plasmodium species must be based on oxopurine nucleobases rather than nucleosides, and accommodate, sequentially: (1) effective transport by hFNT in the erythrocyte membrane; (2) effective uptake by Pf ENT1; (3) efficient conversion by Pf HGXPRT to the corresponding mononucleotide; and (4) the further conversion to ATP or GTP analog.
Medicinal chemistry approaches are essential to exploit both the parasite's purine transporters (i.e., Pf ENTs) and the unique features of P. falciparum purine metabolism. This strategy involves designing purine nucleobase analogs that are both efficiently transported and metabolized by the parasite. For example, modifications of the hENT1 inhibitor draflazine resulted in analogs with varying affinities for ENT1 and ENT2 across different mammalian species (Baldwin et al., 2007), demonstrating the feasibility of tailoring analogs for selective recognition by orthologous transporters, even of closely related species. Building on this principle, el Kouni et al. (1999) demonstrated that 6-(4-nitrobenzyl)-mercaptopurine riboside (NBMPR) could be used to treat T. gondii infections within human cells. Mammalian ENTs are historically distinguished based on their differential sensitivity to NBMPR: hENT1 (equilibrative/sensitive, e.s.), is sensitive to inhibition by sub-nanomolar concentrations of NBMPR, whereas hENT2 (equilibrative/insensitive, e.i.) requires micromolar to millimolar concentrations of NBMPR for complete inhibition (Baldwin et al., 2004; Griffiths et al., 1997). Notably, NBMPR is not ordinarily taken up by human cells, being a selective inhibitor rather than substrate of hENT1. el Kouni et al. (1999) concluded that T. gondii alters host cell membrane permeability, equivalent to the Plasmodium new permeation pathways, and so facilitates NBMPR uptake. This selective uptake of NBMPR was reinforced by the intracellular phosphorylation of the analog by T. gondii to its 5′-monophosphate, maintaining the concentration gradient for the free nucleoside. This example underscores the potential for purine analogs to be optimized for selective targeting of both the parasite transport and metabolic systems. Given that P. falciparum and T. gondii are both apicomplexan parasites, the safe, selective use of NBMPR in T. gondii further supports the plausibility of developing purine-based therapies that are selectively taken up by the Pf ENTs in malaria treatment, yet effective in penetrating the host cells—and potentially only by Plasmodium-infected cells, just like NBMPR was taken up selectively by Toxoplasma-infected host cells and nutrients are selectively accumulated in infected Plasmodium-erythrocytes by the new permeation pathways.
Although purine analogs have demonstrated potent antiparasitic effects (Al Safarjalani et al., 2008; Hulpia et al., 2019b; Mabille et al., 2022; Sosa et al., 2019; Tashie et al., 2025b; Ting et al., 2005), as a chemical class, their reputation for toxicity to human cells, particularly during cancer chemotherapy, remains a significant concern (Berman et al., 1985; Kim et al., 2019; Tseligka et al., 2023; Wang et al., 2016). However, the toxicity observed in cancer treatments often results from prolonged administration of these agents, and at high dosage. In contrast, antimalarial treatments typically involve short-term therapeutic regimens which are less likely to result in the severe side effects associated with long-term chemotherapy. More importantly, the cytotoxicity of anticancer nucleoside prodrugs is derived from their activation, i.e., phosphorylation in cancer cells, and eventual incorporation into nucleic acids, and avoiding the activation by human metabolic enzymes is certainly possible. A classic example is the acyclic class of antiviral guanosine analogs acyclovir, ganciclovir and valacyclovir that require activation by virus-encoded enzymes, thus limiting their toxicity to infected cells (Darby et al., 1981). This pharmacological distinction opens a path for designing purine analogs that are selectively activated within the parasite infected erythrocytes and hence minimizing host toxicity.
Conclusion
5
The absolute dependence of P. falciparum on the purine salvage pathway for proliferation and survival provides a compelling opportunity to target this pathway in the development of antimalarial drugs, particularly in the face of rising resistance to current therapies. The most effective approach will be to exploit Pf ENTs as conduits for purine-based analogs that act as “subversive” substrates of parasite purine-metabolizing enzymes, designed with greater sensitivity for parasite enzymes over human homologs. Purine analogs that efficiently enter P. falciparum-infected erythrocytes, reach Pf ENTs, and undergo selective activation within the parasite will disrupt purine metabolism and nucleic acid synthesis, ultimately leading to parasite death.
By integrating structural insights into Pf ENTs and key enzymes of P. falciparum purine metabolism with advanced medicinal chemistry techniques, it becomes feasible to develop purine-based therapies that exploit the distinct substrate affinities and structural properties of the parasite transporters. Such therapies could provide effective and safe antimalarial treatments, minimizing off-target effects in the host while addressing the urgent need for novel interventions that are not cross-resistant with current antimalarials. Structure-activity relationship studies and molecular docking simulations will be critical in guiding the target-based design of analogs optimized for Pf ENT1-mediated uptake and selective activation within P. falciparum.
In this context, recent work by Tashie et al. (2025b) identified several guanine derivative, 8-azaguanine, 7-deazaguanine, and 6-thioguanine, that inhibited the in vitro growth of P. falciparum at low micromolar concentrations. These compounds provide promising starting points for Pf ENT1-targeted drug development. Future medicinal chemistry approaches should focus on optimizing the antiplasmodial potency of these analogs while improving selectivity. A logical early exploration would involve combining the potent active modifications to guanine, for instance, 6-thio, 8-azaguanine, 6-thio, 7-deazaguanine, and 7-deaza, 8-azaguanine.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Al Safarjalani O. N. Rais R. H. Kim Y. A. Chu C. K. Naguib F. N. el Kouni M. H. (2008). 7-Deaza-6-benzylthioinosine analogues as subversive substrate of Toxoplasma gondii adenosine kinase: activities and selective toxicities. Biochem. Pharmacol. 76, 958–966. doi: 10.1016/j.bcp.2008.07.03518755159 PMC 2581922 · doi ↗ · pubmed ↗
- 2Aldfer M. M. Alfayez I. A. Elati H. A. A. Gayen N. Elmahallawy E. K. Milena Murillo A. . (2022 a). The Trypanosoma cruzi Tcr NT 2 nucleoside transporter is a conduit for the uptake of 5-F 2′-deoxyuridine and tubercidin analogues. Molecules 27:8045. doi: 10.3390/molecules 2722804536432150 PMC 9693223 · doi ↗ · pubmed ↗
- 3Aldfer M. M. Al Siari T. A. Elati H. A. A. Natto M. J. Alfayez I. A. Campagnaro G. D. . (2022 b). Nucleoside transport and nucleobase uptake null mutants in Leishmania mexicana for the routine expression and characterization of purine and pyrimidine transporters. Int. J. Mol. Sci. 23:8139. doi: 10.3390/ijms 2315813935897714 PMC 9331716 · doi ↗ · pubmed ↗
- 4Aldfer M. M. Hulpia F. van Calenbergh S. De Koning H. P. (2024). Mapping the transporter-substrate interactions of the Trypanosoma cruzi NB 1 nucleobase transporter reveals the basis for its high affinity and selectivity for hypoxanthine and guanine and lack of nucleoside uptake. Mol. Biochem. Parasitol. 258:111616. doi: 10.1016/j.molbiopara.2024.11161638401850 · doi ↗ · pubmed ↗
- 5Ali J. A. Creek D. J. Burgess K. Allison H. C. Field M. C. Mäser P. . (2013). Pyrimidine salvage in Trypanosoma brucei bloodstream forms and the trypanocidal action of halogenated pyrimidines. Mol. Pharmacol. 83, 439–453. doi: 10.1124/mol.112.08232123188714 PMC 4857052 · doi ↗ · pubmed ↗
- 6Al-Salabi M. I. de Koning H. P. (2005). Purine nucleobase transport in amastigotes of Leishmania mexicana: involvement in allopurinol uptake. Antimicrob. Agents Chemother. 49, 3682–3689. doi: 10.1128/AAC.49.9.3682-3689.200516127040 PMC 1195421 · doi ↗ · pubmed ↗
- 7Al-Salabi M. I. Wallace L. J. M. Lüscher A. Mäser P. Candlish D. Rodenko B. . (2007). Molecular interactions underlying the unusually high adenosine affinity of a novel Trypanosoma brucei nucleoside transporter. Mol. Pharmacol. 71, 921–929. doi: 10.1124/mol.106.03155917185380 · doi ↗ · pubmed ↗
- 8Alzahrani K. J. H. Ali J. A. M. Eze A. A. Looi W. L. Tagoe D. N. A. Creek D. J. . (2017). Functional and genetic evidence that nucleoside transport is highly conserved in Leishmania species: implications for pyrimidine-based chemotherapy. Int. J. Parasitol. Drugs Drug Resist. 7, 206–226. doi: 10.1016/j.ijpddr.2017.04.00328453984 PMC 5407577 · doi ↗ · pubmed ↗