Crosstalk between vimentin and keratins in viral infection: Implications across the viral life cycle
Xiaolong Lu, Xiaoquan Wang, Xiufan Liu, Xiaowen Liu

TL;DR
This review explores how vimentin and keratin intermediate filaments influence various stages of viral infection and suggest they could be targets for antiviral treatments.
Contribution
The paper provides a comprehensive overview of the multifaceted roles of vimentin and keratin in viral infection processes.
Findings
Intermediate filaments influence viral attachment, entry, replication, and egress.
They contribute to cell-to-cell spread and immune regulation during infection.
Viruses exploit or remodel IF networks, offering potential antiviral intervention targets.
Abstract
Intermediate filaments (IFs) have long been regarded as a static scaffold responsible for maintaining cellular structure and integrity. However, recent studies have revealed that IFs, particularly vimentin and keratin, exert a profound and versatile influence on viral infection. In this narrative review, we summarize how these IFs influence multiple stages of the viral life cycle, including attachment/entry, replication, intracellular trafficking, assembly, and egress. We further discuss their contributions to cell-to-cell spread, host immune regulation, and oncogenic processes. Collectively, these findings illustrate how viruses exploit or remodel the IF network to facilitate propagation, and highlight IF – virus interfaces as potential targets for antiviral intervention.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
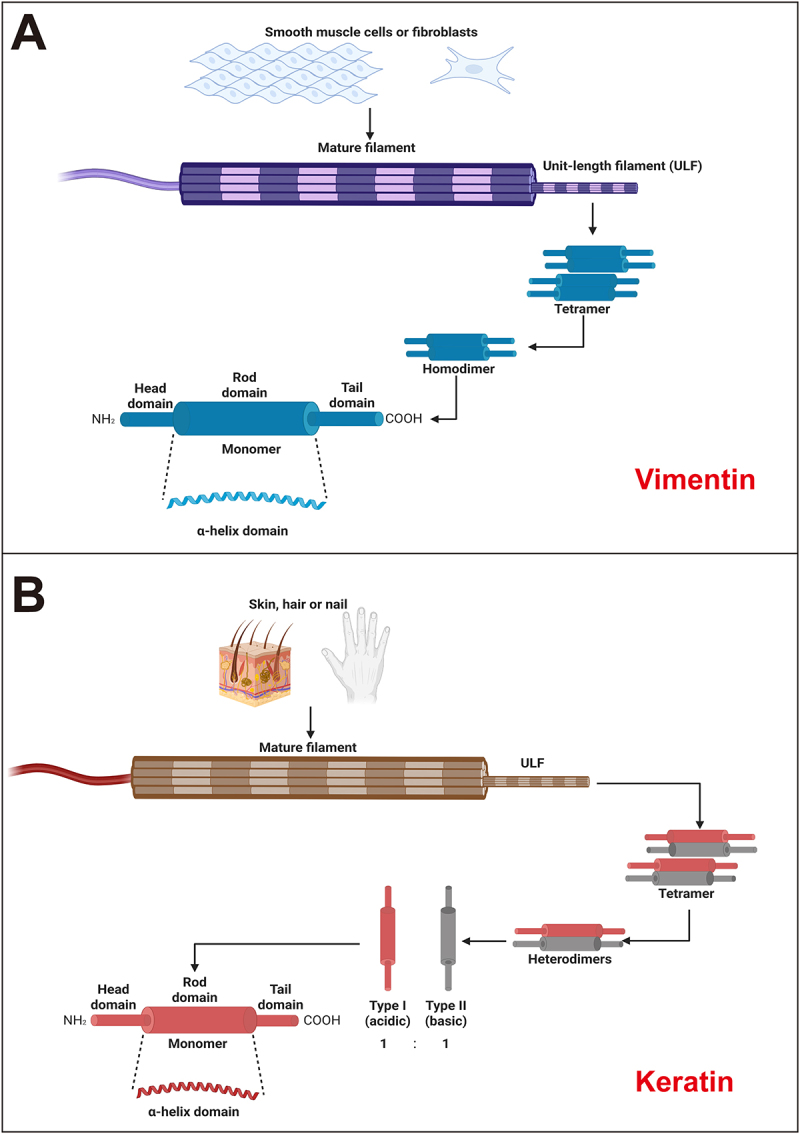 Figure 1
Figure 1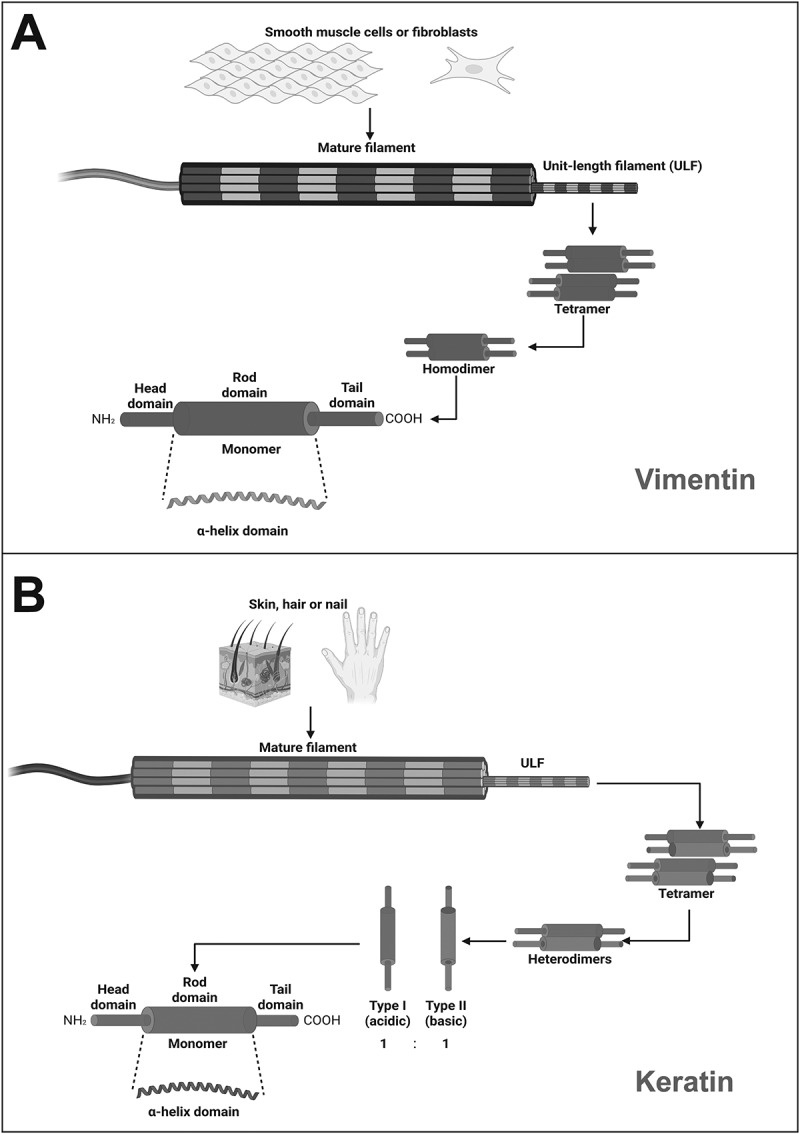 Figure 2
Figure 2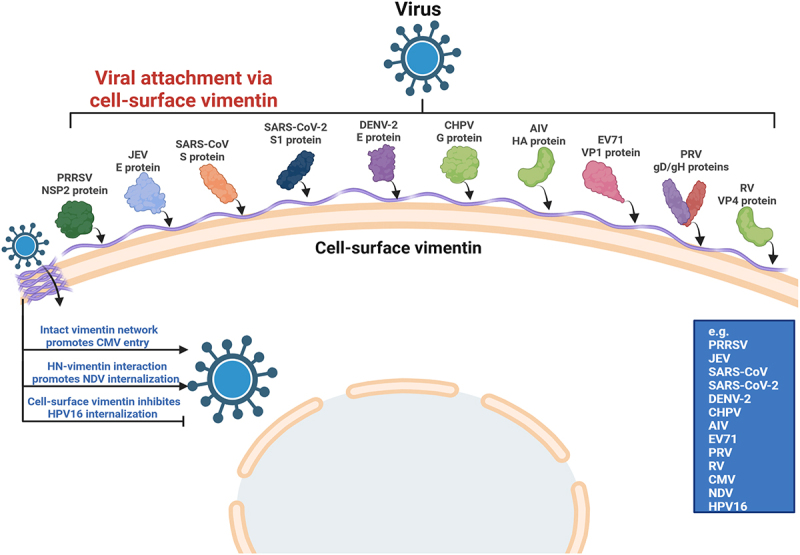 Figure 3
Figure 3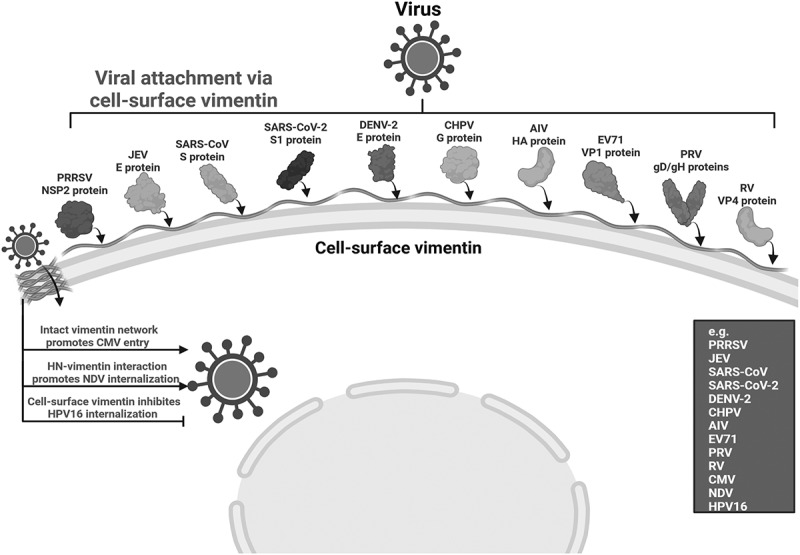 Figure 4
Figure 4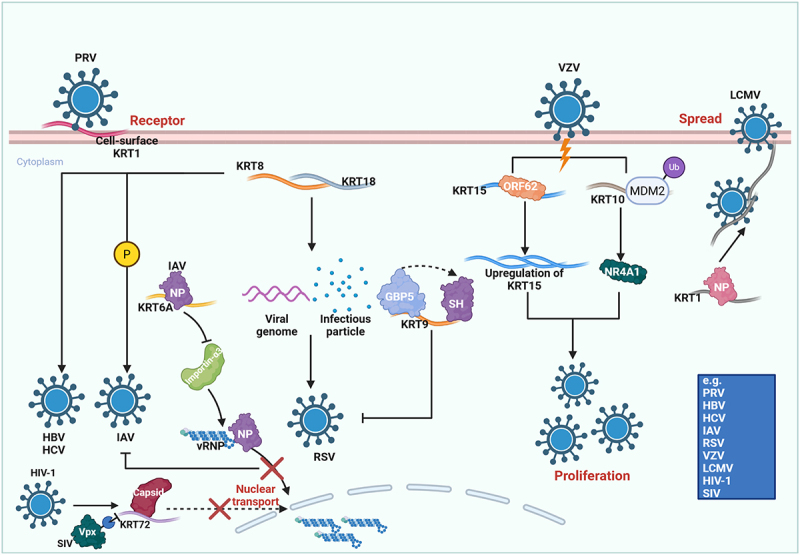 Figure 5
Figure 5 Figure 6
Figure 6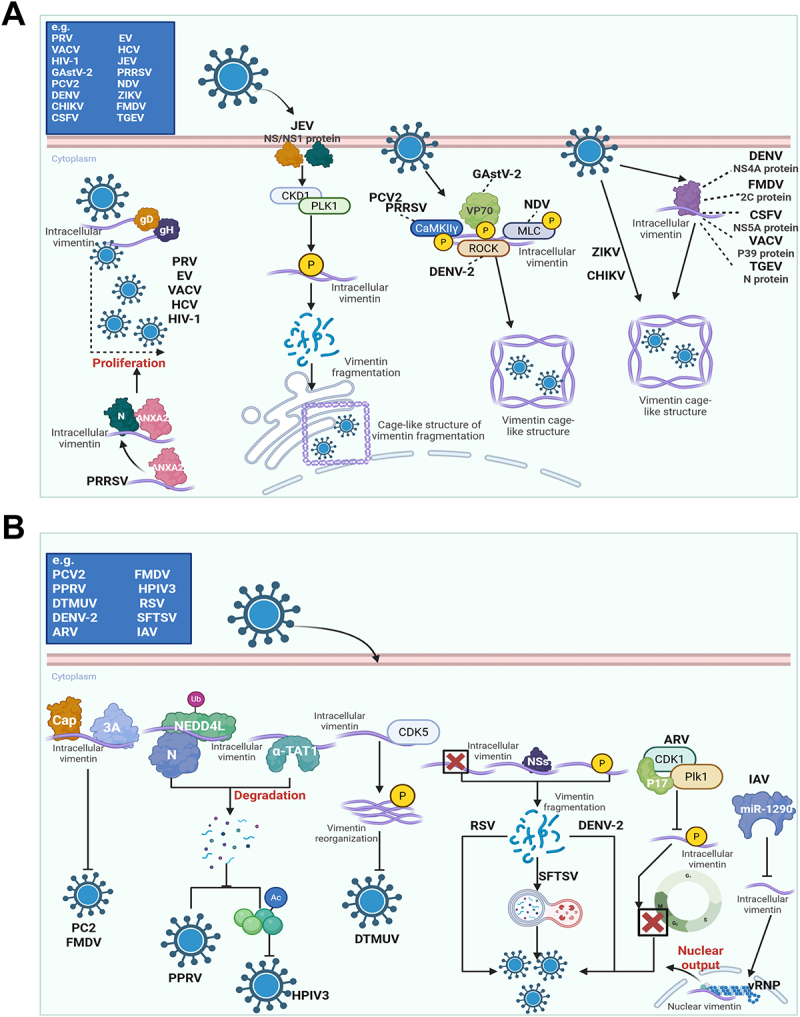 Figure 7
Figure 7 Figure 8
Figure 8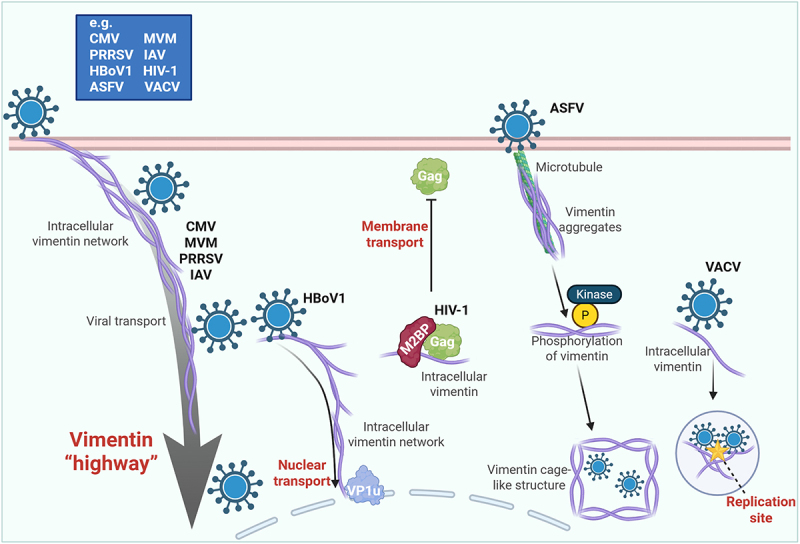 Figure 9
Figure 9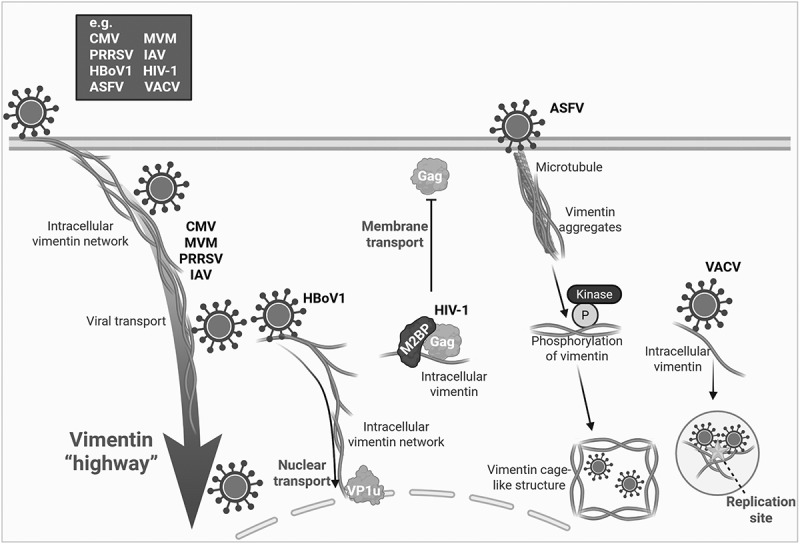 Figure 10
Figure 10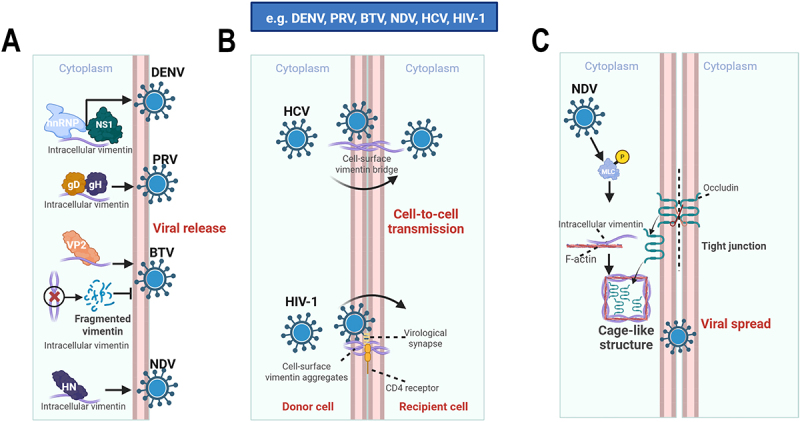 Figure 11
Figure 11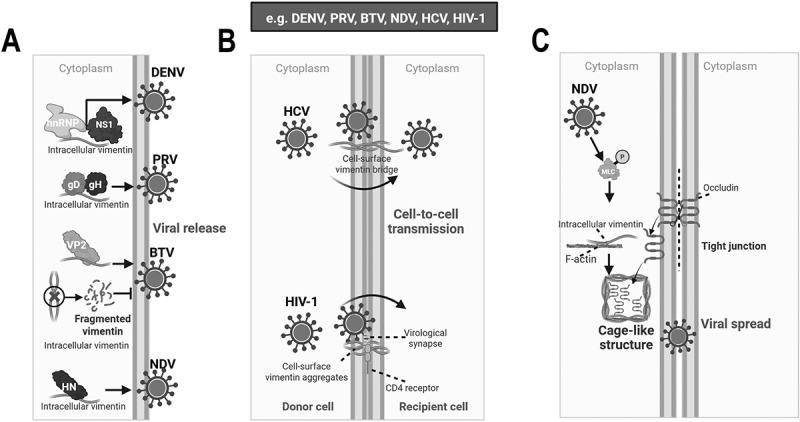 Figure 12
Figure 12- —National Natural Science Foundation of China10.13039/501100001809
- —National Key Research and Development Project of China
- —Open Project Program of Jiangsu Key Laboratory of Zoonosis
- —Priority Academic Program Development of Jiangsu Higher Education Institutions
- —111 Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSkin and Cellular Biology Research · Poxvirus research and outbreaks · Hair Growth and Disorders
Introduction
The successful infection of a virus depends on its ability to precisely hijack and modify host cells [1]. The cytoskeletal system, composed of actin filaments, microtubules, and intermediate filaments (IFs), is a primary target for viral hijacking within the host cell [2,3]. IFs constitute a large, cell type – specific protein superfamily that complements actin and microtubules by providing stress resistance and long-range intracellular connectivity, while also serving as signaling platforms that integrate mechanical and biochemical cues [4,5]. Based on sequence homology and assembly properties, cytoskeletal IF proteins are commonly classified into multiple types: type I and II IFs are acidic and basic keratins, respectively; type III includes vimentin and related proteins (e.g. desmin, GFAP, peripherin); type IV comprises neuron-associated IFs (e.g. neurofilaments and α-internexin); type V corresponds to nuclear lamins; type VI group consists of lens-specific IFs, CP49/phakinin and filen [6,7].
IFs are the most complex and stable type of cytoskeleton. In epithelial cells, the IF network is primarily composed of keratins. Conversely, vimentin is the main IF protein found in mesenchymal cells [8,9]. Vimentin, a type III IF protein, polymerizes from monomers into 10 nm filaments. This assembly proceeds through the formation of parallel, in-register coiled-coil dimers and subsequent antiparallel tetramers, which are the fundamental soluble subunits. A hallmark of the monomer is the central α-helical rod domain, which is bordered by intrinsically disordered head and tail regions [10–12] (Figure 1(A)). By contrast, keratins are obligate heteropolymers. The monomer is characterized by a central α-helical rod domain flanked by intrinsically disordered head and tail regions. Their assembly is initiated by the precise 1:1 pairing of type I (acidic) and type II (basic) polypeptides. This interaction forms coiled-coil heterodimers, which subsequently associate into staggered antiparallel tetramers [13,14]. These tetramers then laterally compact and longitudinally anneal to form unit-length filaments that ultimately mature into robust intermediate filaments, exhibiting extensive type-specific diversification across epithelial tissues [15–17] (Figure 1(B)). Traditionally, these proteins were regarded primarily as structural elements, responsible for maintaining cell shape and resisting mechanical stress [18,19]. Figure 1.Distinct assembly mechanisms of vimentin and keratin intermediate filaments. (A) Vimentin, a type iii IF found in fibroblasts and smooth muscle cells, polymerizes from monomers into parallel, in-register coiled-coil dimers, which form antiparallel tetramers. Vimentin tetramers serve as soluble subunits that laterally compact and longitudinally anneal to generate mature, unit-length filaments. (B) Keratins, the major IFs of skin, hair, and nails, are obligate heteropolymers. Their assembly initiates with the heterodimerization of type I and type ii polypeptides, which then associate to form staggered, antiparallel tetramers. Keratin tetramers laterally associate and longitudinally anneal to form mature, unit-length filaments. The schematic representation is created with Biorender.com.The process begins with a monomer labeled with a head domain, rod domain and tail domain and an alpha-helix domain. Two monomers form a homodimer. Two homodimers assemble into a tetramer. Multiple tetramers form a unit-length filament. Several unit-length filaments combine to create a mature filament. The top of the diagram shows smooth muscle cells or fibroblasts connected by an arrow to the mature filament. The image B showing a schematic diagram of keratin filament assembly in skin, hair, or nail. The process starts with a monomer labeled with a head domain, rod domain, tail domain and alpha-helix domain. There are two types of monomers: type one acidic and type two basic, shown in a one to one ratio. These form heterodimers. Two heterodimers assemble into a tetramer. Multiple tetramers form a unit-length filament. Several unit-length filaments combine to create a mature filament. The top of the diagram shows skin, hair, or nail connected by an arrow to the mature filament.A schematic illustration shows vimentin filament assembly in smooth muscle cells or fibroblasts and keratin filament assembly in skin, hair, or nail, each with a stepwise process from monomer to mature filament.
However, this intracellular structural view is now incomplete. In addition to their filamentous cytoplasmic networks, both vimentin and selected keratins can also be detected at the cell surface or in the extracellular milieu, indicating that IF proteins are not restricted to the cytoskeleton but can also function at the host – environment interface [20–23]. Among IF proteins, extracellular and cell-surface vimentin have been characterized most extensively. In addition to its canonical cytoplasmic filament network, vimentin can be actively released into the extracellular space by activated macrophages and through type III unconventional secretion pathways, and it can also appear as a bona fide cell-surface protein [20,21,24]. Extracellular vimentin is not merely a by-product of cell damage, but can function as an immunologically active molecule, including as a damage-associated molecular pattern (DAMP) and regulator of neutrophil responses, and may also become exposed during inflammatory processes such as NETosis [25]. Although less well studied, keratins can likewise be detected outside the canonical cytoplasmic IF network. Cell-surface expression has been reported for specific keratins, particularly keratin 8 (KRT8) and keratin 1 (KRT1), in stressed or transformed cells, and keratins can also become exposed on apoptotic or necrotic cells, where they participate in immune recognition [22,23,26,27]. Compared with vimentin, however, the mechanisms underlying keratin externalization remain less clearly defined.
However, more and more evidence shows that these two IF proteins act as dynamic signaling platforms. They are deeply involved in many physiological and pathological processes, such as cell migration, signal transduction, and stress responses [28]. Importantly, recognition of extracellular and cell-surface pools of vimentin and keratin substantially broadens the conceptual framework of IF biology. These proteins should now be considered not only intracellular scaffolds but also context-dependent extracellular effectors and surface-accessible ligands/receptors that may shape pathogen attachment, host sensing, inflammatory signaling, and tissue tropism at the earliest stages of infection. In the field of viral infection, the roles of vimentin and keratin have changed from simple structural support to key regulatory factors. Their central importance in the interaction between virus and host is becoming increasingly clear. Although several vimentin-focused reviews have emerged in recent years [29,30], a comprehensive synthesis that jointly integrates vimentin and keratin across the viral life cycle remains lacking. Accordingly, this narrative review is based on the latest research and will critically synthesize the complex functions of vimentin and keratin during the viral life cycle, including virus entry, replication, transport, assembly, release, spread, and immune regulation. Finally, we will look at their future potential as targets for antiviral drugs.
Contribution of intermediate filaments to the early stages of viral infection
The invasion of host cells by viruses begins with their precise recognition and binding to receptors on the cell surface [31]. Current evidence indicates that intermediate filaments, especially vimentin and, to a lesser extent, keratins, are not merely structural components at this stage, but active determinants of viral attachment and entry. Studies have found that IF proteins located on the cell surface can serve as attachment factors, receptors, or co-receptors for various viruses, thereby influencing viral adsorption and internalization. Among them, cell-surface vimentin is the most extensively studied and appears to function as a broadly used but virus-dependent entry factor. In the context of RNA virus infection, cell-surface vimentin has been identified as a host factor that facilitates porcine reproductive and respiratory syndrome virus (PRRSV) infection. It functions as a receptor for PRRSV and interacts with the viral NSP2 protein to form a complex that promotes viral adsorption. Notably, it also serves as a core component of the receptor complex and can confer susceptibility to otherwise non-susceptible cells [32–34]. Similarly, the E protein of Japanese encephalitis virus (JEV) binds cell-surface vimentin as a key receptor, and antibody blockade markedly inhibits infection [35,36]. This role is further modulated by host signaling: D2R stimulation activates phospholipase C (PLC), which increases cell-surface vimentin expression and thereby enhances JEV entry [37]. The cell type-specific distribution of cell-surface vimentin – high in neural progenitor and glial cells but absent in mature neurons – has also been linked to the tropism of JEV [38]. A comparable pattern has emerged for coronaviruses, supporting the idea that cell-surface vimentin often acts as an accessory entry factor that cooperates with canonical receptors. In SARS-CoV infection, the spike protein binds vimentin and promotes its surface accumulation, whereas antibody blockade or genetic inhibition of cell-surface vimentin reduces viral binding and entry [39]. In SARS-CoV-2 infection, cell-surface vimentin likewise functions as an attachment factor or co-receptor. It binds the spike protein, particularly the S1 subunit, and cooperates with ACE2 to promote viral entry in multiple cell types, including endothelial cells [40–43]. The cell-surface vimentin – spike interaction may occur at specialized surface regions such as cilia, further suggesting that cell-surface vimentin helps organize local docking platforms for viral attachment [44]. This entry-promoting role is not restricted to PRRSV, JEV, or coronaviruses. The EDIII domain of the Dengue virus 2 (DENV-2) E protein binds cell-surface vimentin to facilitate adsorption [45]. On neuronal cells, Chandipura virus (CHPV) uses cell-surface vimentin as a co-receptor through its glycoprotein G [46]. H9N2 avian influenza virus (AIV), Enterovirus 71 (EV71), and Newcastle disease virus (NDV) also depend on cell-surface vimentin during early infection [47–51]. DNA viruses exploit the same principle: Pseudorabies virus (PRV) binds the rod domain of cell-surface vimentin through conserved sites in its gD and gH proteins [52], Cytomegalovirus (CMV) entry is facilitated by the vimentin network [53,54], and Hepatitis B virus (HBV) requires vimentin for endocytic entry [55]. Cell-surface vimentin also interacts with Rotavirus (RV) VP4 together with ACTR2 to promote early viral infection [56]. Overall, these findings support the view that cell-surface vimentin serves as a versatile host entry platform exploited by phylogenetically diverse viruses. These findings suggest that cell-surface vimentin is not a virus-specific receptor in a narrow sense, but a versatile host entry platform exploited by diverse viruses. However, the role of vimentin in entry is not uniformly pro-viral. In Human papillomavirus 16 (HPV16) infection, cell-surface vimentin restricts viral internalization after attachment, thereby inhibiting infection [57,58] (Figure 2). Thus, vimentin is better viewed as a multifunctional entry determinant whose effect depends on the entry strategy of each virus. Figure 2.Cell surface vimentin as a critical mediator for viral entry. As a key component on the cell surface, vimentin can function as an attachment point, receptor, or co-receptor for diverse viruses. For PRRSV, vimentin interacts with NSP2 to form a receptor complex that mediates adsorption and confers susceptibility to non-permissive cells. JEV binds directly to vimentin via its E protein; this interaction is enhanced by D2R signaling and contributes to the virus’s cellular tropism. SARS-CoV and SARS-CoV-2 spike proteins also engage vimentin, which acts synergistically with ACE2 to facilitate entry, a process inhibitable by antibody blockade or gene knockdown. Other RNA viruses, including DENV-2, CHPV, H9N2 AIV, EV71, and NDV, similarly exploit vimentin through interactions with their respective envelope or capsid proteins (e, G, HA, VP1, HN) to promote adsorption and internalization. Vimentin also facilitates the entry of DNA viruses such as PRV, CMV, and HBV. PRV binds the rod domain of vimentin via its gD/gH proteins. In contrast to its typical pro-viral role, surface vimentin acts as a restrictive factor for HPV16, impeding viral internalization after attachment and inhibiting infection. The schematic representation is created with Biorender.com.Various virus proteins interact with vimentin: PRRSV NSP2 protein, JEV E protein, SARS-CoV S protein, SARS-CoV-2 S1 protein, DENV-2 E protein, CHPV G protein, AIV HA protein, EV71 VP1 protein, PRV gD/gH proteins and RV VP4 protein. Below, arrows indicate: ‘Intact vimentin network promotes CMV entry‘, ‘HN-vimentin interaction promotes NDV internalization‘ and ‘Cell-surface vimentin inhibits HPV16 internalization‘. A list of viruses includes PRRSV, JEV, SARS-CoV, SARS-CoV-2, DENV-2, CHPV, AIV, EV71, PRV, RV, CMV, NDV and HPV16.Diagram of viral attachment via cell-surface vimentin with various virus proteins.
Compared with vimentin, keratins have been studied less extensively as entry factors, but available data indicate that they also participate in early virus – host interactions. Cell-surface KRT1 has been identified as a key receptor for PRV entry, and both antibody blockade and gene knockout markedly reduce cellular susceptibility [59]. The epithelial-surface keratins contribute to epithelial barrier integrity, indicating that their role at the entry stage is intrinsically dual: they may facilitate viral attachment in some settings while also limiting tissue invasion through maintenance of epithelial structure [60] (Figure 3 and Table S1). Figure 3.Keratins: multifaceted regulators of the viral life cycle. Keratins orchestrate a complex relationship with viruses throughout the infection process. Entry: Keratins can act as essential attachment factors or receptors, exemplified by KRT1 for PRV, where its blockade inhibits infection. Replication: their role is dualistic. While KRT8, KRT15, and KRT18 promote the replication of diverse viruses (HBV, HCV, RSV, VZV), others like KRT6A, KRT9, and KRT10 act as intrinsic antiviral factors by targeting viral proteins (e.g. IAV NP, RSV sh) or modulating host pathways (e.g. NR4A1). Trafficking: Keratins regulate intracellular viral transport; KRT6A impedes nuclear import of IAV vRnps, while KRT72 restricts HIV-1 capsid trafficking, a mechanism counteracted by SIV vpx. Transmission: finally, keratins can facilitate cell-to-cell spread, as LCMV exploits KRT1 to enhance cytoskeletal stability and cell adhesion. The schematic representation is created with Biorender.com.At the top left, the cell-surface KRT1 acts as a receptor for PRV, facilitating viral entry. Below, KRT6A interacts with IAV NP, impeding nuclear import of vRNPs. KRT72 restricts HIV-1 capsid trafficking, counteracted by SIV vpx. In the center, KRT8 and KRT18 promote replication by interacting with the viral genome, leading to the formation of infectious particles. KRT15 is upregulated, influenced by ORF62 and interacts with KRT9, targeting viral proteins like GBP5 and SH. KRT10, modulated by NR4A1, acts as an antiviral factor. On the right, VZV and LCMV exploit keratins for spread, with KRT1 enhancing cytoskeletal stability. The diagram includes a legend at the bottom right listing viruses such as PRV, HBV, HCV, IAV, RSV, VZV, LCMV, HIV-1 and SIV.A diagram showing keratins‘ roles in viral infection processes, including entry, replication and spread.
The central role of intermediate filaments in viral replication process
Upon entry into the host cell, the virus hijacks cellular machinery and resources to initiate genome replication and viral protein synthesis. At this stage, vimentin emerges as a dynamic scaffold that is repeatedly remodeled to support replication, rather than as a passive cytoskeletal bystander. In several systems, an intact vimentin network is essential for efficient viral replication. For example, the synthesis of EV non-structural proteins such as 2A and 3D depends on intact vimentin, and pharmacological disruption of vimentin inhibits Vaccinia virus (VACV) replication [61,62]. A recurrent theme is that many viruses induce vimentin reorganization into perinuclear, granular, or cage-like structures that support replication complexes. JEV activates a CDK1–PLK1 kinase cascade that phosphorylates vimentin, causing its fragmentation into granules that traffic to the endoplasmic reticulum and form a cage-like structure supporting replication [63]. NDV uses a similar strategy by activating MLC kinase to induce vimentin reorganization into a cage-like structure [64]. PRRSV activates CaMKIIγ to phosphorylate vimentin, which assembles a cage around its replication complex to enhance viral efficiency, while Porcine circovirus type 2 (PCV2) utilizes the same pathway to alter vimentin’s cellular distribution for replication, revealing that CaMKIIγ-mediated vimentin phosphorylation is a key replication strategy shared by multiple viruses [65,66]. DENV-2 also induces structural rearrangement of vimentin through ROCK-dependent phosphorylation and perinuclear reorganization [67,68], whereas Zika virus (ZIKV) and Chikungunya virus (CHIKV) likewise induce vimentin cages around replication factories [69,70]. Although the upstream kinases differ, the underlying strategy is similar: viruses exploit vimentin phosphorylation and spatial remodeling to generate a protected replication microenvironment.
In addition to network remodeling, many viruses directly engage vimentin through specific viral proteins. The VP70 protein of Goose astrovirus type 2 (GAstV-2) interacts with vimentin to induce its phosphorylation and reorganization into a complex that encapsulates viral RNA [71]. As noted above, PRV gD and gH interact with the vimentin rod domain, and in PRRSV infection, vimentin first binds ANXA2 and then promotes its interaction with the viral N protein, thereby enhancing replication [72]. Other viral proteins reported to bind vimentin include DENV NS4A [73], Foot-and-mouth disease virus (FMDV) 2C [74], Classical swine fever virus (CSFV) NS5A [75], VACV P39 [76], and Transmissible gastroenteritis virus (TGEV) N [77]. These examples indicate that vimentin supports replication through both structural remodeling and direct virus – host protein interaction. In addition, vimentin has also been reported to be required for efficient replication of Hepatitis C virus (HCV) and Human immunodeficiency virus type 1 (HIV-1) [78,79]. Notably, the contribution of vimentin to HCV infection may be dose-dependent: when highly expressed, vimentin can exert an inhibitory effect by accelerating degradation of the viral core protein [80] (Figure 4(A)). Figure 4.The dual functionality of vimentin in viral replication. (A) Pro-viral functions of vimentin. An intact vimentin network is required for efficient replication of EV and VACV. Many viruses – including JEV, NDV, PRRSV, PCV2, DENV-2, ZIKV, and CHIKV – hijack host kinases to phosphorylate vimentin, reorganizing it into cage-like structures that protect replication sites. Vimentin is also essential for HCV and HIV-1 replication. Numerous viral proteins directly bind vimentin to exploit its scaffolding function, such as VP70 (GAstV-2), gD/gH (PRV), NS4A (DENV), 2C (FMDV), NS5A (CSFV), P39 (VACV), and N (TGEV). For PRRSV, vimentin first binds ANXA2 to promote interaction with the viral N protein. (B) Vimentin as an antiviral factor and viral countermeasures. Vimentin inhibits viruses by directly binding proteins like Cap (PCV2) and 3A (FMDV), recruiting the E3 ligase NEDD4L to degrade PPRSV N protein, or suppressing inclusion body formation in HPIV3. DTMUV replication is inhibited by vimentin phosphorylation via CDK5. To overcome this, viruses have evolved strategies: RSV and SFTSV degrade vimentin; DENV-2 induces its network disassembly; and ARV and IAV manipulate its expression/phosphorylation to disrupt the cell cycle and vRNP trafficking, respectively. The schematic representation is created with Biorender.com.The image A showing a scientific schematic titled by the large letter A, organized around a horizontal cell boundary labeled Cytoplasm, where multiple virus icons connect by arrows to intracellular vimentin and to labeled host and viral proteins that lead to vimentin remodeling and replication support. A boxed list beginning with e.g. contains PRV, VACV, HIV-1, GAstV-2, PCV2, DENV, CHIKV, CSFV, EV, HCV, JEV, PRRSV, NDV, ZIKV, FMDV and TGEV. One pathway shows PRV with two attached labels gD and gH pointing toward intracellular vimentin and a dashed bracketed region ending at the word Proliferation, with nearby stacked virus labels PRV, EV, VACV, HCV and HIV-1. Another pathway shows PRRSV with intracellular vimentin connected to ANXA2 and then to N. A JEV icon connects to the label NS/NS1 protein, then by arrows to CKD1 and PLK1, then to a circle labeled P, then to intracellular vimentin and the label Vimentin fragmentation, ending at a drawn cage-like structure labeled Cage-like structure of vimentin fragmentation. Additional virus icons labeled PCV2 and PRRSV connect to CaMKIIgamma and GAstV-2 connects to VP70, with arrows leading through circles labeled P and through ROCK and MLC toward intracellular vimentin and a labeled Vimentin cage-like structure. A separate cluster shows ZIKV and CHIKV directed toward a second Vimentin cage-like structure. On the far side, a virus labeled DENV points to a protein cluster labeled NS4A protein and a vertical list beside intracellular vimentin reads DENV NS4A protein, FMDV 2C protein, CSFV NS5A protein, VACV P39 protein and TGEV N protein, with arrows from this list toward intracellular vimentin and toward a Vimentin cage-like structure. The image B showing a second scientific schematic titled by the large letter B, again arranged around the Cytoplasm label and intracellular vimentin, depicting inhibitory interactions and viral countermeasures. A boxed list beginning with e.g. contains PCV2, PPRV, DTMUV, DENV-2, ARV, FMDV, HPIV3, RSV, SFTSV and IAV. One chain shows Cap and 3A attached to intracellular vimentin, leading by arrows to NEDD4L with a small label Ub, then to N and then to the word Degradation with particles dispersing below. Another branch shows intracellular vimentin connected to alpha-TAT1 and to a circle labeled Ac. A DTMUV icon connects by an arrow to intracellular vimentin and to CDK5, then to a circle labeled P, then to a vimentin bundle labeled Vimentin reorganization. A boxed region shows RSV and SFTSV associated with intracellular vimentin and a label Vimentin fragmentation, with DENV-2 labeled beside the fragmented vimentin drawing. Another pathway shows ARV connected to CDK1 and Plk1, leading to a circle labeled P and then to intracellular vimentin. A separate IAV branch shows miR-1290 connected toward intracellular vimentin and toward a circular nucleus drawing labeled Nuclear output, with a label vRNP placed along the outgoing arrow and the text Nuclear vimentin placed near the nucleus.A diagram showing vimentin in viral replication, with sub-images A pro-viral and B antiviral pathways.
Importantly, the relationship between vimentin and viral replication is not one-sided. Vimentin can also function as an antiviral factor under specific conditions. It directly binds the PCV2 Cap protein or the FMDV 3A protein to inhibit replication [81,82]. It can recruit the E3 ubiquitin ligase NEDD4L to promote degradation of the Peste des petits ruminant virus (PPRV) N protein [83], and it inhibits Human parainfluenza virus type 3 (HPIV3) inclusion body formation by promoting α-TAT1 degradation and reducing α-tubulin acetylation [84]. In Duck tembusu virus (DTMUV) infection, CDK5-mediated vimentin phosphorylation and reorganization are associated with inhibition of viral replication [85]. Vimentin also exerts antiviral effects through maintenance of intracellular cholesterol homeostasis [86]. Thus, vimentin is better regarded as a contested platform during replication, one that can be exploited by viruses but can also be mobilized by the host. Consistent with this, several viruses have evolved strategies to counteract vimentin. Respiratory syncytial virus (RSV) and Severe fever with thrombocytopenia syndrome virus (SFTSV) promote vimentin degradation [87,88], DENV-2 induces its phosphorylation and solubilization [89], and Avian reovirus (ARV) and Influenza A virus (IAV) manipulate its expression or phosphorylation to favor viral replication and vRNP trafficking [90,91] (Figure 4(B)).
Keratins also display clear functional duality during viral replication, but their effects appear more dependent on the individual keratin member. Keratin 18 (KRT18) and KRT8 are required for RSV replication [92]; KRT8 also promotes Hepatitis B virus (HBV) and HCV replication [93,94]; phosphorylation of KRT8 enhances H1N1 subtype IAV replication [95]; and keratin 15 (KRT15) interacts with the Varicella-zoster virus (VZV) ORF62 protein, leading to the upregulation of its expression and subsequent promotion of viral replication [96]. In contrast, keratin 9 (KRT9) acts as an antiviral factor against RSV by facilitating GBP5-dependent capture and secretion of the viral SH protein [97]. Likewise, VZV promotes replication by inducing MDM2-mediated degradation of keratin 10 (KRT10) [98], and keratin 6A (KRT6A) inhibits IAV replication by binding NP, disrupting vRNP integrity, and blocking nuclear import [99] (Figure 3). Therefore, keratins cannot be generalized as simply pro-viral or antiviral; their effects are strongly member- and virus-specific.
Intermediate filaments as a critical platform for viral transport and assembly
For productive infection, viral components must be efficiently transported to appropriate intracellular sites and assembled into new virions. Current evidence suggests that vimentin contributes to these processes mainly by organizing intracellular trafficking and supporting localized assembly sites. It can act as a “highway” to actively promote infection. The vimentin network facilitates transport of CMV [53], promotes endosomal trafficking of Minute virus of mice (MVM) [100], collaborates with other cytoskeletal structures to assist the transport of PRRSV to the perinuclear region [101], and accelerates the trafficking of IAV from early to late endosomes, thereby promoting viral membrane fusion and genome release [102,103]. In addition, interaction between vimentin and the Human bocavirus 1 (HBoV1) VP1u protein enhances nuclear transport of VP1u and ultimately promotes infection [104]. These observations support the view that vimentin forms part of the intracellular architecture that coordinates viral movement. At the same time, this network can also be used for restriction. The host protein M2BP can capture the HIV-1 Gag protein and anchor it onto vimentin filaments, thereby blocking its transport to the plasma membrane and inhibiting virion production [105]. Thus, as in replication, the role of vimentin in transport is bidirectional. Vimentin also contributes to viral assembly. During African swine fever virus (ASFV) infection, it is reorganized into a perinuclear cage that concentrates viral proteins for assembly [106], and VACV recruits vimentin to the vicinity of viral replication sites to facilitate assembly [76] (Figure 5). Figure 5.Vimentin orchestrates viral intracellular transport and assembly. Vimentin-mediated viral transport. Vimentin acts as a “highway” to facilitate the intracellular trafficking of diverse viruses. It promotes the transport of CMV and the endosomal trafficking of MVM. It collaborates with other cytoskeletal elements to transport PRRSV to the perinuclear region and accelerates IAV trafficking from early to late endosomes, enhancing membrane fusion. The interaction between vimentin and HBoV1 VP1u mediates the nuclear import of VP1u, boosting infection. Conversely, the host can exploit vimentin for defense; the protein M2BP anchors HIV-1 gag to vimentin filaments, blocking its transport to the plasma membrane and inhibiting virion production. Vimentin as a scaffold for viral assembly. Vimentin is frequently co-opted as a dynamic scaffold for viral assembly. During ASFV infection, vimentin is aggregated via microtubules and phosphorylated to form a perinuclear cage that concentrates viral proteins. Similarly, VACV infection recruits vimentin to viral replication sites to facilitate assembly. The schematic representation is created with Biorender.com.At the top left, a blue box lists viruses: CMV, PRRSV, HBoV1, ASFV, MVM, IAV, HIV-1 and VACV. Below, viruses are shown moving along a purple network labeled ‘Intracellular vimentin network,‘ indicating ‘Viral transport.‘ An arrow labeled ‘Vimentin highway‘ points downward, emphasizing the transport role. To the right, HBoV1 is shown with a label ‘Nuclear transport‘ and a protein labeled ‘VP1u‘ interacting with the vimentin network. Further right, HIV-1 is depicted with ‘Gag‘ proteins and ‘M2BP‘ interacting with the vimentin network, labeled ‘Membrane transport.‘ Moving right, ASFV is shown near a ‘Microtubule‘ with ‘Vimentin aggregates‘ and ‘Phosphorylation of vimentin‘ by a ‘Kinase,‘ leading to a ‘Vimentin cage-like structure.‘ At the far right, VACV is shown with ‘Intracellular vimentin‘ leading to a ‘Replication site,‘ indicating the role of vimentin in viral assembly.A diagram showing vimentin-mediated viral transport and assembly with various viruses.
Compared with vimentin, the role of keratins in viral transport has been less extensively characterized and appears mainly restrictive. KRT6A impairs IAV infection by interfering with NP interaction with importin α3, thereby blocking nuclear import [99]. Keratin 72 (KRT72) likewise restricts HIV-1 by hindering capsid transport toward the nucleus, although Simian immunodeficiency virus (SIV) Vpx can counteract this defense by targeting KRT72 for degradation [107] (Figure 3). Whether keratins also make broader contributions to viral assembly remains unclear.
The complex roles of intermediate filaments in viral release and spread
After completing replication, viruses need to be effectively released and spread to new cells or hosts. Although this phase has been studied less extensively than entry or replication, available evidence indicates that intermediate filaments influence both virion release and cell-to-cell spread. For vimentin, several studies support a role in viral egress. NDV engages vimentin through its HN protein to promote release [51]. PRV promotes its release by interacting with the rod domain of vimentin through its gD and gH proteins [52]. In addition to promoting efficient ZIKV replication, vimentin is also crucial for viral release, as its disruption severely hinders the process [69]. Release of Bluetongue virus (BTV) and DENV also depends on interactions with vimentin: the BTV outer capsid protein VP2 binds vimentin through residues glycine 70 and valine 72 [108], whereas DENV NS1 and host hnRNP-containing complexes are associated with vimentin during release [109] (Figure 6(A)). These studies suggest that the proviral functions of vimentin often extend throughout infection, from entry to late-stage egress. Figure 6.Vimentin-mediated viral release and cell-to-cell transmission. (A) Vimentin in facilitating viral release. Vimentin is critical for the release of diverse viruses. NDV HN protein engages vimentin to facilitate release. PRV promotes its release via gD and gH protein interaction with vimentin. ZIKV also requires vimentin for efficient release. For BTV, the outer capsid protein VP2 binds vimentin, and disrupting this interaction or the vimentin network inhibits release. During DENV infection, vimentin forms complexes with host hnRnps and the viral NS1 protein, promoting virus release. (B) Vimentin in cell-to-cell transmission and barrier disruption. Vimentin is crucial for direct viral spread between cells. HCV utilizes vimentin-containing cell bridges for directional transfer, mediated by the E1 protein. HIV-1 recruits vimentin to virological synapses to promote CD4 receptor clustering. (C) Viruses disrupt tissue barriers to accelerate spread. NDV induces reorganization of vimentin and F-actin, leading to the degradation of tight junction proteins and disrupting the cellular barrier. This enhances NDV cell-to-cell spread. The schematic representation is created with Biorender.com.The image A showing a schematic labeled Cytoplasm at the top, with a vertical cell boundary at the right edge and the red text Viral release near the lower right, where four right-pointing arrows run from intracellular components toward virus icons positioned beside the boundary; at the upper left, a curved purple filament labeled Intracellular vimentin sits beneath a light blue label hnRNP and a black arrow points from a dark shape labeled NS1 toward a spiky circular virus icon labeled DENV at the upper right; below, another purple filament labeled Intracellular vimentin sits under two attached shapes labeled gD and gH and a black arrow points right to a spiky circular virus icon labeled PRV; below that, an orange shape labeled VP2 sits above a purple filament and a black arrow points right to a spiky circular virus icon labeled BTV; below, a purple filament labeled Fragmented vimentin is shown as separated pieces with a circled X symbol beside it and a black arrow points right to a spiky circular virus icon labeled NDV, while at the bottom left another label Intracellular vimentin appears near a dark shape labeled HN with a black arrow pointing right toward the same NDV virus icon at the lower right. The image B showing a schematic with Cytoplasm labeled at the top on both sides of a central vertical boundary and a blue banner above reading e.g. DENV, PRV, BTV, NDV, HCV, HIV-1; near the upper center, a spiky circular virus icon labeled HCV is positioned beside the boundary and a curved black arrow arcs across the boundary toward the opposite side, passing over a purple structure labeled Cell-surface vimentin bridge that spans the boundary; near the lower center, a spiky circular virus icon labeled HIV-1 sits beside the boundary and a second curved black arrow arcs across the boundary toward the opposite side, ending near dashed lines labeled Virological synapse, with a purple cluster labeled Cell-surface vimentin aggregates attached near the boundary and a dashed label CD4 receptor placed on the recipient side; the bottom margin labels Donor cell on the left side of the boundary and Recipient cell on the right side and the red text Cell-to-cell transmission appears near the center-right. The image C showing a schematic with Cytoplasm labeled at the top on both sides of a central vertical boundary, where a spiky circular virus icon labeled NDV appears near the upper left and a black arrow points downward to a small circle labeled P attached to a shape labeled MLC, then another black arrow points downward toward a region labeled Intracellular vimentin and a nearby label F-actin; below, a boxed area labeled Cage-like structure contains a coiled filament-like drawing and a black arrow points downward from this region toward a spiky circular virus icon near the lower right; at the boundary, the label Tight junction is placed beside a junction-like drawing and the label Occludin is placed near the upper part of that junction, while the red text Viral spread appears near the lower right.A diagram showing vimentin-mediated viral release, cell-to-cell transmission and barrier disruption in three sub-images.
Vimentin also facilitates direct intercellular spread. HCV exploits vimentin-containing cell bridges for directional transfer of viral components between adjacent cells, mediated by interaction between the HCV E1 protein and the vimentin head domain [110,111]. HIV-1 similarly recruits vimentin during formation of virological synapses, where it promotes CD4 clustering and supports cell-to-cell transmission [112] (Figure 6(B)). Thus, vimentin contributes not only to intracellular infection dynamics but also to intercellular dissemination. A further layer of complexity is that viruses can accelerate tissue spread by remodeling IF-dependent barrier structures. In NDV infection, reorganization of vimentin and F-actin induces degradation of tight-junction proteins such as OCLN and ZO-1, thereby weakening epithelial barrier integrity and favoring viral dissemination [64] (Figure 6(C)). This illustrates that IF remodeling can promote spread not only by acting on virions themselves, but also by dismantling host tissue barriers.
Evidence for keratin involvement in viral spread remains limited but is nonetheless suggestive. Lymphocytic choriomeningitis virus (LCMV) binds KRT1 through its NP protein, stabilizing the cytoskeleton and reinforcing cell – cell adhesion, which in turn promotes cell-to-cell spread [113] (Figure 3). At present, however, the contribution of keratins to release and dissemination remains much less defined than that of vimentin, highlighting an important gap in the field.
The divergent roles of intermediate filaments in antiviral immunity and cancer development
IFs are not only exploited by viruses but also participate in host immune regulation. An important concept emerging from recent studies is that IF proteins regulate antiviral signaling in addition to their structural roles. Studies have found that virus-induced vimentin can inhibit the production of type I interferons by binding to and interfering with TBK1 and IKKε, thereby blocking IRF3 activation and helping the virus evade immune surveillance [114]. In Treg cells, vimentin anchors the IL-18 receptor and inhibits amphiregulin production, thereby impairing tissue repair after viral pneumonia [115]. These findings identify vimentin as a signaling hub that can be subverted to weaken antiviral defense and tissue recovery.
Conversely, IFs can also contribute to antiviral restriction. The interferon-induced protein MXB, for instance, achieves a broad-spectrum antiviral effect by recognizing vimentin and recruiting the kinase AKT to phosphorylate it. This disrupts the vimentin network, thereby blocking the transport of various viral protein complexes [116]. Keratins such as KRT9 and KRT72 likewise participate in host defense [97,107]. Furthermore, studies showing that high-risk HPV suppresses CD8+ T cell immune responses by overexpressing keratin 17 (KRT17) [117], while VZV promotes its own replication by degrading keratin KRT10 [98], further supports the importance of keratins in immune homeostasis.
The relevance of IFs extends beyond infection to cancer biology, particularly in the context of chronic infection, inflammation, and tissue remodeling. Vimentin is a canonical regulator of epithelial-mesenchymal transition (EMT), migration, and metastasis [118]. For example, the HBV HBx protein promotes hepatocellular carcinoma development by downregulating LINC01010 and thereby enhancing vimentin-dependent proliferation and migration [119]. Extracellular vimentin also contributes to communication between endothelial and immune cells within the tumor microenvironment [120]. Keratins, meanwhile, function not only as diagnostic markers but also as active regulators of tumor progression, as illustrated by keratin 7 (KRT7) and KRT17 [121–124]. Together, these observations indicate that IF proteins connect viral infection, immune dysregulation, and cancer progression through shared structural and signaling mechanisms.
Conclusions and perspectives
In summary, vimentin and keratin play dynamic roles throughout the viral life cycle that extend far beyond structural support. Rather than acting as passive cytoskeletal components, they should be regarded as multifunctional host determinants that shape viral entry, replication, transport, assembly, release, spread, and immune responses. Overall, vimentin is more frequently exploited by viruses, whereas keratins display greater functional heterogeneity and often act in a member-specific manner. To facilitate the understanding of IF-mediated regulation during viral infection, we summarized the relevant findings in Supplementary Tables S2–S4.
This mechanistic diversity has important translational implications. First, the recurrent involvement of vimentin in viral attachment and entry supports vimentin-directed approaches as broad-spectrum entry interference strategies, including antibody-based blockade or competitive decoys that disrupt virion binding to surface-exposed vimentin [36,55,125]. Second, because many viruses rely on vimentin remodeling to build replication-competent microenvironments, targeting critical virus – vimentin interfaces or the host kinase circuits that drive vimentin phosphorylation and reorganization (e.g. ROCK and CDK/PLK-related pathways) may provide a rational means to suppress replication without directly targeting viral enzymes [63,68]. Third, given the dual roles of vimentin in innate immune signaling, therapeutic modulation of IF-linked immune regulation may help rebalance antiviral responses and limit immunopathology in selected settings [114,115]. Beyond intervention, IFs also carry biomarker potential: infection-associated changes in IF abundance, post-translational modification, or network architecture often correlate with tissue injury, barrier dysfunction, and disease severity, suggesting that IF remodeling signatures could inform risk stratification and treatment monitoring [126–129].
Despite rapid progress, several key issues remain unresolved. Knowledge of non-vimentin IFs, especially keratins, remains limited; most findings still rely heavily on in vitro systems; and the interplay among different IF proteins during infection is poorly understood. Future studies should therefore move beyond descriptive cataloging and focus more on shared mechanisms, virus-specific differences, and coordinated regulation within IF networks in vivo.
Supplementary Material
Table S1.docx
Table S3.docx
Table S4.docx
Table S2.docx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cvirkaite-Krupovic C-LR V, Tavares P. Virus evolution toward limited dependence on nonessential functions of the host: the case of bacteriophage SPP 1. J Virol. 2015;89(5):2875–15. doi: 10.1128/jvi.03540-1425540376 PMC 4325756 · doi ↗ · pubmed ↗
- 2Radtke K, Döhner K, Sodeik B. Viral interactions with the cytoskeleton: a hitchhiker’s guide to the cell. Cell Microbiol. 2006;8(3):387–400. doi: 10.1111/j.1462-5822.2005.00679.x 16469052 · doi ↗ · pubmed ↗
- 3Zhang Y, Wen Z, Shi X, et al. The diverse roles and dynamic rearrangement of vimentin during viral infection. J Cell Sci. 2020;134(5):jcs 250597. doi: 10.1242/jcs.25059733154171 · doi ↗ · pubmed ↗
- 4Herrmann H, Aebi U. Intermediate filaments: structure and assembly. Cold Spring Harbor Perspectives in Biology. Cold Spring Harb Perspect Biol. 2016;8(11):a 018242. doi: 10.1101/cshperspect.a 01824227803112 PMC 5088526 · doi ↗ · pubmed ↗
- 5Etienne-Manneville S. Cytoplasmic intermediate filaments in cell biology. Annual review of cell and developmental biology. Annu Rev Cell Dev Biol. 2018;34(1):1–28. doi: 10.1146/annurev-cellbio-100617-06253430059630 · doi ↗ · pubmed ↗
- 6Doganyigit Z, Eroglu E, Okan A. Intermediate filament proteins are reliable immunohistological biomarkers to help diagnose multiple tissue-specific diseases. Anat Histol Embryol. 2023;52(5):655–672. doi: 10.1111/ahe.1293737329162 · doi ↗ · pubmed ↗
- 7Eriksson JE, Dechat T, Grin B, et al. Introducing intermediate filaments: from discovery to disease. J Clin Invest. 2009;119(7):1763–1771. doi: 10.1172/jci 3833919587451 PMC 2701876 · doi ↗ · pubmed ↗
- 8Robert A, Hookway C, Gelfand VI. Intermediate filament dynamics: what we can see now and why it matters. Bioessays. 2016;38(3):232–243. doi: 10.1002/bies.20150014226763143 PMC 4772765 · doi ↗ · pubmed ↗