Ultrafast Electron Dynamics of a Ferrocene-Based Butadiyne-Bridged Complex
Kasun C. Mendis, Jesús Valdiviezo, Susannah D. Cox, Peng Zhang, Xiao Li, Tong Ren, David N. Beratan, Igor V. Rubtsov

TL;DR
This paper studies how electrons move in a complex molecule with a ferrocene donor and naphthalimide acceptor, revealing how structural flexibility affects electron transfer dynamics.
Contribution
The study reveals the role of diabatic state coupling and torsional flexibility in controlling ultrafast electron transfer in ferrocene-based donor-bridge-acceptor systems.
Findings
Femtosecond TA measurements identified three relaxation times (0.3–0.5 ps, ∼2.6 ps, and 17–20 ps) in Fc-C4-NAP.
TD-DFT computations showed strong coupling (200–500 cm–1) between acceptor and ferrocene states.
Torsional angle between NAP and cyclopentadienyl ring strongly influences state mixing and energy transfer.
Abstract
Photoinduced electron transfer (ET) in alkyne-linked donor–bridge–acceptor (DBA) compounds is strongly influenced by torsional flexibility, allowing control over ET without altering the donor–acceptor distance. Here, we investigate excited-state dynamics in Fc-C4-NAP, a DBA compound featuring a ferrocene (Fc) donor, a butadiyne bridge (C4), and a 1,8-naphthalimide (NAP) acceptor. Unlike analogues DBA compounds with fully organic planar donors, Fc-C4-NAP exhibits a complex excited-state manifold. Femtosecond transient absorption (TA) measurements in the visible and mid-IR regions found three characteristic relaxation times (0.3–0.5 ps, ∼2.6 ps, and 17–20 ps) following its excitation at 402 nm, which prepares NAP-centered excited states.TD-DFT computations indicate that the acceptor-based locally excited (LE) and the charge separated (CS) diabatic states are well coupled to the Fc states…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1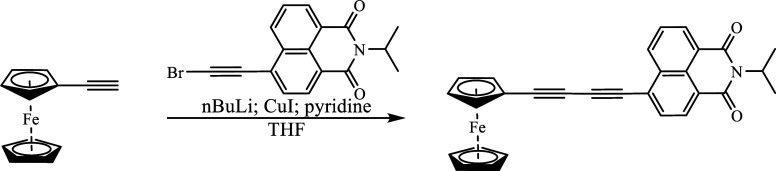 1
1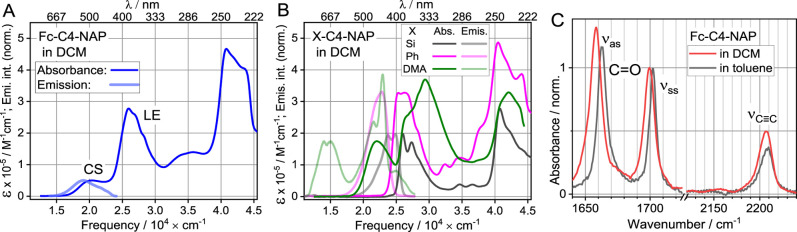 2
2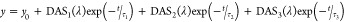 3
3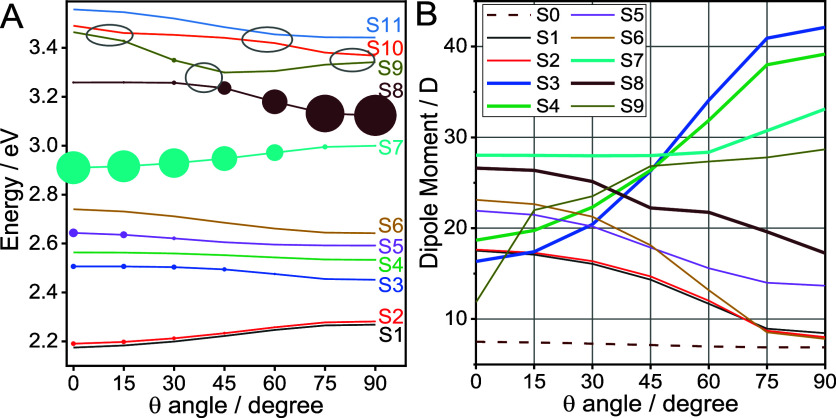 4
4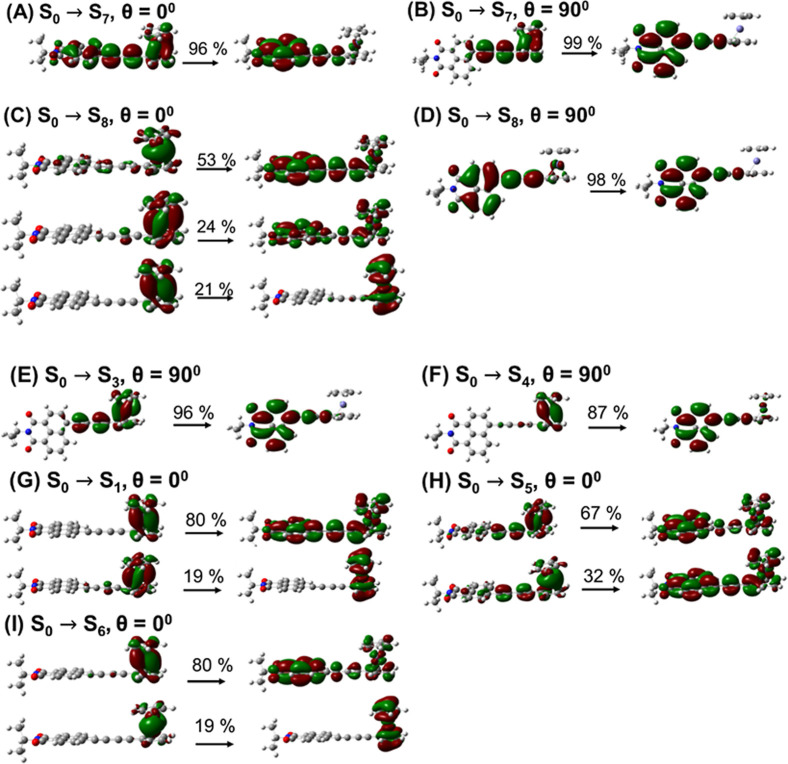 5
5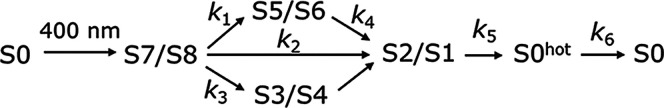 6
6- —Division of Chemistry10.13039/100000165
- —Division of Chemistry10.13039/100000165
- —Division of Chemistry10.13039/100000165
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and characterization of novel inorganic/organometallic compounds · Synthesis and Properties of Aromatic Compounds · Molecular Junctions and Nanostructures
Introduction
1
Photoinduced electron transfer (ET) in donor-bridge-acceptor (DBA) compounds plays a central role in biological and synthetic photophysical systems. DBA compounds with conjugated bridges are particularly attractive because these bridges enhance the coupling between local electronic states, thus accelerating ET. As a result, DBAs with conjugated bridges have found wide use in applications including light-harvesting chromophores, ?−? ? optical limiting, and nonlinear absorption. ?−? ?
A variety of conjugated DBA bridges have been utilized for DBA constructs, which result in D–π–A complexes,? including alkenes, ?−? ? ? ? ? stilbenes,? phenylenes, ?−? ? alkynes, ?−? ? ? ? ? ? and motifs with more complex π structures. ?−? ? ? ? ? Alkyne bridges feature linearity, rigidity, and compactness. These structures also allow torsional motion between the donor (D) and acceptor (A)planes (the angle θ). Since the energy barrier for torsional motion is often small, a wide range of θ-conformers is typically accessed.? The torsion angle can strongly influence bridge-mediated couplings between D and A, ?−? ? thus providing an opportunity to influence the rates of charge separation (CS) and charge recombination (CR) as well as the outcome of optical excitation. ?−? ? ?
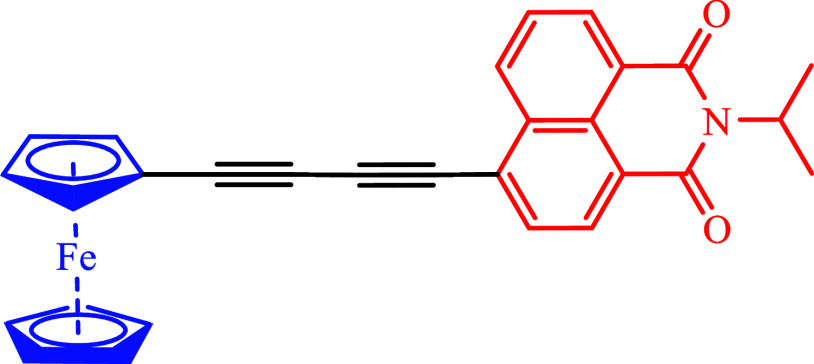
In some θ-conformers, for example, strong coupling between the CS state and a D- or A-localized state may strongly reduce the CS character of the lowest-energy state.? Elimination of strong coupling can be achieved by increasing the energy gap between the diabatic CS and local-excited (LE) states by using a more strongly reducing electron donor, a more strongly oxidizing electron acceptor, or a more polar solvent, thus lowering the diabatic CS state energy and producing the CS state with nearly complete charge separation.? Here, we studied the influence of a strong electron donor, ferrocene (Fc), on the excited state dynamics in the Fc-C4-NAP compound, where C4 is a butadiyne bridge, and NAP is N-isopropyl-1,8-napthalimide acceptor (Figure). We compare our findings with the results of our recent studies of DMA-C4-NAP with a dimethyl aniline (DMA) donor. The DMA donor studies showed a fast CS (0.63 ps) in the strongly coupled D–A conformers (θ∼15–45°) and a slow ET (4.3 ps) in the weakly coupled D–A conformers (θ∼0–15°, and 63–90°).?
Structure of Fc-C4-NAP.
Electrochemical studies of Yuan and co-workers found that the oxidation potential for Fc/Fc^+^ in DCM of 0.46 V (measured with respect to Ag/AgCl) varies only slightly when an alkyne bridge is attached to Fc, reaching a potential of 0.5–0.6 V.? Similarly, the oxidation potential of DMA (0.88–0.96 V) also increases slightly when it is attached to an alkyne moiety, reaching 1.0–1.1 V.? The lower oxidation potential of Fc indicates that it is a stronger donor than DMA. In addition to being a stronger donor, the sandwich structure of Fc? establishes a lower energy barrier for tortional θ motion. ?,? Furthermore, ferrocene-based DBA complexes are appealing because they may be suitable for future applications associated with their excellent electrochemical performance? in redox probes, ?,? surface electrochemical applications, ?,? molecular electronics, ?,? stimuli-responsive molecular systems, ?,? and solar cells. ?,?
We combine steady-state UV–vis, FTIR, and ultrafast UV/vis and UV/mid-IR transient absorption (TA) spectroscopies with TD-DFT analysis, to investigate the photoinduced dynamics of Fc-C4-NAP (Figure) in DCM. Despite the simplicity of the compound’s UV–vis spectrum, our findings reveal complex excited-state dynamics involving both charge separation and energy transfer pathways. Notably, the coupling between the low-lying Fc states, primarily associated with the d orbitals of Fe, and the LE and CS states produces complex state mixing. The low energy barrier for torsional motion allows access to a broad range of torsional angles and coupling of torsional motion to electron and energy transfer processes, enriching the light-driven dynamics.
Synthesis
1.1
The Cadiot–Chodkiewicz coupling reaction? between 4-bromo-ethynyl-N-isopropyl-1,8-naphthalimide (BrC_2_NAP^iPr^) and ferrocene substituted ethyne (Scheme) was used to synthesize FcC4NAP. UV–vis, FTIR and ^1^H NMR spectroscopic techniques were used for the characterization. More details of synthesis and characterization can be found in the Supporting Information (Section S1).
Synthesis of Fc-C4-NAPiPr, Denoted as Fc-C4-NAP
Time-Resolved Measurements
1.2
An in-house built apparatus was used for the transient UV–vis pump and Vis or m-IR probe measurements, as described recently. ?,? The fundamental beam at 804 nm is generated by a Ti:Sapphire fs oscillator (Vitesse, Coherent Inc.) and regenerative amplifier (Spitfire, Spectra Physics), with a 44 fs duration at a 1 kHz repetition rate. UV pulses at 402 nm of 1–1.5 μJ were generated via a frequency doubling process in a 1 mm BBO crystal; this light source provided electronic excitation of the compound. The mid-IR pulses, tunable from 1000 to 4000 cm^–1^, were generated by an optical parametric amplifier and difference-frequency generation unit featuring pulse energies of ca.1 μJ and spectral width of ca. 200 cm^–1^. The white light continuum, serving as a probe beam in the visible and near-IR regions, was generated by a portion of the fundamental beam (ca.7 μJ per pulse) in a c-cut sapphire wafer. Transient spectra in the visible and near-IR regions were measured with a CCD camera (PIXIS-100, Princeton Instrument) mounted to a monochromator (TRIAX-190, Horiba), while the mid-IR spectra were measured with a single-channel MCT detector (Infrared Associates). The sample was placed in a flow cell with 100 μm optical path length and 2 mm-thick CaF_2_ windows. The measurements were performed at room temperature (22 ± 1 °C).
Computational Details
1.3
DFT and TD-DFT calculations for Fc-C4-NAP were performed at the B3LYP/Def2-SVP level of theory as implemented in Gaussian 16 (Rev C.01). The polarizable continuum model (PCM) was employed to simulate the solvent effects of dichloromethane. Computations at several fixed torsion angles of 0°, 15°, 30°, 60°, 75°, and 90° between the donor (Fc) and the acceptor (NAP) were performed to track θ dependences of the spectroscopic parameters and couplings. The CAM-B3LYP, wB97X-D, and MN15 functionals were tested as well, finding B3LYP optimal for reproducing spectroscopic signals. B3LYP has been successfully used to reproduce absorption spectra of ferrocene compounds. ?−? ?
Results and Discussion
2
Linear
Spectroscopy Results
2.1
UV–Vis Absorption
and Emission Spectra
2.1.1
The UV–vis absorption spectrum of Fc-C4-NAP in DCM (FigureA) has two lowest energy peaks that are similar to those in the absorption spectrum of DMA-C4-NAP (FigureB). The peak at 385 nm is the characteristic lowest energy π–π absorption of the NAP moiety; it appears at 385 nm in DMA-C4-NAP and Si–C4–NAP and at 395 nm in Ph–C4-NAP (FigureB).? This state is referred to as an NAP-based local excited (LE) state. Higher energy peaks at 250 and 282 nm are also assigned to the NAP-based states.
Linear absorption and emission spectra of (A) Fc-C4-NAP in DCM and (B) Si–C4–NAP, Ph–C4-NAP, and DMA-C4-NAP (λexc = 340 nm). (C). Solvent-subtracted FTIR absorption spectra of Fc0C4-NAP in DCM and in toluene.
The broad peak at ca. 500 nm (20,000 cm^–1^) is assigned to the charge-separated (CS) state (FigureA). Such peak was not observed in the spectra of the individual components, Fc and NAP, and in the spectra of Ph–C4-NAP and Si–C4–NAP but was observed in DMA-C4-NAP at 455 nm (22,000 cm^–1^), FigureB. Notice that the CS peak in Fc-C4-NAP is ca. 2000 cm^–1^ lower than that in DMA-C4-NAP, as expected due to stronger donor properties of Fc compared to DMA. ?,?
The emission spectrum of Fc-C4-NAP (FigureA, light blue line) matches closely the CS state spectrum, featuring a small Stokes shift of ca. 1000 cm^–1^. Such a small Stokes shift for a highly polarized CS state in a sufficiently polar solvent (DCM) is unusual and suggests that the CS state is short-lived, not offering much time for the state solvation and internal reorganization processes. Note that the Stokes shift for the CS state for DMA-C4-NAP in DCM is ca. 8000 cm^–1^ (FigureB).?
FTIR Spectra
2.1.2
FigureC shows selected regions of the FTIR spectrum of Fc-C4-NAP in DCM and in toluene. The symmetric CC stretching mode at ca. 2207 cm^–1^, denoted as ν_CC_, is much stronger than the antisymmetric CC stretching mode locates at ca. 2155 cm^–1^. The carbonyl region of the spectrum shows symmetric (∼1700 cm^–1^, ν_ss_) and antisymmetric (∼1660 cm^–1^, ν_as_) carbonyl stretching modes of NAP. ?,? There is a considerable solvent-dependent shift for the CO peaks (∼5 cm^–1^ for ν_as_), which indicates significant polarization of the carbonyl bonds in the ground electronic state (GS). The solvent-dependent peak shift of ν_CC_ is also considerable at ca. 2 cm^–1^ (FigureC), indicating a polarization of the whole molecule in the GS.
Transient Absorption Measurements
2.2
UV/Vis TA Results
2.2.1
Transient UV/vis absorption spectra at selected probe delay times, t, following the 402 nm excitation, are shown in FigureA. The 402 nm pulses excite predominantly the NAP-based LE state. The transient spectrum at short delay time (t = 0.2 ps) has the strongest amplitude across the visible region, peaking at ca. 475 nm. The spectrum decays rapidly at all wavelengths, except for the region of 455–520 nm, where the decay is significantly slower. We performed a decay-associated spectral analysis by fitting the data globally with a multiexponential function (Figure caption). Three exponential components were required to fit the data, resulting in three characteristic times of τ_1_ = 0.5 ± 0.1 ps, τ_2_ = 2.6 ± 0.1 ps, and τ_3_ = 17 ± 0.5 ps. The amplitudes associated with each of the three components, DAS_1_(λ), DAS_2_(λ), and DAS_3_(λ), are shown in FigureB. Notice that the slowest component of ca. 17 ps results in a complete GS recovery within ca. 50 ps, likely indicating the presence of rapid energy transfer (EnT) processes, which were reported previously for Fc-containing compounds.? The 17 ps decay time matches a typical vibrational cooling time of medium-size organic molecules to a non-hydrogen bonding solvent, ?−? ? ? but can also be caused by a relaxation of an electronically excited state to the GS. In other words, the state that decays with the 17 ps characteristic time could be an electronically excited state or a vibrationally hot GS. Transient UV/mid-IR spectroscopy unequivocally shows that this state is an electronically excited state (vide infra). The fastest decay component of 0.5 ps corresponds to a decay across the whole spectral region (DAS_1_ in FigureB). The DAS_2_, associated with the characteristic time of 2.6 ps, shows a differential shape that could be due to solvation and/or vibronic relaxation processes. It can also be due to the formation of a new state, including the CS state.
Transient spectra in the visible (A) and mid-IR (C, D) regions for Fc-C4-NAP in DCM measured at indicated time delays following 402 nm excitation. Panel C also shows a scaled linear absorption spectrum (FTIR). The results of the global analysis are shown in panels B (UV/vis) and E (UV/m-IR). A three-exponential function was used for the global analysis of the UV/vis data in the form of y=y0+DAS1(λ)exp(−tτ1)+DAS2(λ)exp(−tτ2)+DAS3(λ)exp(−tτ3) . A two-exponential function was used to fit the UV/mid-IR data in the whole region from 1400 to 2300 cm–1 (E) and a three-exponential function was used for the narrow spectral region around 2200 cm–1, as shown in panel F. The resulting characteristic times are shown in panels B, E, and F as insets.
UV/mid-IR
Results
2.2.2
Transient UV/mid-IR spectra measured over a broad range of frequencies from 1400 to 2250 cm^–1^ (FigureC) provide insight for assigning intermediate states. Surprisingly, the transient spectra measured at small delay times (0.2 and 0.6 ps) feature unusually broad absorption, spanning the entire detection region (FigureC). Such broad absorption cannot be caused by vibronic transitions, but rather is assigned to an electronic transition or several electronic transitions, similar to related findings in the literature.? Note that no corresponding broad absorption was found in DMA-C4-NAP in DCM,? suggesting involvement of the Fc states of Fc-C4-NAP. Featureless, broad mid-IR absorption bands have been previously reported for oxidized Pt(II) acetylide complexes, attributed to acetylide-centered oxidation. In these systems, the CC ESA spans ∼1800–2000 cm^–1^ (fwhm ∼150 cm^–1^) with amplitudes smaller than those of carbonyl ESA.? In contrast, in Fc–C4–NAP, the absorbance is significantly broader (1400–2300 cm^–1^) and comparable in amplitude to that of carbonyl ESA, indicating its electronic origin.
As the broad absorption is peaking at t = 0, the state from which the broad absorption originates is the bright NAP-based LE state. The broad absorption decays rapidly, with a time constant of ca. 0.25 ps (FigureC, E), indicating that the NAP-based LE state is short-lived. Surprisingly, spatially separated NAP-centered and Fc-centered states are coupled so well to result in such fast relaxation; TD-DFT analysis sheds light on the origin of these observations (see Section 3.3).
The global fit of the UV/mid-IR transient spectra involving the whole frequency range resulted in only two different DAS components, shown in FigureE. A three-exponential global analysis performed for the whole frequency range produced DAS components where two components are superimposable while differing by a sign and carrying essentially the same characteristic times, indicating that they are not independent. Note that the spectral changes in the 2200 cm^–1^ spectral region (FigureD) are clearly more complex than can be described by two exponential components, showing continuous blue shift of ESA peak apparent for t ≥ 5 ps, which results in a shift of the zero-crossing point. Such changes, however, are too small in amplitude to be identified in the global fit of the overall data set. To circumvent the difficulty, we performed a three-component global fit for the narrower spectral region, shown in FigureF.
The characteristic times obtained in the three-component fit are similar to those obtained for the global fit of the TA spectra in the visible (FigureA,B). The DAS spectrum and the characteristic time of the fast component are essentially the same as for the wide fit of FigureE. The third component has a similar shape to the slow component of FigureE with negative GSB and positive ESA parts. Such a shape of the DAS reports on the decay to zero of the whole spectrum as the positive portion of DAS_3_ matches the positive TA signal and thus reporting on its decay, while the negative portion of DAS_3_ matches the negative TA signal, also reporting on its decay to zero. Again, the DAS_3_ component reports on the disappearance of the TA signals with ca. 22 ps characteristic time. The middle component describes a shift of the transient spectrum to higher frequencies as its negative and positive parts both report an increase of the TA amplitudes and the increase is to the blue with respect to the features at earlier times. The characteristic time of DAS_2_ is ca. 5 ps, which seems to be too short for vibrational cooling (∼15 ps is expected) and could involve electronic relaxation as well as vibrational cooling. Because the characteristic time of the DAS_2_ component is rather short and there is a similar component in UV/vis spectral changes (FigureB), we assign it to electronic relaxation.
The ESA of ν_CC_ peaks at 2185 cm^–1^ (FigureF, red line), which is shifted by 22 cm^–1^ from that in the GS. So large frequency shift of the relatively long-lived state (ca. 20 ps) clearly indicates that the state is an electronically excited state, not a hot GS. In fact, formation of the hot GS is apparent at larger delays, most clearly seen in the spectra at t of 20 and 50 ps (FigureD), characterized by a small (<3 cm^–1^) ν_CC_ frequency red shift caused by small vibrational anharmonicities of ν_CC_ and other modes in the compound. ?−? ? This shift has not been captured by the three-component global fit, likely because of the small amplitudes of the changes and significant noise. A similarity of the characteristic time of electronic relaxation and vibrational relaxation makes the separation of different signal contributions more difficult.
While this long-lived state is clearly an electronic state, the ν_CC_ frequency shift of ca. 22 cm^–1^ is too small to assign the state as the CS state. A red shift of ca. 150 cm^–1^ was observed for ν_CC_ in DMA-C4-NAP where a nearly pure CS state is formed. ?,?,?−? ? ? Such strongly red-shifted absorption of ν_CC_ was not observed in Fc-C4-NAP (FigureE, red line). In addition, the IR intensity of its ν_CC_ ESA peak is only 1.3 times greater than that of the GS, compared to the 10-fold increase for the CS state in DMA-C4-NAP.? The frequency shifts of the carbonyl modes (ν_ss_ and ν_as_) in the long-lived (∼20 ps) state are small (<3 cm^–1^, FigureC, E, F), which suggests that the long-lived state can have only a small contribution derived from the NAP moiety, thus indicating that it is predominantly Fc-based. TA measurements were also performed in toluene solvent. The characteristic times were found to be only slightly slower than those in DCM, caused by the lower polarity of toluene (see Section S7).
The experimental linear and nonlinear absorption data provide limiting insight for important questions regarding the nature of the peak at 500 nm in the linear absorption spectrum, the broad transient absorption in mid-IR, or the nature of the long-lived excited state with the 17–20 ps lifetime. The data also do not provide insight into how the states vary with the torsion angle, θ. TD-DFT analysis reveals a complex but trackable state pattern as a function of θ.
TD-DFT Computations
2.3
TD-DFT analysis was performed to characterize how the energies, dipole moments, and oscillator strengths of the 12 lowest-energy electronic states depend on θ. The energy difference for θ rotation in the GS was computed to be small, ca. 10 meV, which makes all θ angles thermally accessible at room temperature (Figure S3).
FigureA shows how the energies of the lowest 11 excited states depend on θ. The diameter of the circles in FigureA is proportional to the oscillator strength; Figure S16 shows the computed values of the oscillator strength directly. The lowest energy bright state of Fc-C4-NAP is expected to be the NAP-based LE state, observed at ca. 385 nm (FigureA). A large oscillator strength is found for state S7 at small θ angles and for state S8 at large θ angles (FigureA). Such complementary behavior as a function of θ can be interpreted as the coupling of two or more site states, producing their θ-dependent mixing. Because of the presence of orbitals that are fully delocalized across the compound (FigureA), the coupling pattern is complex for the S7 and S8 states and for many other states. Natural transition orbitals (NTO) show this complexity clearly (Figures S4–S14). For example, at small θ angles, state S7 is mostly NAP-based, although it involves the C4 bridge and partially involves Fc. As such, the S7 state has some charge transfer character, although its dipole moment is not very high at ca. 26D (FigureB). The state character at large θ values changes, and the state becomes essentially the CS state (FigureB), confirmed by its increased dipole moment of 33D compared to its dipole moment at small θ. Similar changes of the state character are found for S8 (FigureC, D), which at small θ angles has a large Fc → NAP CS contribution (53 + 24 = 77%, FigureC) as well as a Fc → Fc contribution of 21%. However, at large θ angles, the state becomes predominantly NAP-based, manifested in its small dipole moment (FigureB) and large oscillator strength (FigureA). Thus, the orbital delocalization results in efficient θ-dependent mixing of different localized states, so that the resulting state character changes drastically as a function of θ.
TD-DFT computed excited state energies (A) and permanent dipole moments (B) as a function of torsion angle, θ. The symbol size (diameter) in panel A represents the oscillator strength of the transition from the ground state.
Natural transition orbitals (NTO) for the transitions to selected excited states from the ground state at selected θ angles (see more data in Figures S4–S14). The numbers above arrows indicate the contributions to the transition.
A CS state can be identified by its large dipole moment (FigureB). Such states at low energies are S3 and S4, featuring large dipole moments at θ = 90° of 43 and 39 D, respectively (FigureB), which corresponds approximately to a charge-separated state with a hole on Fc and an electron on NAP, FiguresE,F, S5 and S6. Interestingly, there are two such states, supported by two orthogonally polarized orbitals of Fc (FigureG) with similar energies. State pairs S1, S2 and S5, S6 also feature orthogonally polarized hole states of Fc (see FigureH,I for states S5 and S6).
The TD-DFT analysis finds that the lowest energy excited states, S1 and S2, have very small oscillator strengths for all torsion angles (θ), and are localized primarily on the Fc moiety, associated with d-orbitals of Fe. Nevertheless, at small θ angles both states have Fc → NAP contributions (FigureG), which leads to higher dipole moments of ca. 17D, compared to 8D at θ = 90°, where such contributions were not found. The diabatic states’ coupling and mixing are apparent at higher energies as avoided crossings (S8–S11), labeled in FigureA with ovals. Oscillator strength sharing, as for example observed among states S8 and S9 at θ = 30°, is a clear indication of such coupling.
The TD-DFT computed linear absorption spectrum is shown in Figure S15. It describes fairly the low-frequency peak amplitudes, although the frequency of the NAP-based LE state was predicted to occur at a lower energy than that observed experimentally, likely caused by an overemphasis of orbital delocalization in the calculations.
The TD-DFT analysis describes the origin of the main experimental observations. The broad absorption in the 1400–2400 cm^–1^ spectral region (FigureC) is explained by the transition from states S7 and S8 to the states S9–S11. States S7–S8 and S9–S11 have components localized on the same moieties (Figures S11–S13), which increases the oscillator strength for these transitions. The light-induced transition from S7 to S8 may also account for some of the broad absorption in Fc-C4-NAP.
Figure summarizes the processes that occur upon excitation of the S7/S8 bright states. Electronic relaxation to the S3/S4 states, which can be thought as two CS states, is not clearly observed as the states formed feature very small shifts of the ν_CC_ peak frequency compared to that in the GS. Therefore, we conclude that k 3 ≪ k 1 + k 2. The S7/S8 state relaxation can populate S5/S6 and/or S1/S2 states. The fast decay of the broad absorption is assigned to these two processes together: k 1 + k 2 = (0.25 ps)^−1^. The observation of the mixed characters of states S7 and S8 makes it unsurprising that their relaxation rate is so fast. The existing data do not allow us to determine the relative contributions of these relaxation channels (k 1 vs k 2). The middle component observed in the UV/vis and UV/m-IR data, that is about 3–5 ps, can be assigned to either k 1, k 2, or k 4. We concluded that this component is due to electronic relaxation or vibronic cooling rather than due to vibrational cooling. The vibrational cooling time is expected to be about 15 ps (k 6), which approximately matches the electronic relaxation component of ca. 20 ps, attributed to S2/S1 → S0^hot^ relaxation (k 5). This rate match results in a stronger influence of the two processes on one another, leading to an increased error in determining k 5, the relaxation rate of the lowest energy excited state of Fc-C4-NAP to the GS.
Summary of the processes occurring upon Fc-C4-NAP excitation by 402 nm pulses.
Interestingly, Fc-C4-NAP exhibits an additional higher-energy charge-separated state at ca. 330 nm. This state involves a transition from a different hole state of Fc, as revealed by the TD-DFT analysis.
The presence of Fe atom can potentially result in intersystem crossing (ISC) as fast as a few hundred femtoseconds, populating triplet excited states. Triplet state formation often results in longer lived ligand-localized states, as reported for Fe(II) complexes, ?−? ? Pt(II) donor–acceptor systems, ?−? ? and cobalt(III) cyclam complexes.? Notice, however, that relaxation of the initially excited NAP-based singlet state is significantly faster than a few hundred femtoseconds. There is no indication that an NAP-based triplet state is formed as a significant frequency shifts of ν_ss_ and ν_as_, expected in this case, ?−? ? were not observed with actual shifts at less than 3 cm^–1^. The lower energy Fc-centered states can undergo fast ISC. However, we do not have any evidence of such process, and the final excited states (S1/S2) remain short-lived (20 ps), suggesting that ISC may not play essential role in Fc-C4-NAP.
Concluding Remarks
3
The extent of charge separation in Fc-C4-NAP is influenced by the coupling of the pure LE and CS states, which was also observed in other DBA compounds, such as DMA-C4-NAP. Strong coupling, which occurs when the two pure states are in resonance, produces their strong mixing, leading to incomplete charge separation in each of the eigenstates. By tuning the energy of the pure CS state away from the LE state, such state mixing can be avoided, resulting in essentially pure CS eigenstates. The opportunity of manipulating the mixing motivated our selection of the electron donor, Fc, which is stronger than DMA, thus decreasing the energy of the pure CS state and ensuring, as expected, a higher polarization of the CS states. The Fc-C4-NAP compound, however, demonstrated complexity in the nature of its excited states, which was dictated by the presence of several dark excited states associated with the Fc moiety in the vicinity of the pure CS and LE states. Surprisingly, these Fc-localized states are found to be coupled strongly to both pure CS and LE states. The large coupling, estimated to be 200–500 cm^–1^ for different states pairs, and high density of Fc-based pure states, resulted in several strong coupling situations involving pure CS and LE states, occurring at different torsion angles (FigureA). The energies of pure LE and CS states vary significantly with the torsion angle, as for DMA-C4-NAP,? while the energies of the Fc-based states do not. As a result, resonances with strong coupling cases occur at different θ values, linking pure LE and CS states to several pure Fc-based states. Some other pure Fc-based states, such as S1 and S2, are coupled weakly to the pure LE and CS states, nevertheless, leading to their wave function mixing, although the mixing is much smaller than in the case of S9–S11. Such mixing, strong and weak, results in fast energy relaxation from the bright, predominantly LE states, to the lowest energy excited states of Fc (S1 and S2), passing the CS eigenstates (S3–S4). The lifetime of the S1–S2 states is found to be ca. 20 ps.
The reason for the mixing of various diabatic states lies in the strength of their electronic coupling, which is mediated by the π-conjugated alkyne bridge and the high density of low-lying excited states associated with the Fc moiety. Such conditions were avoided in earlier DBA triads involving a Fc donor because of the use of saturated bridges, which greatly reduce the through-bond coupling, thus minimizing electronic communication between the donor and acceptor. The reduced communication results in slower charge separation but also avoids state mixing. For example, Albinsson and co-workers reported a diporphyrin system with a ferrocene donor linked via a nonconjugated bridge, where, despite the presence of low-lying Fc-based states, the charge-separated (CS) state was long-lived and exhibited little evidence of state mixing, suggesting weak coupling between the local and CS states.? Similarly, Fukuzumi and colleagues studied Fc–Ph–C_60_ triads with alkyl linkers and observed efficient photoinduced electron transfer followed by formation of long-lived CS states, again consistent with weak donor–acceptor coupling arising from spatial separation.? In another study, Imahori et al. found that in Fc–oligophenylene–C_60_ systems, improved conjugation slightly enhanced electronic communication, but state mixing remained minimal.? Taken together, these examples show how the combination of a conjugated bridge and the electronic structure of the Fc donor in our system creates the conditions needed to couple diabatic excited states. Fast energy relaxation between excited ZnTPP and Fc directly tethered to one of the phenyl moieties of ZnTPP was reported by Wasielewski and co-workers, indicating dominance of the energy transfer to low-laying Fc states compared to the CS process.?
Experimental observations of this study, combined with TD-DFT analysis, offer a clear picture of the excited-state characters and dynamics in Fc-C4-NAP. Our findings demonstrate how the structural flexibility offered by the alkyne bridge and Fc-based dark diabatic excited states produce a distinctly different excited states compared to DBA species with simpler, planar, fully organic donor moieties. The findings underscore the utility of incorporating both electronic and conformational factors to understand and to control the photoinduced electron transfer in donor–bridge–acceptor systems. The insights gained provide guidance toward formulating design principles for ultrafast charge separation modules of utility in molecular materials for solar energy, photocatalysis, and optoelectronic applications.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Duncan T. V.Ghoroghchian P. P.Rubtsov I. V.Hammer D. A.Therien M. J.Ultrafast excited-state dynamics of nanoscale near-infrared emissive polymersomes J. Am. Chem. Soc.2008130309773978410.1021/ja 711497 w 18611010 PMC 2737527 · doi ↗ · pubmed ↗
- 2Shan B.Nayak A.Williams O. F.Yost D. C.Polizzi N. F.Liu Y.Zhou N.Kanai Y.Moran A. M.Therien M. J.Excitation energy-dependent photocurrent switching in a single-molecule photodiode Proc. Natl. Acad. Sci. U. S. A.201911633161981620310.1073/pnas.190711811631366631 PMC 6697812 · doi ↗ · pubmed ↗
- 3Shi Y.Frattarelli D.Watanabe N.Facchetti A.Cariati E.Righetto S.Tordin E.Zuccaccia C.Macchioni A.Wegener S. L.Ultra-high-response, multiply twisted electro-optic chromophores: influence of π-system elongation and interplanar torsion on hyperpolarizability J. Am. Chem. Soc.201513739125211253810.1021/jacs.5b 0463626360110 · doi ↗ · pubmed ↗
- 4Bai Y.Rawson J.Roget S. A.Olivier J.-H.Lin J.Zhang P.Beratan D. N.Therien M. J.Controlling the excited-state dynamics of low band gap, near-infrared absorbers via proquinoidal unit electronic structural modulation Chem. Sci.2017895889590110.1039/C 7SC 02150 J 28989620 PMC 5619129 · doi ↗ · pubmed ↗
- 5Fenenko L.Shao G.Orita A.Yahiro M.Otera J.Svechnikov S.Adachi C.Electrical properties of 1, 4-bis (4-(phenylethynyl) phenylethynyl) benzene and its application for organic light emitting diodes Chem. Commun.2007222278228010.1039/b 700466 d 17534516 · doi ↗ · pubmed ↗
- 6Nayak A.Park J.De Mey K.Hu X.Duncan T. V.Beratan D. N.Clays K.Therien M. J.Large hyperpolarizabilities at telecommunication-relevant wavelengths in donor–acceptor–donor nonlinear optical chromophores ACS Cent. Sci.201621295496610.1021/acscentsci.6b 0029128058285 PMC 5200929 · doi ↗ · pubmed ↗
- 7JovaišaitėJ.Baronas P.Jonusauskas G.Gudeika D.Gruodis A.Gražulevičius J. V.Juršėnas S.TICT compounds by design: comparison of two naphthalimide-π-dimethylaniline conjugates of different lengths and ground state geometries Phys. Chem. Chem. Phys.20232532411241910.1039/D 2CP 04250 A 36598166 · doi ↗ · pubmed ↗
- 8Dumur F.Gautier N.Gallego-Planas N.Şahin Y.Levillain E.Mercier N.Hudhomme P.Masino M.Girlando A.Lloveras V.Novel fused D– A dyad and A– D– A triad incorporating tetrathiafulvalene and p-benzoquinone J. Org. Chem.20046962164217710.1021/jo 035689 f 15058966 · doi ↗ · pubmed ↗