Chronic viral infections and their role in shaping the tumor immune microenvironment
Huizi Li, Xiulin Jiang, Quanan Zhang, Yihang Yuan

TL;DR
Chronic viral infections like HBV and HCV shape the tumor immune environment, promoting cancer growth and metastasis through immune suppression and altered signaling pathways.
Contribution
The paper identifies key immunoregulatory mechanisms and signaling pathways linking chronic viral infections to tumor progression and metastasis.
Findings
Chronic viruses promote tumor growth by increasing immunosuppressive cells and cytokines.
Viruses activate pathways like NF-κB and PD-1/PD-L1 to support tumor survival and metastasis.
Combining antiviral therapy with immune checkpoint inhibitors may improve outcomes in virus-related cancers.
Abstract
Chronic viral infections, such as HBV, HCV, EBV, and HPV, contribute to tumorigenesis not only through direct oncogenic effects but also by reshaping the tumor immune microenvironment (TIME) via complex immunoregulatory mechanisms. These infections enhance immune suppression and promote metastasis. Viruses induce the accumulation of regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and immunosuppressive cytokines, while driving CD8+ T cell exhaustion and impairing NK cell function, creating an immune environment favorable for tumor survival. Chronic inflammation, pro-angiogenic factors, and signals mediated by exosomes and microvesicles further remodel local and distant microenvironments, forming a “pre-metastatic niche” that supports tumor cell colonization and metastasis. Key signaling pathways, including NF-κB, STAT3, PD-1/PD-L1, and TGF-β, are persistently…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
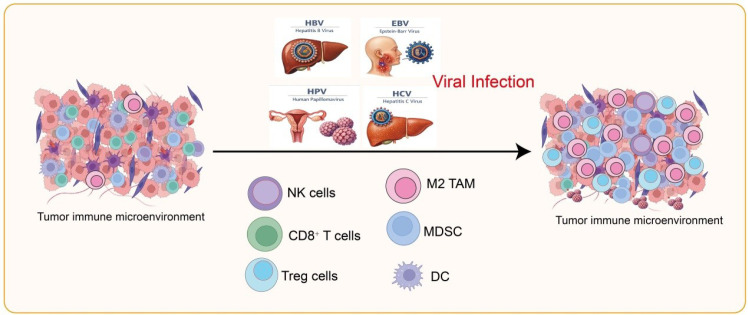 Figure 1
Figure 1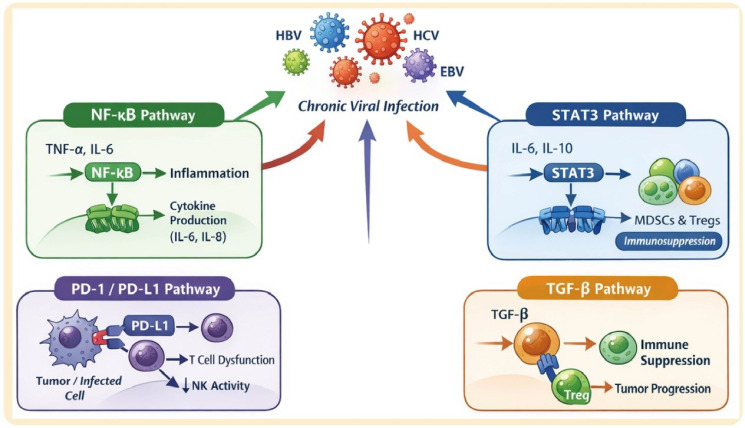 Figure 2
Figure 2| Feature/virus | HBV | HCV | HPV | EBV |
|---|---|---|---|---|
| Virus Type | DNA virus, partially double-stranded | RNA virus, single-stranded positive-sense | DNA virus, double-stranded | DNA virus, double-stranded |
| Genome Size | ~3.2 kb | ~9.6 kb | ~8 kb | ~170 kb |
| Virus Family | Hepadnaviridae | Flaviviridae | Papillomaviridae | Herpesviridae |
| Replication Site | Nucleus (via reverse transcription) | Cytoplasm | Nucleus | Nucleus |
| Transmission | Blood, sexual, perinatal | Blood, needle sharing, transfusion | Sexual, skin-to-skin contact | Saliva, blood, organ transplant |
| Target Cells | Hepatocytes | Hepatocytes | Epithelial cells (cervix, anogenital) | B cells, epithelial cells |
| Oncogenic Mechanism | Integration into host genome, chronic inflammation | Chronic inflammation, oxidative stress | E6/E7 proteins inactivate p53/Rb | Latent infection, immortalization of B cells, LMP1/EBNA proteins |
| Associated Cancers | HCC | HCC | Cervical cancer, anogenital cancers, oropharyngeal cancer | Burkitt lymphoma, Hodgkin lymphoma, nasopharyngeal carcinoma |
| Immune Cell Changes | Chronic infection: exhausted CD8+ T cells, impaired NK cells | Chronic infection: exhausted CD8+ T cells, altered NK cells, increased Tregs | Local immune evasion: reduced antigen presentation, altered Langerhans cells | Altered B cell activation, T cell exhaustion, immune evasion via latent proteins |
| Chronic Infection Risk | High (~5–10% of adults) | Moderate (~50–85% of infections become chronic) | Low (most cleared, persistent infection with high-risk types) | Low for symptomatic disease, lifelong latent infection common |
| Vaccine Availability | Yes (effective) | No (treatment available) | Yes (HPV vaccine) | No |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmune cells in cancer · Cancer Immunotherapy and Biomarkers · Extracellular vesicles in disease
Introduction
1
Chronic viral infections are a major global public health concern and significant risk factor for multiple cancers (1). Persistent infections with viruses such as hepatitis B virus (HBV), hepatitis C virus (HCV), Epstein-Barr virus (EBV), and human papillomavirus (HPV) are strongly linked to specific cancers (2). For example, HBV and HCV are closely associated with hepatocellular carcinoma (HCC), EBV is linked to nasopharyngeal carcinoma and certain lymphomas, and high-risk HPV types are major drivers of cervical and other reproductive tract cancers (3). Chronic viral infection provides a molecular basis for tumorigenesis by directly modulating host cell proliferation, inhibiting apoptosis, and inducing genomic instability (4, 5).
However, the impact of viruses on cancer development extends beyond direct cellular changes. Increasing evidence indicates that chronic viral infections significantly reshape the tumor immune microenvironment (TIME) through sustained immune stimulation and suppression (6). Persistent viral antigens drive CD8^+^ T cell exhaustion, impair natural killer (NK) cell function, and enable immune evasion, while promoting the accumulation of regulatory T cells (Tregs), Myeloid-Derived Suppressor Cells (MDSCs), and immunosuppressive cytokines (7). This creates an immune milieu conducive to tumor survival and progression. In addition, chronic viral infection can modulate immune cell infiltration and function through pro-inflammatory cytokines, exosomes, or microvesicle-mediated signaling, thereby facilitating tumor cell colonization and distant metastasis (8).
This mini-review focuses on how chronic viral infections shape the tumor microenvironment via immune regulatory mechanisms and drive tumor metastasis. We summarize viral effects on innate and adaptive immunity, dissect the molecular mechanisms of CD8^+^ (Cluster of Differentiation 8–positive) T lymphocyte cell exhaustion and immune evasion, and discuss the role of virus-driven immunosuppressive microenvironments in tumor metastasis. This analysis aims to provide insights into the immune pathogenesis of virus-associated cancers and support the development of novel therapeutic strategies targeting the virus–immune–metastasis axis.
Chronic viral infection shapes the tumor immune microenvironment
2
Chronic viral infections promote tumor development not only by directly altering host cell functions but also by reshaping the TIME through complex immunoregulatory mechanisms, creating conditions favorable for tumor growth and metastasis (9, 10). This remodeling primarily involves establishing an immunosuppressive environment, chronic inflammation with pro-tumor signaling, impaired antigen presentation and immune evasion, and regulation mediated by exosomes and microvesicles (11). HBV,HCV, HPV, and EBV are major oncogenic viruses that differ in genome type, target cells, transmission routes, and mechanisms of immune modulation, ultimately contributing to distinct virus-associated cancers (Table 1) (12). As illustrated in Figure 1, these viral infections suppress the activity of cytotoxic NK and CD8^+^ T cells while enhancing immunosuppressive populations including regulatory Tregs, M2 tumor-associated macrophages, and myeloid-derived suppressor cells, thereby reshaping the immune landscape in favor of tumor survival and growth.
Impact of viral infections on the tumor immune microenvironment. Viral infections, including HBV, HCV, HPV, and EBV, modulate immune cell composition and function within the tumor microenvironment. NK cells, CD8+ T cells, and DCs show downregulated activity, whereas Treg cells, M2-TAMs, and MDSCs are functionally upregulated.
Establishment of an immunosuppressive environment
2.1
Chronic viral infection significantly promotes the accumulation and activity of immunosuppressive cells, generating “cold” tumor microenvironments (13). Tregs and MDSCs are markedly increased in various virus-associated tumors. For instance, Tregs are elevated in HBV-related hepatocellular carcinoma, suppressing CD8^+^ T cell and NK cell cytotoxicity (14). Viral infection also induces anti-inflammatory cytokines, such as IL-10 and TGF-β, which further enhance immunosuppression by inhibiting effector T cell function and promoting the expansion of suppressive cells (15). Concurrently, CD8^+^ T cells display an exhausted phenotype, characterized by high expression of Programmed Cell Death Protein 1 (PD-1), T-cell Immunoglobulin and Mucin-Domain Containing Protein 3 (TIM-3), and Lymphocyte-Activation Gene 3 (LAG-3), while NK cell activity declines, leading to impaired antitumor immunity and facilitating immune escape (16).
Chronic inflammation and pro-tumor signaling
2.2
Chronic viral infection often induces persistent local or systemic low-grade inflammation. This prolonged inflammatory state not only causes DNA damage and increases mutation risk but also supports tumor proliferation through pro-growth factors such as vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and hepatocyte growth factor (HGF) (17). Key inflammatory signaling pathways, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and signal transducer and activator of transcription 3 (STAT3), are continuously activated in chronic infection-associated tumors, driving both pro-tumor and immunoregulatory gene expression (18). Tumor-associated inflammatory cells, such as tumor-associated macrophages (TAMs) and neutrophils, secrete chemokines including C-C motif chemokine ligand 2 (CCL2) and C-X-C motif chemokine ligand 8 (CXCL8/IL-8), which recruit immunosuppressive immune populations—such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs)—into the tumor, thereby promoting immune evasion (19). For example, Interleukin-21 (IL-21) can enhance MDSC-mediated immunosuppression, reducing liver inflammation in chronic HBV infection, suggesting that the IL-21–MDSC axis may regulate hepatic immune responses and optimize anti-inflammatory therapy (20).
Antigen presentation and immune evasion
2.3
Viral infections interfere with antigen presentation and immune recognition through multiple mechanisms. Chronic viral or tumor cells often downregulate MHC I/II expression, impairing antigen processing and presentation, and reducing CD8^+^ T cell recognition and cytotoxicity (21). For instance, EBV LMP1 disrupts antigen-processing pathways and decreases surface antigen presentation in tumor cells (22). Viruses also induce inhibitory receptors and cytokines and modulate immune checkpoint signaling, establishing immune evasion pathways that protect tumor cells from host immunity (23). Antigen-presenting cells (APCs), including macrophages, dendritic cells (DCs), Langerhans cells (LCs), and B cells, are the primary effector cells in peripheral immune microenvironments (24). EBV infects autoreactive antinuclear antigen B cells in systemic lupus erythematosus (SLE), reprogramming them into activated antigen-presenting cells that drive T peripheral helper cell activation and systemic autoimmunity. This mechanistic link provides direct evidence that EBV can promote SLE by expanding and functionally altering nuclear antigen–reactive B cells (24). In peripheral immune microenvironments, macrophages polarize into a proinflammatory M1 phenotype under VEGF-, IL-6-, and IL-8, rich conditions, supporting CD4^+^ T cell infiltration (25). Extending these principles to virus-associated tumor microenvironments, during cervical lesion progression, macrophages shift to an immunosuppressive M2 phenotype, inhibiting CD8^+^ T cell function and recruiting regulatory Tregs and MDSCs (26). In high-grade serous ovarian cancer (HGSOC) tumor cells, B7-H3 (an immune checkpoint protein with immunosuppressive functions) expression promotes tumor immune evasion via the CCL2-CCR2–M2 macrophage axis, reducing IFNγ^+^CD8^+^ T cell infiltration. Targeting B7-H3 or the CCL2-CCR2 pathway may therefore represent a promising strategy to overcome immune suppression in PD-L1–low, nonimmunoreactive HGSOC (26). Similarly, high-risk HPV reduces chemokine and E-cadherin expression in keratinocytes, limiting Langerhans cell (LC) migration to infection sites and impairing LC and DC retention within the infected epidermis, thereby weakening local antiviral responses (27).
Exosome- and microvesicle-mediated immune regulation
2.4
Exosomes and microvesicles secreted by virus-infected cells play a critical role in TIME modulation (28). These vesicles can carry viral proteins, miRNAs, or immunoregulatory molecules that directly suppress CD8^+^ T cell and NK cell activity. For example, exosomes from HBV-infected hepatocytes carrying HBx protein and miR-21 inhibit T cell proliferation and promote MDSC recruitment (29). For example, CircCCAR1 promotes HCC growth and metastasis through a circCCAR1/miR-127-5p/WTAP feedback loop and enhances PD-L1 transcription via CCAR1–β-catenin interaction. Exosomal circCCAR1 induces CD8^+^ T cell dysfunction and resistance to anti-PD-1 therapy, representing a potential target to overcome immunosuppression in HCC (29). Beyond remodeling the local tumor environment, exosomes can form pre-metastatic niches in distant tissues, altering immune cell composition and function to facilitate tumor colonization and metastasis (30). Notably, plant-derived GELN exosomes carrying osa-miR164d can reprogram macrophage polarization by targeting TAB1 and downregulating NF-κB, suppressing intestinal inflammation (31). Based on this concept, engineered biomimetic exosomes (osa-miR164d-MGELNs) effectively reprogram macrophages and alleviate colitis symptoms, representing an innovative cross-species miRNA delivery strategy (31). Similarly, circPRKD3 in glioma stem cell (GSC) exosomes binds HNRNPC in an m6A-dependent manner, accelerates IL6ST mRNA degradation, and inhibits STAT3 signaling, reprogramming tumor-associated macrophages to secrete CXCL10 and recruit CD8^+^ T cells against glioblastoma (32). Brain-targeted lipid nanoparticles delivering circPRKD3 combined with immune checkpoint blockade show strong antitumor synergy, highlighting a promising RNA-based immunotherapeutic approach for GBM (32).
Immune mechanisms of chronic viral infection–driven tumor metastasis
3
Chronic viral infections not only promote local tumor growth but also facilitate distant metastasis through complex immune regulatory mechanisms (33). By altering antitumor immunity and remodeling both the tumor microenvironment and distant tissue microenvironments, viruses enable tumor cells to evade immune surveillance and successfully colonize distant organs.
Immune escape facilitates metastasis
3.1
CD8^+^ T cells and NK cells are central effectors of antitumor immunity. In virus-associated tumors, these effector cells often display exhaustion or functional impairment. Persistent antigen stimulation and immunosuppressive factors produced by oncogenic viruses (e.g., HBV, HCV, HPV, EBV), such as viral PD-L1, IL-10, and TGF-β, reduce cytokine secretion and proliferation of CD8^+^ T cells while decreasing NK cell cytotoxicity (34). This immune escape allows circulating or distant tumor cells to evade host surveillance, increasing metastatic potential (34). For instance, CD8^+^ T cell exhaustion in HBV-related hepatocellular carcinoma correlates closely with intravascular tumor dissemination (35). Compared with uninfected samples, HPV-infected and cervical cancer tissues show significant increases in myeloid and NK/T cell populations, while epithelial cell proportions decrease (36). Functional enrichment analysis reveals activation of αβ T cells, T cell receptor signaling, and neutrophil chemotaxis, highlighting substantial immune remodeling during HPV-driven tumor progression. SPP1^+^ macrophages interact with tumor cells and exhausted T cells via the SPP1–CD44 axis, shaping an immunosuppressive microenvironment (37). Moreover, HPV16-driven cervical cancer releases IL-1 family cytokines that skew bone marrow myelopoiesis toward immunosuppressive neutrophils, dampening systemic T cell immunity. Blocking IL-1 signaling (e.g., anti-IL1RAP) inhibits this neutrophil expansion, restoring the efficacy of otherwise ineffective HPV16 E7 vaccines, which can be further enhanced by combining with anti- Cytotoxic T-Lymphocyte–Associated Protein 4 (CTLA-4) therapy (38).
Immune cell–mediated metastatic microenvironment
3.2
Chronic viral infection indirectly promotes metastasis by regulating immune cell populations. MDSCs and tumor-associated macrophages (TAMs) are expanded in virus-associated tumors and secrete pro-migratory factors, including VEGF, matrix metalloproteinases (MMPs), and chemokines (39). VEGF supports tumor angiogenesis, providing channels for tumor cells to enter circulation, while MMPs degrade the extracellular matrix, facilitating invasion and migration (40). TAMs also secrete chemokines such as CCL2 and CCL5, recruiting immune cells to the metastatic microenvironment and further enhancing tumor invasiveness (41). For example, MARCO is a key regulator of MDSC differentiation and immunosuppression in breast cancer (42). Genetic deletion or antibody-mediated inhibition of MARCO suppresses tumor growth, reduces immunosuppressive MDSCs and TAMs, and enhances CD8^+^ T cell and NK cell infiltration (42). Combining MARCO inhibition with PD-1 blockade produces synergistic antitumor effects, highlighting MARCO as a promising immunotherapy target.
Chronic inflammation and angiogenesis
3.3
Persistent low-grade inflammation induced by chronic viral infection maintains a pro-tumor microenvironment and promotes metastasis via angiogenesis (43). Continuous inflammatory signaling activates NF-κB and STAT3, inducing VEGF, IL-6, and other pro-angiogenic factors that drive new vessel formation. These vessels supply nutrients and oxygen while providing physical routes for tumor cells to enter circulation (43). HPV16 E6 upregulates hypoxia-inducible factor 1α (HIF-1α) and promotes VEGF expression, strongly enhancing angiogenesis (44). MMPs, particularly MMP-2 and MMP-9, degrade the vascular basement membrane, exposing VEGF-A receptors on endothelial cells and stimulating further angiogenesis (45). Tumor cells exploit these vascular gaps to intravasate, facilitating distant metastasis. HPV16 E6/E7 promotes HIF-1α and GLUT1 expression in lung cancer cells by downregulating the tumor suppressor RRAD, which activates NF-κB via nuclear translocation of p65. This HPV-RRAD-p65-HIF-1α-GLUT1 axis highlights RRAD as a critical regulator in HPV-related lung cancer and a potential therapeutic target (46).
Exosomes and cytokines establish pre-metastatic niches
3.4
Chronic viral infection also establishes pre-metastatic niches in distant tissues via exosomes and cytokines (47). Tumor and virus-infected cells release exosomes carrying viral proteins, miRNAs, or immunosuppressive factors, reshaping the immune landscape of distant organs (47). Exosomes can recruit MDSCs or inhibit NK cell activity, providing immune protection for circulating tumor cells to colonize and proliferate (48). Inflammatory mediators such as TNF-α and IL-6 can act remotely to modulate stromal and endothelial cell functions, optimizing the metastatic “landing” environment (48). For example, during chronic viral infections, a subset of virus-specific CD8^+^ T cells expressing Tcf1 sustains T cell responses despite terminal exhaustion, displaying memory-like features and inhibitory receptors such as PD-1 and Lag-3 (49). This population represents a key target for interventions aimed at enhancing antiviral immunity and may be influenced by viral-derived factors, including exosomes (49). In chronic viral infections, PD-1+ Tcf1+ stem-like CD8^+^ T cells are not irreversibly exhausted and can be reprogrammed by PD-1 blockade combined with IL-2 to generate highly functional effector CD8^+^ T cells (50). This synergy relies on IL-2 signaling through the high-affinity CD25 receptor and highlights a potential strategy for enhancing antiviral immunity and informing cancer immunotherapy (50).
Chronic viral infection–associated signaling pathways: NF-κB, STAT3, PD-1/PD-L1, and TGF-β
4
Chronic viral infections regulate the tumor immune microenvironment through multiple signaling pathways, promoting tumor growth and metastasis. Key pathways include NF-κB, STAT3, PD-1/PD-L1, and TGF-β. Specific viral proteins can directly modulate these pathways, enhancing pro-tumor and immunosuppressive signaling (Figure 2).
Chronic viral infection–associated signaling pathways modulating immune responses. Schematic illustration showing the key signaling pathways activated during chronic infections with oncogenic viruses such as HBV, HCV, HPV, and EBV. TNF-α and IL-6 activate the NF-κB pathway, promoting inflammation and cytokine (IL-6, IL-8) production. The STAT3 pathway, activated by IL-6 and IL-10, drives immunosuppression via the expansion of MDSCs and Tregs. PD-1/PD-L1 interactions between infected or tumor cells and T cells induce T cell dysfunction and reduce NK cell activity. TGF-β signaling promotes immune suppression and tumor progression through Treg activation.
NF-κB: chronic inflammation and pro-metastatic factors
4.1
The NF-κB pathway is persistently activated in chronic virus-associated tumors and serves as a central regulator of chronic inflammation and pro-metastatic factor expression (51). Chronic infections, including HBV, HCV, and EBV, activate IκB kinase (IKK), maintaining NF-κB activity. Activated NF-κB upregulates pro-inflammatory cytokines (e.g., TNF-α, IL-6), chemokines (e.g., CCL2, CXCL8), and pro-angiogenic factors such as VEGF (51). This sustains chronic inflammation, enhances MDSC and TAM activity, and promotes tumor invasion and metastasis. For example, EBV LMP1 mimics CD40 signaling to continuously activate NF-κB, driving expression of pro-inflammatory and pro-migratory genes (52).
STAT3: regulator of immunosuppressive cells and metastasis
4.2
STAT3 is another key node persistently activated in virus-associated tumors, linking immunosuppression and metastasis (53). Chronic viral infections activate STAT3 through pro-inflammatory cytokines such as IL-6 and IL-10, promoting Treg and MDSC proliferation and function while suppressing CD8^+^ T cell activity (53). STAT3 activation also enhances VEGF and MMP expression, facilitating angiogenesis and extracellular matrix degradation, thereby supporting metastasis (54). HBV HBx protein further promotes STAT3 activation via PI3K/Akt and JAK/STAT3 pathways, driving hepatocellular carcinoma progression.
PD-1/PD-L1: central mechanism of immune escape
4.3
The PD-1/PD-L1 immune checkpoint is a key mechanism of immune escape in virus-associated tumors (55). Chronic viral infection induces high PD-1 expression on CD8^+^ T cells, while tumor or infected cells upregulate PD-L1, inhibiting T cell proliferation and cytokine production through PD-1/PD-L1 binding (55). Persistent activation of this pathway weakens local antitumor immunity and allows circulating tumor cells to evade immune surveillance in distant organs, facilitating metastasis. In EBV-associated tumors, LMP1 upregulates PD-L1, directly contributing to immune evasion (56).
TGF-β: immunosuppression and epithelial–mesenchymal transition
4.4
TGF-β is a critical mediator of immunosuppression and metastasis in chronic virus-associated tumors (57). TGF-β promotes Treg expansion, suppresses effector T cell and NK cell function, and establishes an immunosuppressive microenvironment (58). Simultaneously, TGF-β drives EMT, enhancing tumor cell migration and invasiveness. Viral proteins such as HBV HBx and EBV LMP1 upregulate TGF-β expression or enhance downstream signaling, further promoting immune escape and metastasis (59). In pediatric EBV infection, macrophage PD-L1 expression is regulated by viral antigens, with CD163^+^ M2 macrophages correlating with latent LMP1 in primary infection and CD68^+^ M1 macrophages correlating with lytic BMRF1 in reactivation. These results suggest that EBV modulates macrophage-mediated immune exhaustion early in infection, highlighting potential targets for EBV-associated tumor therapy (60).
Potential therapeutic strategies and research prospects
5
Recent studies have proposed several intervention strategies targeting the immunosuppressive and pro-metastatic features of chronic virus-associated tumors. These approaches aim to restore antitumor immunity, block virus-mediated pro-metastatic pathways, and improve clinical outcomes. Immune checkpoint inhibitors (ICIs), such as PD-1/PD-L1 and CTLA-4 blockers, have demonstrated efficacy in various virus-associated cancers (61). For example, patients with EBV-positive nasopharyngeal carcinoma or HBV-related hepatocellular carcinoma often show higher response rates to PD-1 inhibitors than those with non-virus-associated tumors, likely due to virus-driven PD-L1 overexpression and unique immune microenvironment characteristics (62). ICIs can partially restore CD8^+^ T cell and NK cell activity, reverse immunosuppression, and inhibit tumor progression and metastasis. Viral clearance or suppression can reduce persistent antigen stimulation, alleviate CD8^+^ T cell exhaustion, and improve the immune microenvironment (63). For instance, combining antiviral therapy against HBV or HCV with immunotherapy can lower viral replication while enhancing antitumor immunity, providing a novel approach for liver cancer prevention and treatment. Such combination strategies may also reduce the risk of distant metastasis (64).
Exosomes play a pivotal role in immune regulation and pre-metastatic niche formation in virus-associated tumors. Targeting or inhibiting exosome secretion, transport, or function could serve as a novel immunotherapeutic approach (65). Inhibiting exosome release from tumor or virus-infected cells may reduce the accumulation of immunosuppressive miRNAs or proteins in the microenvironment, restore CD8^+^ T cell and NK cell activity, and suppress metastasis. Changes in the immune microenvironment of virus-associated tumors can also serve as biomarkers for early diagnosis, therapeutic response monitoring, and prognostic prediction (66). Indicators such as serum exosomal miRNAs, proportions of circulating immunosuppressive cells, and PD-L1 expression levels can dynamically reflect immune microenvironment status and guide personalized immunotherapy. Future development of high-throughput, multi-modal biomarker profiling will facilitate precise assessment of the immune status and metastatic risk in virus-associated cancers.
Conclusion
6
Chronic viral infections play a critical role in tumor initiation, immune evasion, and distant metastasis through multi-layered immunoregulatory mechanisms. Key processes include the accumulation of immunosuppressive cells, CD8^+^ T cell exhaustion, chronic inflammation, angiogenesis, and exosome-mediated local and distant immune modulation (67). Viral proteins, such as EBV LMP1 and HBV HBx, further enhance immunosuppression and pro-metastatic signaling by modulating key pathways including NF-κB, STAT3, PD-1/PD-L1, and TGF-β. Future research should focus on elucidating the molecular and cellular mechanisms of the virus–immune–metastasis axis, including key factors driving pre-metastatic niche formation. Comprehensive analysis of exosomes, immune cells, and cytokines in distant tissues will provide insights for metastasis intervention. Developing novel therapies that combine antiviral and immunotherapeutic strategies or target exosomes and immunosuppressive pathways may restore antitumor immunity and prevent metastasis. By integrating mechanistic studies with clinical translation, these approaches have the potential to provide more precise immunotherapeutic strategies for virus-associated tumors, reduce metastasis incidence, and improve patient prognosis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Volpe S Irish J Palumbo S Lee E Herbert J Ramadan I . Viral infections and chronic rhinosinusitis. J Allergy Clin Immunol. (2023) 152:819–26. doi: 10.1016/j.jaci.2023.07.018. PMID: 37574080 PMC 10592176 · doi ↗ · pubmed ↗
- 2Deigendesch N Stenzel W . Acute and chronic viral infections. Handb Clin Neurol. (2017) 145:227–43. doi: 10.1016/B 978-0-12-802395-2.00017-1, PMID: 28987172 · doi ↗ · pubmed ↗
- 3Singh G Ansari S Yadav S Aran KR . Gut microbiota's role in NAFLD- and HBV/HCV-related hepatocellular carcinoma: Mechanisms and therapeutic implications. Microb Pathogen. (2026) 211:108273. doi: 10.1016/j.micpath.2025.108273. PMID: 41475279 · doi ↗ · pubmed ↗
- 4Seo W Gao Y He Y Sun J Xu H Feng D . ALDH 2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J Hepatol. (2019) 71:1000–11. doi: 10.1016/j.jhep.2019.06.018. PMID: 31279903 PMC 6801025 · doi ↗ · pubmed ↗
- 5Nevola R Beccia D Rosato V Ruocco R Mastrocinque D Villani A . HBV infection and host interactions: The role in viral persistence and oncogenesis. Int J Mol Sci. (2023) 24:7651. doi: 10.3390/ijms 24087651. PMID: 37108816 PMC 10145402 · doi ↗ · pubmed ↗
- 6Auriti C De Rose DU Santisi A Martini L Piersigilli F Bersani I . Pregnancy and viral infections: Mechanisms of fetal damage, diagnosis and prevention of neonatal adverse outcomes from cytomegalovirus to SARS-Co V-2 and Zika virus. Biochim Biophys Acta Mol Basis Dis. (2021) 1867:166198. doi: 10.1016/j.bbadis.2021.166198. PMID: 34118406 PMC 8883330 · doi ↗ · pubmed ↗
- 7Brezgin S Kostyusheva A Bayurova E Volchkova E Gegechkori V Gordeychuk I . Immunity and viral infections: Modulating antiviral response via CRISPR-Cas systems. Viruses. (2021) 13:1373. doi: 10.3390/v 13071373. PMID: 34372578 PMC 8310348 · doi ↗ · pubmed ↗
- 8Andreatta M Tjitropranoto A Sherman Z Kelly MC Ciucci T Carmona SJ . A CD 4(+) T cell reference map delineates subtype-specific adaptation during acute and chronic viral infections. e Life. (2022) 11:e 76339. doi: 10.7554/e Life.76339.sa 2. PMID: 35829695 PMC 9323004 · doi ↗ · pubmed ↗