krepp: a k-mer-based maximum pseudo-likelihood method for estimating read distances and genome-wide phylogenetic placement
Ali Osman Berk Şapcı, Siavash Mirarab

TL;DR
krepp is a new method that uses k-mers to accurately place sequencing reads on large phylogenies, improving metagenomic analysis.
Contribution
krepp introduces a scalable, alignment-free method for genome-wide phylogenetic placement using k-mers.
Findings
krepp computes accurate read distances comparable to alignment-based methods.
krepp enables phylogenetic placement at scale on ultra-large reference trees.
The method improves metagenomic sample comparison and characterization.
Abstract
Comparing each sequencing read in a sample to a reference database is a fundamental step in wide-ranging applications. Results of these comparisons can enable phylogenetic characterization. However, phylogenetic placement is currently only possible at scale for marker genes, a small fraction of the genome. We introduce krepp, an alignment-free k-mer-based method that enables placing reads from anywhere on the genome on an ultra-large reference phylogeny (e.g., 123,853 leaves). We show that krepp is scalable and computes accurate distances that approximate those using alignments, leading to accurate placements. These precise phylogenetic identifications improve our ability to compare and characterize metagenomic samples. The online version contains supplementary material available at 10.1186/s13059-026-03999-y.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
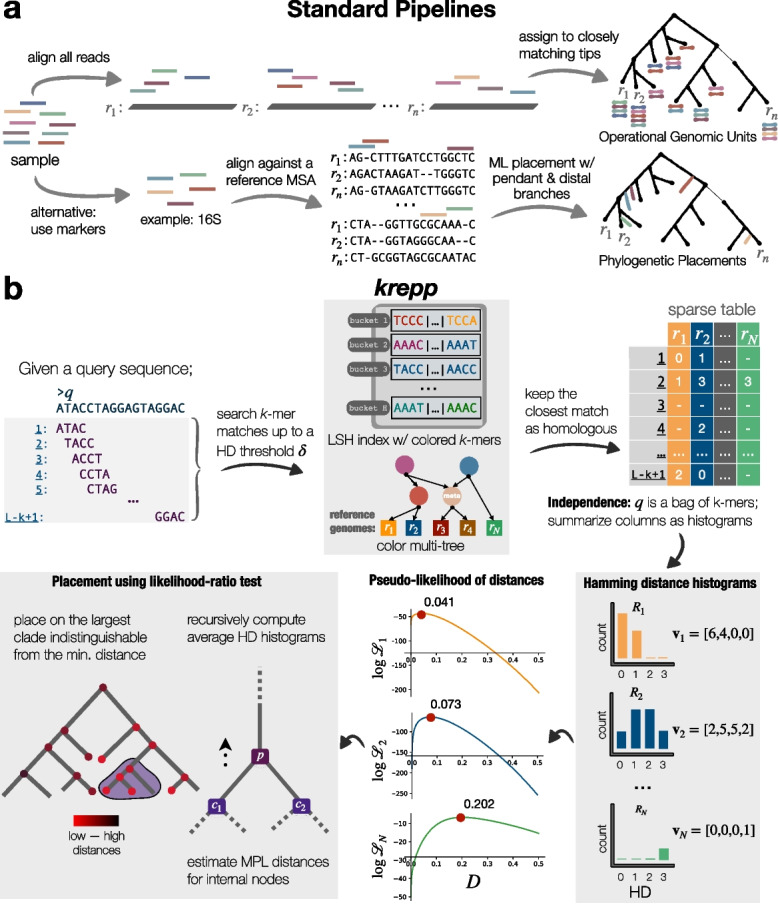 Figure 1
Figure 1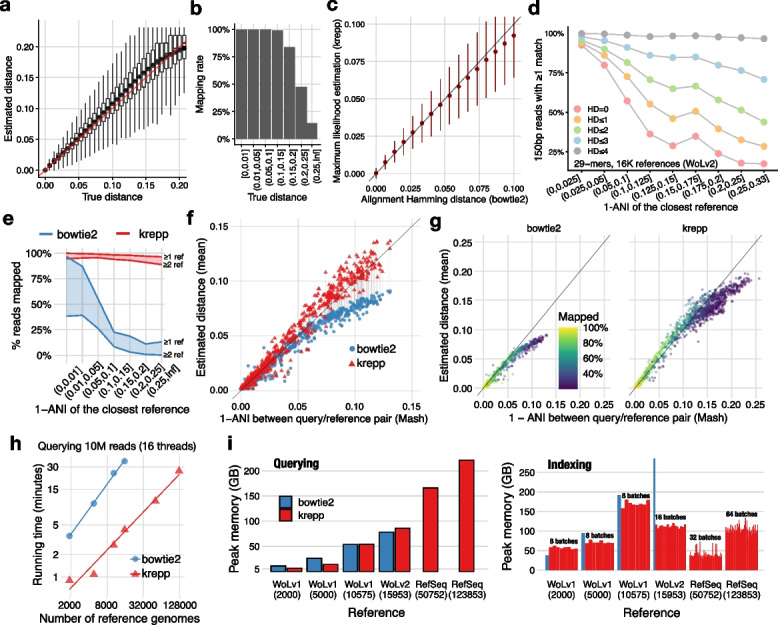 Figure 2
Figure 2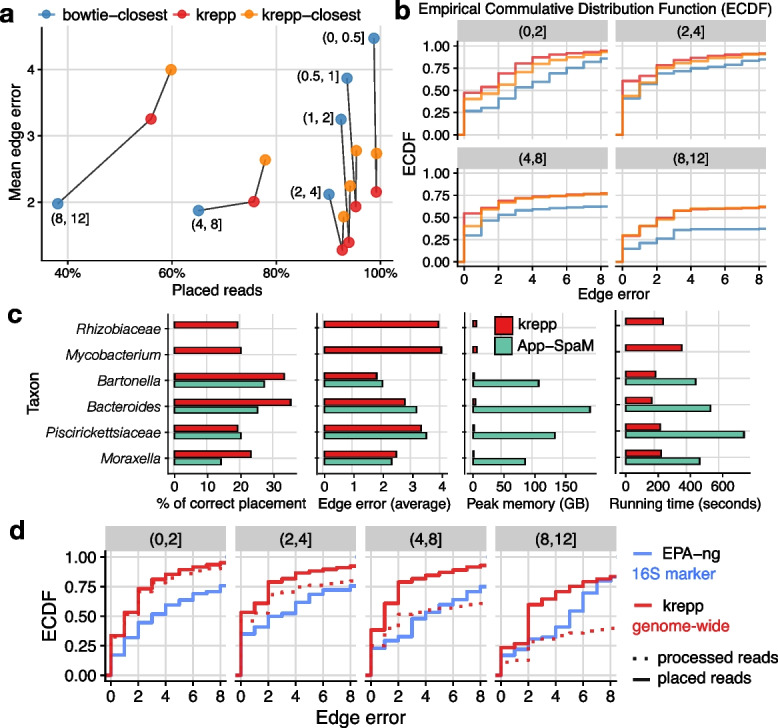 Figure 3
Figure 3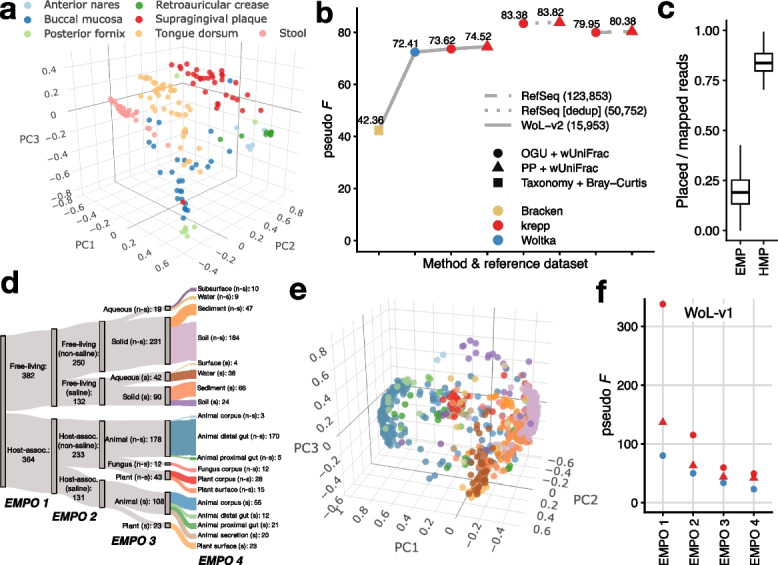 Figure 4
Figure 4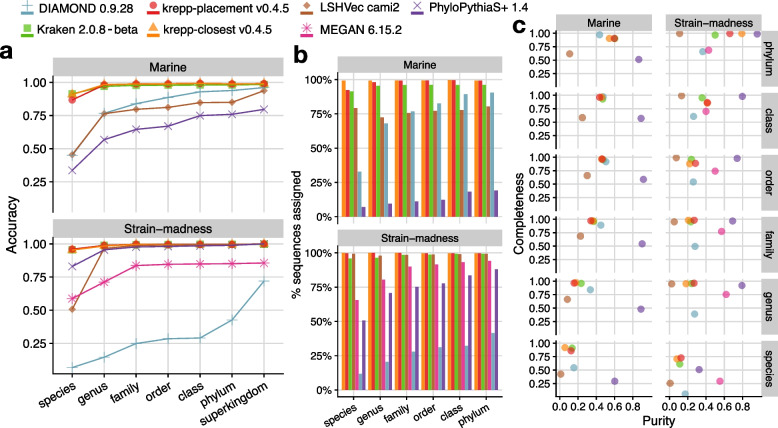 Figure 5
Figure 5 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Genome Rearrangement Algorithms · Fractal and DNA sequence analysis