To Kill a Macrophage: Targeted Strategies to Eliminate Macrophage Reservoirs of HIV
Laura Rikard-Bell, Morgane Brunton-O’Sullivan, Sushama Telwatte, Anthony Jaworowski, Anna C. Hearps

TL;DR
This paper reviews challenges in eliminating HIV-infected macrophages and explores strategies to overcome their resistance to death.
Contribution
The paper highlights novel strategies and methodological improvements needed to target macrophage HIV reservoirs.
Findings
HIV-infected macrophages are more resistant to death than CD4+ T cells.
HIV modulates apoptotic pathways and uses survival mechanisms like autophagy.
Improved pre-clinical models are essential for eliminating macrophage reservoirs.
Abstract
Persistent HIV reservoirs in long-lived macrophages pose a unique and formidable challenge to achieving HIV cure. HIV-infected macrophages are more resistant than CD4+ T cells to both virus- and immune-mediated death pathways including apoptosis, facilitating their persistence in tissue sanctuary sites and potential to contribute to viral rebound upon therapy cessation. This resistance is driven by HIV-induced modulation of both intrinsic and extrinsic apoptotic pathways, alongside survival mechanisms including autophagy. In this review, we examine the biological mechanisms promoting macrophage survival and explore novel translational strategies aimed at subverting this resistance. Crucially, we highlight the methodological limitations hindering progress, including the scarcity of robust in vitro macrophage models, the influence of culture conditions, and physiological relevance to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
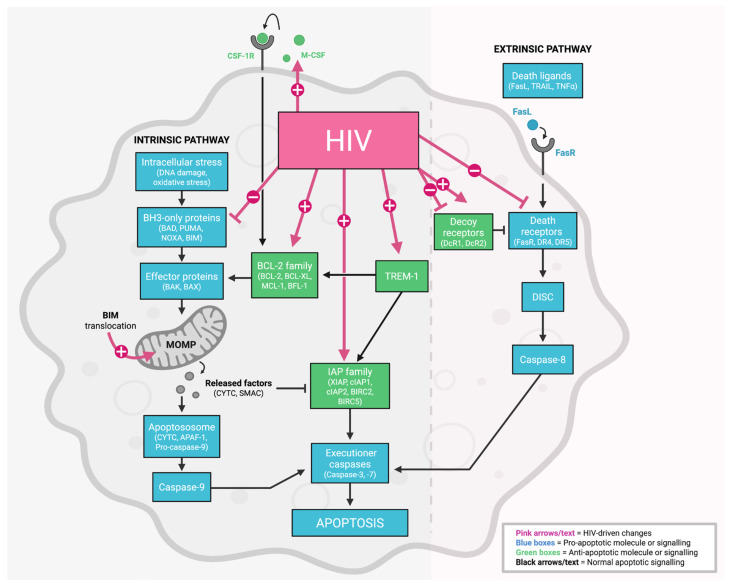 Figure 1
Figure 1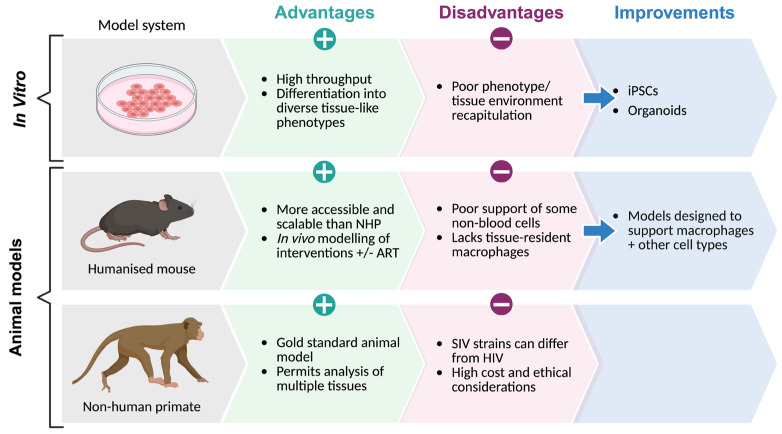 Figure 2
Figure 2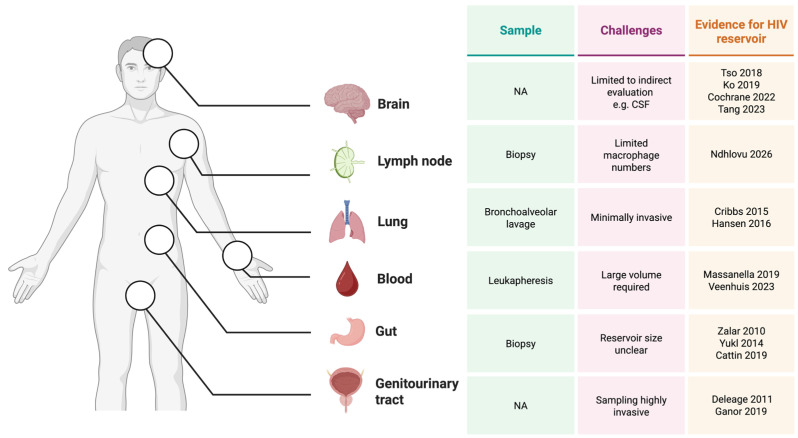 Figure 3
Figure 3| HIV Exposure | Cell Type | Culture Supplements | Impact on Viability | Mechanism | Ref. |
|---|---|---|---|---|---|
|
| |||||
| BaL (R5) | MDM | Human serum + M-CSF | NA | ↑ | Guillemard 2004 [ |
| LAI (X4) | MDM | Human serum + M-CSF | ↓ TRAIL-induced apoptosis (DNA fragmentation assay) | ↓ | Swingler 2007 [ |
| BaL (R5) | MDM | Not described | ↓ LPS+CHX-induced cell death (DNA viability stain) | NA | Chugh 2007 [ |
| NL4-3 (X4) | Microglial cell line (HMC3) | Bovine serum | ↓ LPS+CHX-induced cell death (DNA viability stain) | NA | Chugh 2007 [ |
| SG3 with transmitter founder Env | MDM | Bovine serum + M-CSF | NA | ↓ TRAIL decoy receptors (protein) | Zhu 2011 [ |
| ADA (R5) | MDM | Human serum + M-CSF | ↑ apoptosis in p24+ vs. uninfected cells (TUNEL assay) | ↑ | Castellano 2017 [ |
| Not described | MDM | Human serum | NA | ↑ TREM-1 (mRNA, protein) | Yuan 2017 [ |
| Transmitter founder virus (R5) | Macrophage cell line (THP-1) | PMA | ↓ apoptosis in HIV+ THP-1 macrophages vs. HIV+ THP-1 monocytes (Annexin-V, DNA viability stain) | ↑ | Xue 2017 [ |
| BaL (R5) | MDM | Bovine serum + M-CSF | ↓ apoptosis (ssDNA) | ↑ TREM-1, | Campbell 2019 [ |
| Pseudotyped BaL | MDM | Human serum + M-CSF | No Δ apoptosis in HIV+ vs. bystander (cell viability dye, caspase-3/7 protein) | ↑ anti-apoptotic long noncoding RNA (mRNA) | Boliar 2019 [ |
| BaL (R5) | MDM | Human serum + M-CSF | NA | ↑ TREM-1 (protein) | Hyun 2019 [ |
| IIIB (X4) | Kupffer cells | Bovine serum | NA | ↑ TREM-1 (protein, mRNA) | Hyun 2019 [ |
| BaL (R5) | MDM | Bovine serum + M-CSF | NA | ↑ | Campbell 2020 [ |
| CS204 (R5X4) | MDM | Bovine serum + M-CSF | ↑ apoptosis with | NA | Caballero 2021 [ |
| BaL(R5) | Monocyte-derived microglia | M-CSF, GM-CSF, NGFβ, CCL2, and IL-34 | ↓ apoptosis (ssDNA, LDH assay) | ↑ | Campbell 2023 [ |
|
| |||||
| Tat | Microglial cell line (HMC3) | Bovine serum | ↓ LPS+CHX-induced cell death (DNA viability stain) | Activation of the PI3K/AKT pathway | Chugh 2007 [ |
| Vpr | Macrophage cell line (THP-1) | PMA | ↓ apoptosis (Annexin-V, DNA viability stain) | No Δ cIAP1, BCL-2 (protein) | Busca 2012 [ |
| Vpr | MDM | Bovine serum + M-CSF | No Δ in viability (Annexin-V, DNA viability stain) | No Δ cIAP1, BCL-2 (protein) | Busca 2012 [ |
| Tat, Gp120 | Mouse macrophage cell line (Raw264.7) | Bovine serum | NA | ↑ TREM-1 (mRNA, protein) | Yuan 2017 [ |
| Tat, Gp120 | Mouse macrophage cell line (Raw264.7) (TREM-1 KD) | Bovine serum | ↑ apoptosis (TUNEL assay) | ↑ caspase 3 ↓ BCL-2 (protein) | Yuan 2017 [ |
| Tat, Gp120 | Mouse bone marrow-derived macrophage | Bovine serum + M-CSF | NA | TREM-1 KO ↑ caspase 3 vs. WT cells (protein) | Yuan 2017 [ |
| Tat, Gp120, RNA40 | MDM | Bovine serum + M-CSF | ↓ apoptosis (ssDNA) | ↑ TREM-1, BCL-2, BCL-XL (protein) | Campbell 2019 [ |
| Env, Gp120, Pro, Rev | MDM | Human serum + M-CSF | NA | ↑ TREM-1 (receptor expression) | Hyun 2019 [ |
| Tat | Monocyte-derived microglia | M-CSF, GM-CSF, NGFβ, CCL2, and IL-34 | ↓ apoptosis (ssDNA, LDH assay) | ↑ | Campbell 2023 [ |
|
| |||||
| Post-mortem brain tissue [CD68+ macrophages/microglia] | ↓ apoptosis (TUNEL assay) | NA | Cosenza 2004 [ | ||
| Post-mortem brain tissue [macrophage/microglia] | NA | ↑ | Castellano 2017 [ | ||
| CD68+ PBMCs (PWH | NA | ↑ TREM-1 (receptor expression) | Hyun 2019 [ | ||
- —National Health and Medical Research Council of Australia
- —Doherty Institute for Infection and Immunity Locarnini Fellowship in Virology
- —University of Melbourne Department of Infectious Diseases Research Support Package
- —National Institutes of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHIV Research and Treatment · Immune cells in cancer · Autophagy in Disease and Therapy
1. Introduction
The persistence of HIV reservoirs within latently infected cells despite antiretroviral therapy (ART) remains the main barrier to HIV cure [1,2,3]. While the majority of the HIV reservoir in blood resides in CD4+ T cells, peripheral blood monocytes can also harbour HIV [4,5,6,7] with an estimated 40% of people with HIV (PWH) on ART found to contain intact proviral DNA within blood monocytes [8]. There is extensive evidence of HIV infection of myeloid cells in tissue (reviewed comprehensively by Wong et al. and Veenhuis et al. [9,10]) with viral reservoirs persisting in myeloid cells in lungs, brain, seminal vesicles, and urethra despite ART [8,11,12,13,14,15,16,17,18,19,20,21]. Myeloid reservoirs have been shown to contribute to viral rebound after cessation of ART in humans [22] as well as mouse models [23] of HIV. Despite this, cure strategies to date have largely overlooked the myeloid reservoir. It remains unclear whether macrophage reservoirs persisting in PWH are truly latent or whether low-level viral transcription and translation may occur. This is relevant not only for viral recrudescence but also for the pathogenesis of comorbidities in PWH, as HIV-infected macrophages may contribute to disease pathology in tissues such as the brain [24,25] and lung [26]. Hence, strategies to eliminate this reservoir are important for not only HIV cure but also to improve the healthy lifespan of people with HIV. Due to their distinct biology and location in diverse anatomical locations, HIV-infected macrophages represent unique challenges for elimination. Furthermore, they appear to resist cell death, hence strategies to overcome this resistance may provide an effective approach for elimination. Across HIV elimination strategies, latency reversal is likely to be a critical precursor for reservoir elimination. While latency reactivation may differ substantially between macrophages and CD4+ T cells [27,28,29], a detailed discussion of these differences is beyond the scope of this review. This review focuses on macrophage biology and host–viral interactions that promote the survival of HIV-infected macrophages, explores novel translational approaches to eliminate this reservoir, and emphasises the importance of developing methods to evaluate cure interventions targeting macrophages in humans.
2. Macrophage HIV Reservoirs Pose Unique Challenges
Macrophages are a unique and formidable target for HIV cure due to their long lifespan [30,31], heterogeneity, location in tissue sites, and abundance in immunological sanctuary sites like the brain and testes. These features make the macrophage reservoir a significant barrier to cure, but clinically relevant for the pursuit of ART-free viral control.
2.1. Heterogeneity and Anatomical Location
Macrophages exist as a heterogenous population shaped by their ontogeny and tissue microenvironment, resulting in diverse phenotypes and functions. They are present in virtually all tissues, including the brain, heart, gut-associated lymphoid tissue (GALT), lung, liver, spleen, and bone [32]. Tissue-resident macrophages are derived from yolk-sac progenitors and foetal liver and are capable of self-renewal, whereas monocyte-derived macrophages (MDM) originate from circulating bone marrow-derived monocytes. The relative proportions of tissue- and monocyte-derived macrophages varies between different tissue locations and is further influenced by factors such as inflammation [33,34,35]. Many tissue sites harbour multiple subsets of macrophages from distinct progenitor sources including yolk-sac-derived microglia and blood monocyte-derived perivascular macrophages in the brain [36] as well as yolk-sac/foetal-liver-derived alveolar macrophages and blood monocyte-derived interstitial macrophages in the lung [37,38,39,40]. An additional layer of heterogeneity is provided by the ability of macrophages to adopt different functional phenotypes known as polarisation states [41]. In HIV, primary infection has been demonstrated to skew macrophages in urethral tissue towards an intermediary macrophage polarisation state and away from traditional M1 or M2 phenotypes [16]. While HIV reservoirs exist in diverse tissue macrophage types, the influence of macrophage heterogeneity and ontogeny on the formation and maintenance of macrophage reservoirs is currently unknown. These anatomical niches pose specific challenges for cure (reviewed by Ferreira et al. [42]), as they can limit immune access and treatment delivery.
2.2. Distinct Infection Mechanisms and Cellular Compartmentalisation in Macrophages
The importance of macrophages as HIV reservoirs has been controversial, due to previous data from macaque studies demonstrating HIV can be detected in macrophages following engulfment or phagocytosis of infected CD4+ T cells [43,44]. However, more recent evidence of integrated HIV DNA and HIV RNA in macrophages has convincingly demonstrated their ability to be productively infected (reviewed in [10,45]). Macrophages can be infected by cell-free HIV or through cell-to-cell transmission. Additionally, HIV can be taken up by macrophages via non-traditional HIV entry receptors involving lectin-mediated interactions, such as by Siglec-1 [46]. Within HIV-infected macrophages, nascent virions can accumulate within intracellular vesicles known as virus-containing compartments (VCCs) [47,48]. While these VCCs remain connected to the plasma membrane by narrow microchannels, they can enable virions to be shielded from HIV-specific neutralising antibodies [48] and potentially immune-mediated killing, allowing HIV to survive in these compartments for weeks [49]. In addition to virus budding from the cell surface, viral particles contained in VCCs can be released upon stimulation or cell death. Thus, this unique feature of HIV-infected macrophages can provide a source of replication-competent virus that is hidden from immune surveillance and is an important consideration for targeting HIV reservoirs in these cells.
2.3. Resistance to Death
A fundamental characteristic of HIV-infected macrophages is their increased resistance to cell death, the mechanisms of which are discussed in detail below. The elimination of HIV-infected macrophages following latency reversal will require therapeutic strategies that specifically target the underlying host–viral mechanisms that promote macrophage survival.
3. Why HIV-Infected Macrophages Do Not Die: HIV-Induced Alterations to Cell Survival Pathways in Macrophages
3.1. HIV-Infected Macrophage Resistance to Immune-Mediated Clearance
HIV-infected macrophages are less susceptible to cytotoxic CD8+ T-cell- and natural killer (NK) cell-mediated killing [50,51], consistent with findings from simian immunodeficiency virus (SIV) models [52,53]. This resistance leads to inefficient elimination by immune cells which, if translated in vivo, is likely to enhance persistence. Currently, little is known about the mechanisms associated with resistance to immune-mediated killing of HIV-infected macrophages. The budding of HIV into VCCs may be a mechanism that limits exposure of HIV antigens such as the envelope (Env) protein on the cell surface, and potentially protects macrophages from immune-mediated recognition and killing [51]. Additionally, antibodies targeting specific Env epitopes exhibit differential binding to HIV-infected macrophages as compared to CD4+ T cells [54], which may influence the efficacy of macrophage targeting through antibody-dependent cellular cytotoxicity and other antibody-mediated mechanisms.
Typically, cytotoxic T lymphocytes (CTLs) [55] andNK cells [56] are able to kill virus-infected target cells via the extrinsic pathway and through Fas ligand (FasL) and Fas receptor (FasR) interactions (see Section 3.2.2). However, HIV-1 accessory proteins including Nef down-modulate major histocompatibility complex class I (MHC-I) from the surface of virus-infected cells, including in primary human macrophages [57] to evade CTL-mediated immunity. In theory, this downregulation should make these cells targets for NK cell-mediated cytotoxicity, however (as stated above) in vitro HIV-infected macrophages can resist efficient NK cell-mediated killing. The increased resistance of HIV-infected macrophages to both CTL and NK cell-mediated killing is associated with a heightened inflammatory response from both cytotoxic effector cells and macrophage targets [50,51], which may contribute to the pathogenesis of HIV and associated comorbidities [58]. Further work is required to fully elucidate the mechanisms underpinning this resistance to immune-mediated killing to inform the development of immune-mediated strategies to eliminate HIV-infected macrophages.
3.2. HIV-Infected Macrophage Resistance to Virus-Mediated Death
HIV-infected cells, as well as bystander T cells, may undergo programmed or lytic cell death induced by viral replication [59,60]. However, macrophages have evolved distinct mechanisms to resist virus-induced cell death, enabling them to persist as long-lived reservoirs (reviewed in Le Douce et al. [61]). Apoptosis is a key cell death pathway for both virus- and immune-mediated clearance of HIV-infected cells. In the context of HIV cure, targeting apoptosis provides a mechanism to eliminate HIV-infected cells while importantly avoiding inflammatory responses, which is especially crucial in sanctuary sites like the brain. Hence, this review focusses on the evidence for HIV-induced resistance to apoptotic cell death in macrophages.
3.2.1. Evidence for HIV-Mediated Resistance to Apoptosis in Macrophages
Upon differentiation from monocytes, macrophages have been reported to acquire an anti-apoptotic signature [62,63,64]. This intrinsic resistance, alongside the slow replication of HIV in macrophages [65,66], and their ability to sustain long-term HIV infection without significant cell loss [67] is consistent with observations of resistance to apoptosis during HIV infection [68,69]. Macrophages are known to undergo activation-induced cell death, which, alongside apoptosis, is a fundamental host mechanism for resolving infection and limiting inflammation [70]. The impact of HIV infection on apoptosis in monocytes is less clear, with in vitro and SIV models demonstrating enhanced apoptosis of monocytes in response to infection [71], while other reports show monocytes from viraemic PWH exhibit an anti-apoptosis profile [72]. The relative extent to which intrinsic myeloid cell survival mechanisms and specific effects of HIV infection and viral proteins contribute to apoptosis resistance is unclear but has implications for HIV cure strategies.
Despite the long-held dogma that HIV-infected macrophages are resistant to apoptosis, robust in vitro evidence, particularly using primary human-derived cells, remains scarce. Several early mechanistic studies of HIV-infected macrophages did not include definitive functional cell death assays [73,74]. Initial support for this resistance phenotype came from observations that HIV-infected MDM cultures resisted apoptosis following Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) [68] or lipopolysaccharide (LPS)/cycloheximide (CHX) [75] stimulation. In vivo evidence further supported this concept, demonstrating reduced apoptosis in microglia from post-mortem brain tissue of HIV-seropositive donors compared to seronegative controls [76]. The first clear in vitro demonstration of primary HIV-infected macrophage protection from apoptosis emerged a decade later [77,78,79], with apoptosis resistant profiles observed in macrophage cell lines [64,79,80]. Furthermore, HIV viral proteins such as Tat, Gp120, and Vpr have been consistently implicated in conferring this resistance phenotype [64,75,77,78,80]. However, not all studies report impaired apoptosis in HIV-infected macrophages, with some studies finding no change [81] or even enhanced [82] apoptosis. These key findings are shown in Table 1 and summarised below.
A major limitation of this work is that almost all studies examine bulk infected cultures, making it impossible to distinguish whether observed resistance reflects truly infected cells or impacts on uninfected bystanders. To date, only two studies—by Boliar et al. [81] and Caballero et al. [83]—have directly compared HIV+ (p24+) macrophages with bystander cells and parallel uninfected cultures; further studies with this design are required to fully determine the extent and mechanism of apoptosis resistance in HIV-infected and bystander cells.
3.2.2. Viral Mechanisms Implicated in Inhibiting Apoptosis in Macrophages
Mechanistic studies primarily implicate the intrinsic apoptosis pathway as a target of HIV-inhibition of macrophage apoptosis, specifically factors within the BCL-2 family [64,68,73,77,79,80,85], although evidence also supports the involvement of the extrinsic pathway (e.g., FasL, TRAIL, tumour necrosis factor alpha (TNFα)) [68,74] (Figure 1).
Intrinsic Pathway. There is consistent and substantial evidence that HIV reprograms the mitochondrial (intrinsic) apoptotic machinery in macrophages (Table 1). Based on current in vitro and ex vivo data, this occurs most commonly through the upregulation of anti-apoptotic BCL-2 family members (BCL-2, BCL-XL, MCL-1) [68,73,77,78,79] and inhibitor of apoptosis (IAP) proteins (cIAP1, cIAP2, XIAP) [68,86]. Collectively, these data support a HIV-driven mitochondrial protection phenotype that promotes macrophage survival [64,68,75,76,77,78,79].
While anti-apoptotic BCL-2/IAP-centred survival signalling represents the most consistent findings, mitochondrial localisation of the pro-apoptotic BIM protein is also observed [77,82] even without accompanying HIV-mediated cytopathogenesis [77]. This suggests that pro-apoptotic signalling or mitochondrial priming could be engaged, but that progression to mitochondrial depolarisation is constrained, potentially by increased anti-apoptotic buffering by BCL-2 or IAP proteins.
Triggering Receptor Expressed on Myeloid Cells 1 (TREM-1) is a member of the immunoglobulin superfamily expressed on neutrophils, monocytes and macrophages that amplifies inflammatory responses to pattern recognition receptor signalling [87]. Several studies have shown upregulated expression of TREM-1 in primary HIV-infected MDMs [77,80,84] and Kupffer cells [84] compared to uninfected cells. In mechanistic studies, HIV proteins Tat and Gp120 were shown to inhibit apoptosis in primary MDMs and macrophage cell lines via TREM-1 signalling [80], linking TREM-1 expression to changes in BCL-2 and caspase activity, reinforcing survival. Additionally, as TREM-1 amplifies pattern recognition receptor signalling, viral activation of these receptors in the liver may also contribute to TREM-1 associated inflammatory responses in HIV infection [84]. Given its role in amplifying pattern recognition receptor signalling, TREM-1 represents a potential therapeutic target to both sensitise HIV-infected macrophages to apoptosis and reduce virus-induced inflammation and comorbidities.
Extrinsic Pathway. Across studies in Table 1, the impact of HIV on death-receptor signalling in macrophages is heterogeneous. Findings diverge regarding macrophage sensitivity to TRAIL-induced apoptosis, reflecting variable expression of death receptors (DR4/DR5) that induce apoptotic signalling versus decoy receptors (DcR1/DcR2) which inhibit signalling by binding to TRAIL, across primary MDMs and macrophage cell lines [68,74]. Macrophage colony-stimulating factor (M-CSF) is elevated in HIV-infected primary MDMs and microglia compared to their uninfected counterparts [68,88]. M-CSF is implicated in dampening TRAIL-mediated apoptosis, with evidence of Env-dependent reductions in DR4/DR5 death receptors, increased anti-apoptotic mediators (e.g., BFL-1, MCL-1), and elevated M-CSF in infected MDMs [68]. Overall, while HIV does modulate extrinsic death-receptor pathways, the evidence is less abundant, more model-dependent, and less consistent than evidence implicating mitochondrial (intrinsic) alterations [77,78,79].
3.2.3. Other (Virus-Mediated) Mechanisms of Macrophage Survival
Beyond apoptosis resistance, macrophage survival during HIV infection can be influenced by other mechanisms such as autophagy and cell cycle regulation. Autophagy can be modulated by HIV proteins to favour viral persistence (reviewed in [89]), but its role in survival of HIV-infected macrophages is less clear. Autophagy may play a role in inhibiting Vpr-induced apoptosis in macrophages [90], while the ability of second mitochondria-derived activator of caspases (SMAC) mimetics to increase apoptosis of HIV-infected macrophages involves enhancing the early stages of autophagy [86]. Further work is required to fully elucidate the interaction of autophagy and apoptosis pathways in HIV-infected macrophages to determine the potential for modulating these pathways to enhance infected cell elimination.
While many macrophage types are considered to be non-dividing, cell cycle pathways also regulate important stress and survival responses [91]. HIV Vpr induces cell cycle arrest in HIV-infected macrophages to enhance replication (reviewed in [92]). While HIV-induced cell cycle arrest has been associated with apoptosis in T cells (reviewed in [93]), whether this occurs also in macrophages in unclear.
The anti-apoptotic long noncoding RNA SAF is highly upregulated in MDMs infected with pseudotyped HIV, but not bystander cells [81]. SAF downregulation increases capase-3/7 protein levels and cell death of HIV+ macrophages, suggesting a role for this long noncoding RNA in conferring apoptosis resistance. Within MDMs, HIV induces degradation of lincRNA-p21, which is involved in mediating apoptosis in response to double stranded DNA breaks, thereby evading apoptosis that may be triggered by HIV integration [94]. Furthermore, this apoptosis-evading mechanism may be unique to macrophages, highlighting key cell-type differences in HIV reservoir cells regarding cell survival mechanisms.
3.2.4. Limitations and Methodological Considerations
While in vitro studies have substantially advanced our understanding of HIV infection dynamics and cell survival in macrophages, several methodological and translational limitations exist which complicate the ability to draw definitive conclusions from multiple studies.
Varying and Non-Physiological Virus and Cell systems. Relatively few studies investigating apoptosis pathways in macrophages explicitly demonstrated that HIV protects macrophages from death using validated experimental indicators of apoptosis. Furthermore, some studies rely on immortalised cell lines, such as HMC3 or THP-1, which are transcriptionally different to primary cells in vitro [95,96] and may not reflect long-lived macrophage reservoirs in vivo. Although chronically HIV-infected myeloid cell lines, including U1 and OM10.1 [97], alongside adapted THP-1 cell lines [28,98], are highly useful models for studying HIV latency in myeloid cells, these models may have similar limitations. Additionally, viral gene expression has been shown to differ significantly between cell lines and ex vivo cells from PWH [99]. Viral strain selection may further complicate interpretation, as T-cell-adapted strains such as LAI and macrophage-tropic strains such as BaL engage distinct entry receptors, differ in their capacity to infect macrophages, and may have different replication kinetics and host–viral interactions [100,101]. Furthermore, laboratory HIV strains may not accurately reflect the behaviour of tissue-resident quasi-species found in compartments such as the central nervous system (CNS) and GALT. Consequently, further studies employing diverse clinical isolates are required to specifically identify altered apoptosis pathways in HIV-infected primary macrophages to more accurately recapitulate the dynamics of the viral reservoir.
Microenvironmental Influence and Macrophage Heterogeneity. Current in vitro models often fail to capture the diversity of tissue-resident macrophages. There is substantial macrophage heterogeneity not only between but also within various anatomical compartments [102,103,104,105] which may have implications for HIV reservoirs and how they can be eliminated [92]. Unlike MDMs, tissue-resident macrophages are long-lived and self-renewing [106], properties that may confer intrinsic differences in apoptotic thresholds and survival programmes. Cytokine-driven polarisation further modulates apoptotic resistance, with M-CSF promoting upregulation of anti-apoptotic mediators including MCL-1, BFL-1, and BCL-2, while granulocyte-macrophage colony-stimulating factor (GM-CSF) downregulates TRAIL death receptors [68]. Together, these observations suggest that sensitivity to apoptosis modulation is likely phenotype- and tissue-dependent. However, to date, relative apoptosis resistance between macrophages derived from different tissues, or between distinct macrophage subtypes within the same tissue, has not been explored. A further limitation to these studies is the investigation of productively-infected cells. Virtually no studies have specifically investigated the apoptotic resistance phenotype of reactivated latently infected myeloid reservoirs.
Confounding Factors in In Vitro Culture. The overwhelming majority of studies employ bulk analysis of HIV-infected macrophage cultures that includes both uninfected bystander and infected cells. This likely masks the resistance phenotype of the persistently infected cells, since studies in CD4+ T cells have demonstrated distinct patterns of cell death in infected versus bystander cells [107]. Differences in culture conditions, such as the use of human versus bovine serum, have been implicated in altering the phenotype and surface receptor expression of MDMs [108]. This variability could contribute to discrepancies in apoptotic signalling, particularly for the extrinsic pathway, potentially explaining conflicting results regarding death receptor expression [68,74].
Methodological Challenges in Apoptosis Measurement. A major limitation of studies to date is the disconnect between mechanistic analyses and concurrent apoptosis measurements. Many studies rely heavily on techniques such as proteomics or gene expression profiling without confirming that the observed molecular changes result in measurable resistance to cell death, underscoring the need for integrated functional assays. Additionally, because apoptotic factors undergo extensive post-translational modifications, including phosphorylation of pro-survival enzymes or the cleavage of pro-caspases, mRNA levels may not accurately reflect the abundance or activation state of the proteins that ultimately execute the apoptotic programme [109].
Detachment of macrophages required for flow cytometric analysis can mechanically cleave surface receptors such as FasR [110] and potentially modify receptor-signalling pathways and impact background cell death [111]. Therefore, microscopy or other assays not requiring cell detachment for analysis of macrophage cell death are preferable. Finally, the choice of apoptosis marker is critical; since different indices evaluate different stages of apoptosis, with some being earlier and potentially more transitory (e.g., Annexin-V) while others reflect a later and more irreversible stage (e.g., TUNEL assay or caspase activity). Using multiple complementary assays is recommended to definitively distinguish apoptosis from other forms of programmed cell death [112].
Moving forward, robust, physiologically relevant in vitro models that employ integrated functional assays coupled with specific analysis of HIV-infected as compared to bystander cells are essential for accurately studying apoptosis and evaluating cure strategies targeting the macrophage HIV reservoir.
4. Translational Strategies for Macrophage Reservoir Elimination
Given HIV-infected macrophages exhibit heightened resistance to multiple apoptotic stimuli, targeting this resistance may represent a promising strategy for their elimination. Indeed, a number of approaches have been investigated, with some promising strategies involving small molecule inhibitors like SMAC mimetics, modulating the phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT) pathway as well as combination strategies. These avenues for inducing apoptosis in HIV-infected macrophages are summarised in Table 2.
4.1. Small Molecule Inhibitors
AKT inhibitors. HIV infection of macrophages constitutively activates the PI3K/AKT cell survival pathway [113] and AKT inhibition using perifosine promotes cell death and restricts viral production in HIV-infected MDMs and microglial cell lines [114]. The efficacy of more contemporary AKT inhibitors such as capivasertib to modulate death of HIV-infected macrophages remains to be evaluated.
IAP inhibitors/SMAC mimetics. SMAC mimetics are small molecules that mimic endogenous SMAC proteins and trigger the degradation of IAPs, which normally sequester caspases to prevent apoptosis. These agents are also among the most commonly used compounds for targeting HIV-infected macrophages in vitro. SMAC mimetics LCL161 and birinapant have demonstrated efficacy in vitro in inducing selective apoptosis of HIV-infected MDM from uninfected controls [86] and PWH, regardless of ART status [83]. Beyond their pro-apoptotic role, these compounds have shown dual potential as latency reversing agents in CD4+ T cells [121]. However, dose-limiting toxicity issues including adverse neurological events and cytokine release syndrome may limit the clinical use of many SMAC mimetics [122,123,124,125].
CSF-1R antagonists. HIV-1 infection induces production of the pro-survival cytokine M-CSF (CSF-1), which drives resistance to apoptotic stimuli [68]. Consequently, inhibiting the M-CSF receptor (CSF-1R) presents a potential strategy for macrophage elimination. Imatinib and high-affinity CSF-1R inhibitors such as PLX647, PLX3397, and PLX5622, have shown efficacy in selectively killing HIV-infected macrophages by removing these critical survival signals [68,119].
TREM-1 blockade. Given that TREM-1 is upregulated in HIV-infected macrophages (Section 3.2.2) and contributes to an anti-apoptotic phenotype, blocking this receptor or its downstream signalling is an emerging avenue for inducing selective apoptosis [80]. Campbell et al. demonstrated that TREM-1 silencing in HIV-infected macrophages leads to decreased anti-apoptotic proteins and increased cell death, specifically through disruption of mitochondrial membrane potential [77]. TREM-1 silencing increases caspase 3 activation and reduces BCL-2 expression, making macrophages more susceptible to apoptosis [80]. Importantly, silencing of TREM-1 causes HIV-infected microglia to undergo cell death without triggering broader inflammatory responses [78].
4.2. Combination Strategies
The failure of initial “shock and kill” strategies to eliminate HIV-infected cells despite efficient viral reactivation has prompted the development of ‘prime, shock, and kill’ approaches which aim to sensitise (“prime”) latently infected cells to cell death by targeting intrinsic survival pathways [126,127]. This strategy has been successfully modelled in CD4+ T cells with the BCL-2 inhibitor Venetoclax which, when combined with latency reversing agents, led to the selective clearance of HIV-infected CD4+ T cells in vitro and ex vivo [126]. Further synergy has been observed when targeting multiple BCL-2 family members simultaneously, such as combining Venetoclax with the MCL-1 inhibitor S63845 in a mouse model of HIV [128]. While the efficacy of these models has not been tested in macrophages, they hold significant potential provided toxicity issues observed in mouse models can be overcome [129].
A similar approach combining latency reversal with inhibition of autophagy and agents to enhance apoptosis has demonstrated efficacy in inducing death of HIV-infected cells both in vivo in humanised mouse models and ex vivo in peripheral blood mononuclear cells (PBMCs) from PWH [130]. This approach was shown to reduce HIV reservoirs in CD4+ T cells, CD11b+ macrophages in the brain, and PBMCs harbouring intact provirus in humanised mice [131]. Chen et al. further confirmed the ability of this approach to clear HIV-infected PBMCs from both ART-experienced and naïve rhesus macaques ex vivo [132], establishing combination prime, shock and kill approaches as promising strategies for potential HIV eradication. However, these approaches have not yet been specifically assessed in human macrophage reservoirs, which will be important to determine their translational relevance for comprehensive HIV reservoir eradication.
4.3. Delivery Challenges
The clinical translation of these strategies is hindered by the anatomical sequestration of macrophages in sanctuary sites such as the CNS and GALT [12,17,133,134]. Penetrating the blood–brain barrier remains a major hurdle, necessitating the development of specialised delivery platforms such as nanoparticles. Recent advances have seen nanoparticle technologies adapted for the targeted delivery of ART, including to macrophages [135], and have demonstrated significantly enhanced accumulation within lymph node resident populations [136]. The delivery of mRNA via lipid nanoparticles to reverse latency in HIV-infected CD4+ T cells in vitro has been recently shown to be highly potent with minimal toxicity [137]. Additionally, SMAC-loaded, T-cell membrane-coated nanoparticles selectively killed HIV-infected macrophages via autophagy-dependent apoptosis without off-target effects on bystander cells [116]. This strategy could also enhance the targeting of other drugs that may show promise at eliminating primary macrophages in vitro. Leveraging these platforms to deliver pro-apoptotic priming agents could fundamentally transform HIV cure strategies by ensuring these compounds reach the deep-tissue reservoirs where macrophages persist. However, the pre-clinical validation of these novel strategies is constrained by the limited availability of high-fidelity human macrophage models and standardised measurement assays.
5. Limitations in Discovery and Pre-Clinical Targeting of Macrophage Reservoirs
The study of macrophage reservoirs remains a major challenge for HIV cure research due to the difficulty of isolating rare, tissue-resident HIV+ macrophages from PWH [2]. Consequently, most mechanistic and therapeutic studies rely on in vitro, ex vivo, and animal models, each with its own distinct advantages and limitations, which are summarised in Figure 2 below. Important advancements to improve pre-clinical investigations of HIV macrophage reservoirs include the use of induced pluripotent stem cell (iPSC)-derived models [138,139,140] and more complex organoid systems, which offer three-dimensional, tissue-like microenvironments and have recently been developed to model HIV infection in the brain [141,142,143,144].
6. Clinical Evaluation of Cure Interventions Within Myeloid Reservoirs
Cure interventions have historically prioritised analysis of peripheral blood CD4+ T-cell reservoirs, often overlooking monocyte and tissue-resident macrophage reservoirs. Given sustained, ART-free remission is likely to require comprehensive targeting of diverse anatomical sites and cell types [151], incorporating analysis of myeloid populations in tissue-based sampling will be important to achieve this goal.
A limitation in current cure research is the difficulty in characterising the source of rebound virus, specifically their anatomical origins and cellular lineages, however the latter is possible and has been used to demonstrate the macrophage reservoirs can contribute to viral rebound upon ART interruption [22]. While profiling the cellular reservoir prior to intervention has been applied to T cells [152], this precedent has not yet been extended to myeloid cells and is challenged by low infection frequency and poor yields from tissue biopsies. Despite compelling arguments for including myeloid reservoirs as primary or secondary trial endpoints [153], current protocols rarely incorporate such sampling. Furthermore, observations in peripheral blood often fail to capture the effects of interventions within deep-tissue sanctuaries [154], where viral compartmentalisation and restricted drug penetration may facilitate persistence [155]. Robust evaluation requires pre- and post-intervention measurements of myeloid reservoir size, distribution, and functionality using macrophage-specific assays. The following section briefly discusses tissue sites that represent key priorities for quantifying the macrophage reservoir in clinical trials (Figure 3).
6.1. Potential Tissue Site Sampling for Myeloid Reservoir Studies
Blood. Peripheral blood sampling offers accessible means to interrogate the monocyte reservoir but necessitates large-volume leukapheresis to achieve adequate sensitivity. Utilisation of myeloid-adapted versions of the intact proviral DNA assay and quantitative viral outgrowth assay provide a robust and compartment-specific measurement [8] and demonstrate the feasibility of evaluating the monocyte reservoir pre-and post-intervention provided sufficient blood sampling is available. This approach possibly represents the least invasive and most feasible approach for assessing myeloid reservoirs in HIV cure studies.
Gut. While routine gut biopsies, primarily obtained from the colon, enable sampling of tissue-resident immune cells in the GALT, there is limited evidence supporting the existence of a HIV reservoir in GALT macrophages [157]. Despite this, advancements in lamina propria leukocyte isolation, combined with sensitive techniques such as droplet digital polymerase chain reaction or in situ hybridisation, permit high-resolution detection of HIV nucleic acids in gut tissues [158,159,160] and may advance our understanding of myeloid reservoirs in the gut. Importantly, patient acceptability of gut tissue sampling is high when paired with routine cancer screenings [161]. While gut sampling has been incorporated in a number of HIV cure studies, these investigations have not systematically interrogated macrophage populations [162,163,164].
Lung. Bronchoalveolar lavage provides an accessible and high-yielding source of primary tissue macrophages, typically recovering a population of >90% alveolar macrophages [13,26,165]. Given that these macrophages harbour replication-competent HIV during ART [13,18], the lung is a critical and clinically feasible compartment for studying the myeloid reservoir [166].
Other sites. Lymph node sampling has been used to characterise HIV T-cell reservoirs in PWH [167]. Recently, HIV proviral DNA and inducible, multiply spliced RNA has been detected in germinal centre macrophages within the lymph node of PWH with sustained viral suppression [156]. Lymph node biopsies and fine needle aspirates are feasible and well-tolerated [168] but often yield limited macrophage numbers [169]. Similarly, while hepatic macrophages (Kupffer cells) constitute a potential reservoir, there is currently no evidence for replication-competent virus in the liver [170]. Direct assessment of the brain HIV reservoir is not feasible in living participants; consequently, cerebrospinal fluid is often used as a surrogate for CNS viral activity [171] but cannot be attributed to HIV cell type. Substantially more work is required to evaluate whether myeloid HIV reservoirs can be detected or reliably quantified in different tissue samples to determine the clinical utility of including these biopsies in interventional studies, given the invasive nature and associated risk of tissue sampling.
6.2. Advanced Analytical Frameworks and Success Criteria
Emerging multi-omic approaches are transforming the capacity to define the macrophage HIV reservoir. In situ hybridisation approaches allow HIV reservoir characterisation within the native tissue environment [172] while spatial transcriptomics (reviewed in [173,174]) offers the potential for high-resolution mapping of the viral microenvironment, including cell-to-cell interactions and viral integration sites [175], although this approach is yet to be fully applied to the analysis of HIV reservoirs. Single-cell RNA sequencing approaches enable cell-specific identification of HIV reservoirs. Recent advancements significantly improve the sensitivity for detecting HIV transcripts and can be combined with surface phenotyping for unparalleled characterisation of the HIV reservoir [176]. When combined with epigenetic profiling, cell phenotyping, viral sequencing, and integration site analysis, these single-cell approaches provide critical insight into reservoir characteristics including clonality and transcriptional competence [177,178,179]. In parallel, the identification of surrogate biomarkers—such as macrophage-specific inflammatory signatures in the brain or lung—could offer indirect but reliable measures of reservoir dynamics and the in vivo efficacy of cure interventions.
7. Conclusions and Future Perspectives
The resistance of HIV-infected macrophages to both virus- and immune-mediated elimination mechanisms highlight the critical need to understand cellular and molecular determinants that confer resistance to death. This understanding is essential for developing effective therapeutic strategies to target this persistent reservoir. HIV-infected macrophages exhibit resistance to apoptosis through both intrinsic and extrinsic pathways; thus, therapeutic targeting of these survival mechanisms represents a promising strategy for their elimination and for advancing the HIV cure agenda. Achieving this will require a shift toward tissue-resident macrophage models, such as induced pluripotent stem cell derived systems and organoids, to accurately capture the impact of the tissue microenvironment and macrophage heterogeneity on viral persistence and develop interventions able to penetrate tissue sanctuary sites.
Finally, the implementation of robust myeloid reservoir sampling and measurements before and after therapeutic intervention will be essential for targeting all cellular reservoirs of HIV and achieving sustained HIV remission and thus improving long-term health outcomes for PWH.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Finzi D. Hermankova M. Pierson T. Carruth L.M. Buck C. Chaisson R.E. Quinn T.C. Chadwick K. Margolick J. Brookmeyer R. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy Science 19972781295130010.1126/science.278.5341.12959360927 · doi ↗ · pubmed ↗
- 2Sengupta S. Siliciano R.F. Targeting the Latent Reservoir for HIV-1Immunity 20184887289510.1016/j.immuni.2018.04.03029768175 PMC 6196732 · doi ↗ · pubmed ↗
- 3Siliciano J.D. Siliciano R.F. In Vivo Dynamics of the Latent Reservoir for HIV-1: New Insights and Implications for Cure Annu. Rev. Pathol. Mech. Dis.20221727129410.1146/annurev-pathol-050520-11200134736342 · doi ↗ · pubmed ↗
- 4Crowe S.M. Sonza S. HIV-1 Can Be Recovered from a Variety of Cells Including Peripheral Blood Monocytes of Patients Receiving Highly Active Antiretroviral Therapy: A Further Obstacle to Eradication J. Leukoc. Biol.20006834535010.1189/jlb.68.3.34510985250 · doi ↗ · pubmed ↗
- 5Ellery P.J. Tippett E. Chiu Y.-L. Paukovics G. Cameron P.U. Solomon A. Lewin S.R. Gorry P.R. Jaworowski A. Greene W.C. The CD 16+ Monocyte Subset Is More Permissive to Infection and Preferentially Harbors HIV-1 In Vivo J. Immunol.20071786581658910.4049/jimmunol.178.10.658117475889 · doi ↗ · pubmed ↗
- 6Massanella M. Bakeman W. Sithinamsuwan P. Fletcher J.L.K. Chomchey N. Tipsuk S. Chalermchai T. Routy J.-P. Ananworanich J. Valcour V.G. Infrequent HIV Infection of Circulating Monocytes during Antiretroviral Therapy J. Virol.201994 e 01174-1910.1128/JVI.01174-1931597764 PMC 6912110 · doi ↗ · pubmed ↗
- 7Shiramizu B. Gartner S. Williams A. Shikuma C. Ratto-Kim S. Watters M. Aguon J. Valcour V. Circulating Proviral HIV DNA and HIV-Associated Dementia AIDS 200519455210.1097/00002030-200501030-0000515627032 PMC 1557628 · doi ↗ · pubmed ↗
- 8Veenhuis R.T. Abreu C.M. Costa P.A.G. Ferreira E.A. Ratliff J. Pohlenz L. Shirk E.N. Rubin L.H. Blankson J.N. Gama L. Monocyte-Derived Macrophages Contain Persistent Latent HIV Reservoirs Nat. Microbiol.2023883384410.1038/s 41564-023-01349-336973419 PMC 10159852 · doi ↗ · pubmed ↗