NRF2 as a Therapeutic Target in Dermatological Disorders: Mechanisms and Molecules
Ismael Khiar-Fernández, Nora Khiar-Fernández, José-Juan Pereyra-Rodríguez, Inmaculada Fernández

TL;DR
This paper reviews how activating the NRF2 pathway could help treat skin conditions like psoriasis and vitiligo by reducing oxidative stress and inflammation.
Contribution
The paper provides a comprehensive analysis of various NRF2-activating compounds and their potential for dermatological therapies.
Findings
Multiple classes of NRF2 activators show antioxidant and anti-inflammatory effects in skin cells.
Dimethyl fumarate and tapinarof have advanced to clinical stages for dermatological use.
Further research is needed to optimize formulations and safety for clinical practice.
Abstract
The nuclear factor erythroid 2–related factor 2 (NRF2) is a master transcription factor that orchestrates cellular defense against oxidative and electrophilic stress. Dysregulation of the KEAP1–NRF2–ARE pathway has been implicated in several dermatological disorders, including vitiligo, psoriasis, atopic dermatitis, photoaging, and radiation dermatitis. This review summarizes recent advances in the understanding of NRF2 activation mechanisms and highlights pharmacological and natural compounds with potential dermatological applications. A comprehensive analysis of natural, semisynthetic, and synthetic NRF2 modulators is provided, describing their chemical structures, synthetic approaches, mechanisms of action, preclinical and clinical evidence, and therapeutic relevance for skin disorders. Multiple classes of NRF2 activators, including isothiocyanates such as sulforaphane, triterpenoids…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
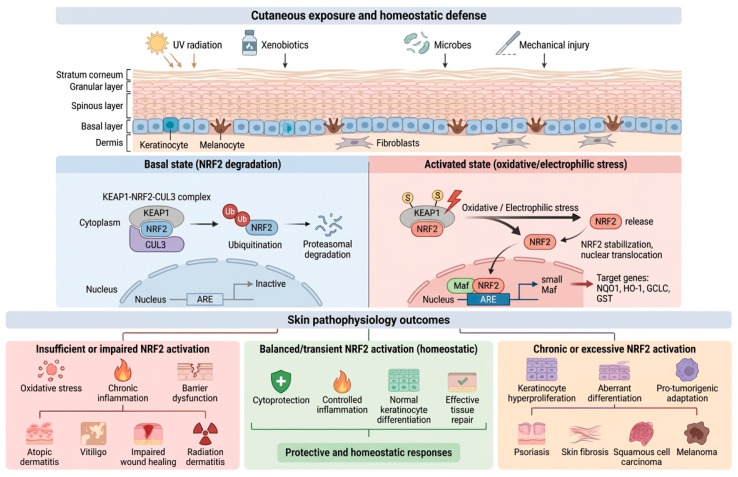 Figure 1
Figure 1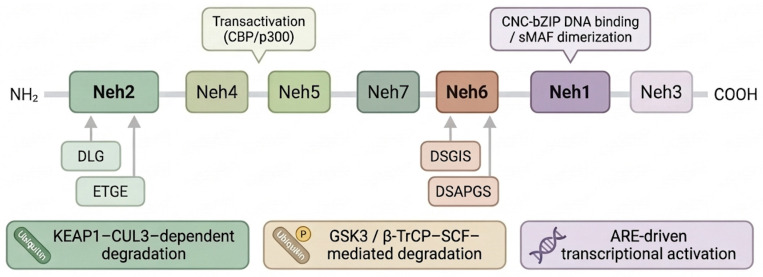 Figure 2
Figure 2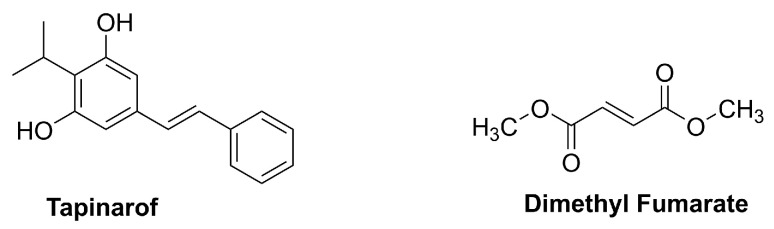 Figure 3
Figure 3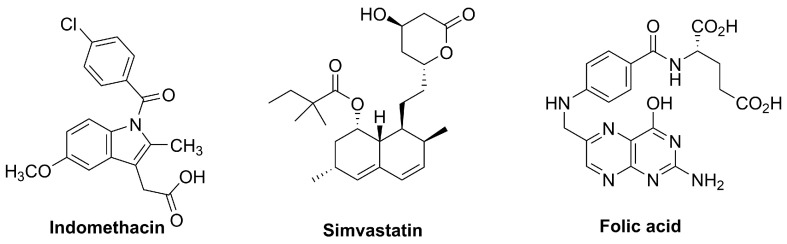 Figure 4
Figure 4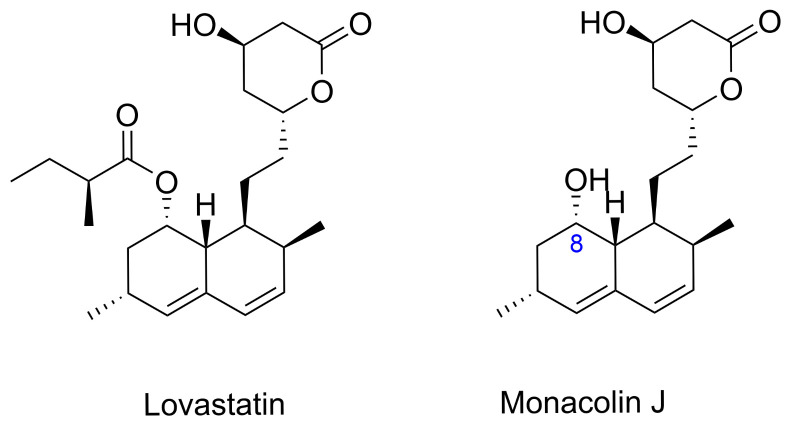 Figure 5
Figure 5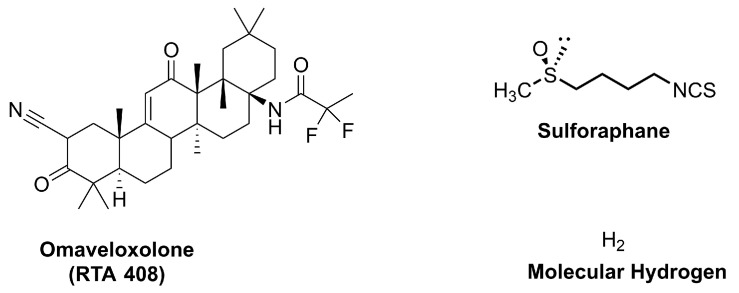 Figure 6
Figure 6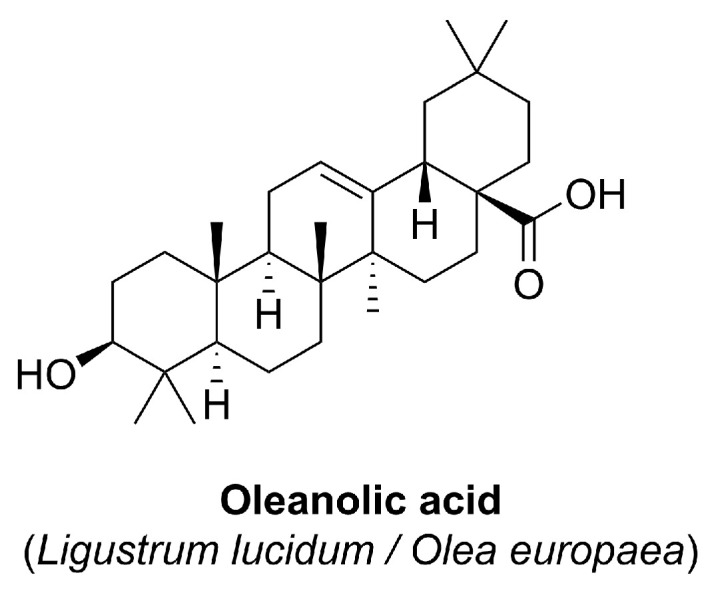 Figure 7
Figure 7 Figure 8
Figure 8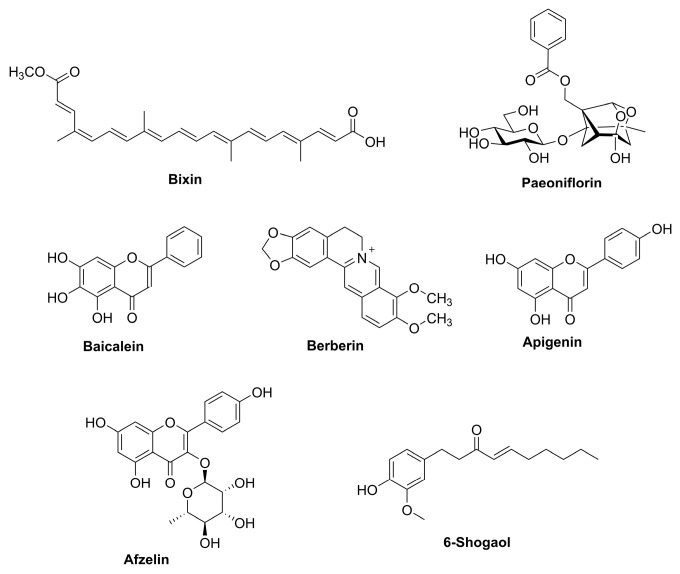 Figure 9
Figure 9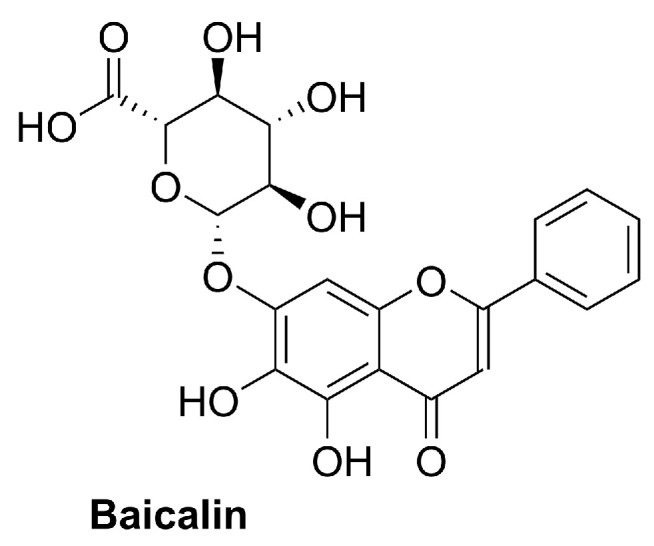 Figure 10
Figure 10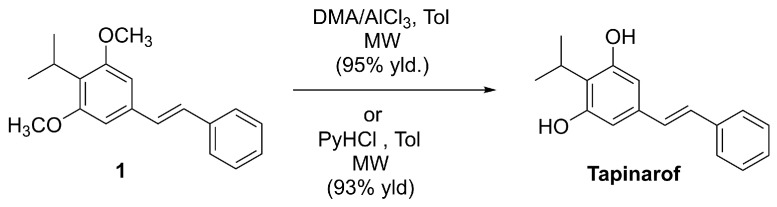 Figure 11
Figure 11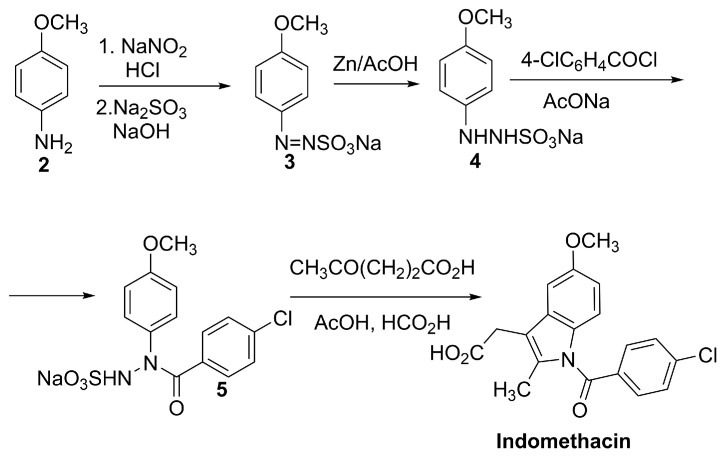 Figure 12
Figure 12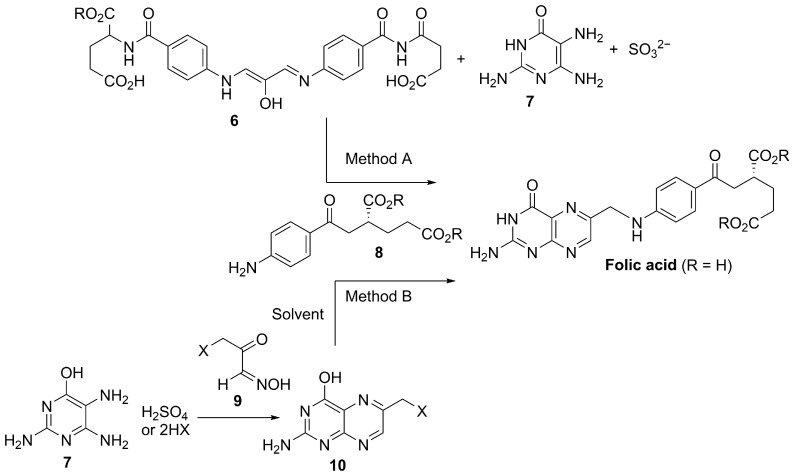 Figure 13
Figure 13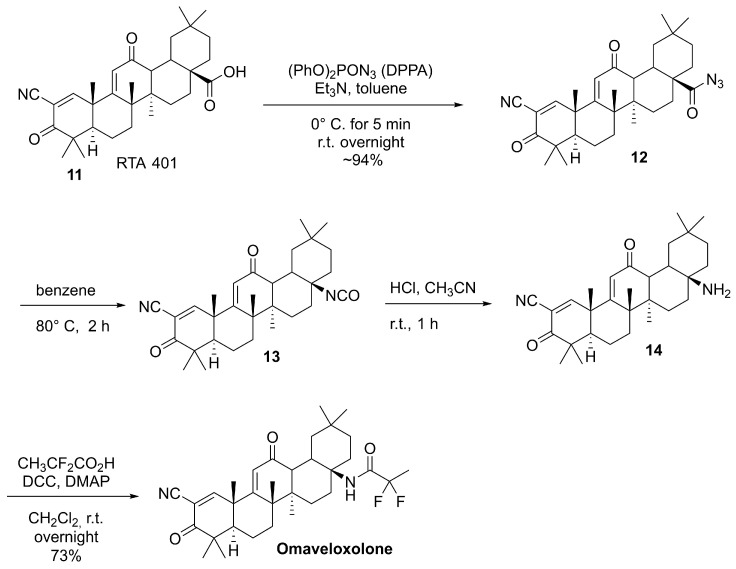 Figure 14
Figure 14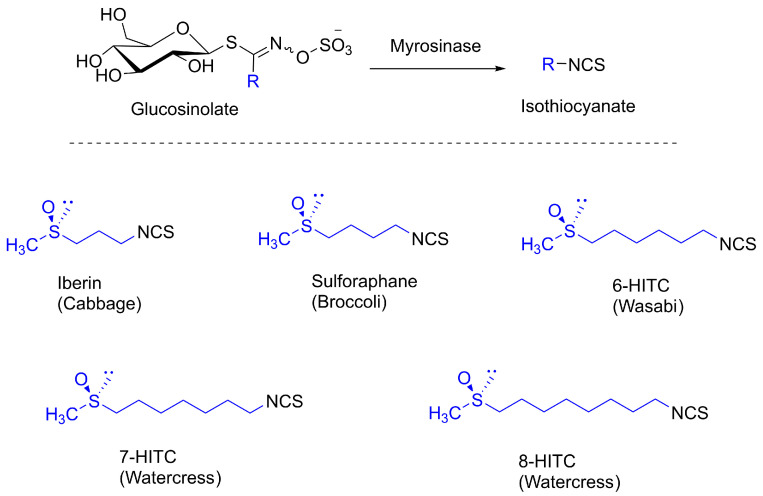 Figure 15
Figure 15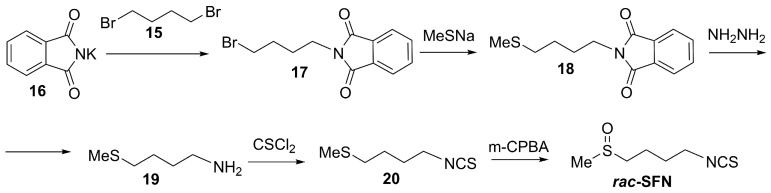 Figure 16
Figure 16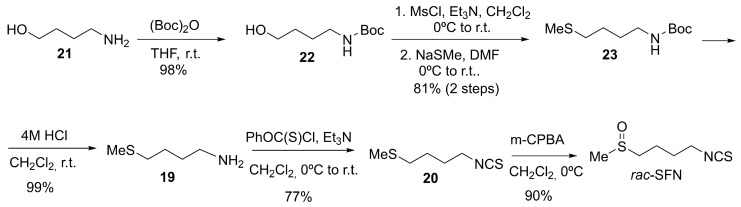 Figure 17
Figure 17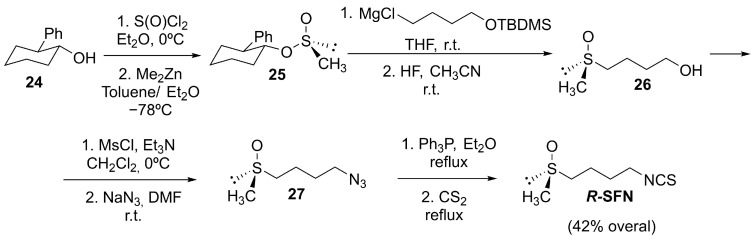 Figure 18
Figure 18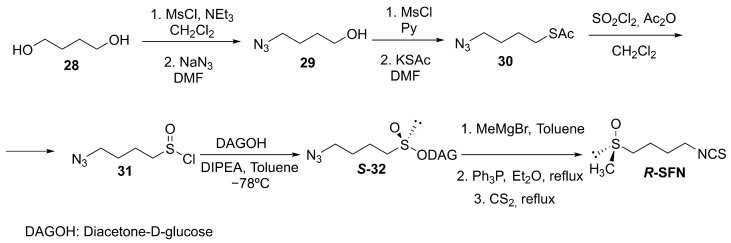 Figure 19
Figure 19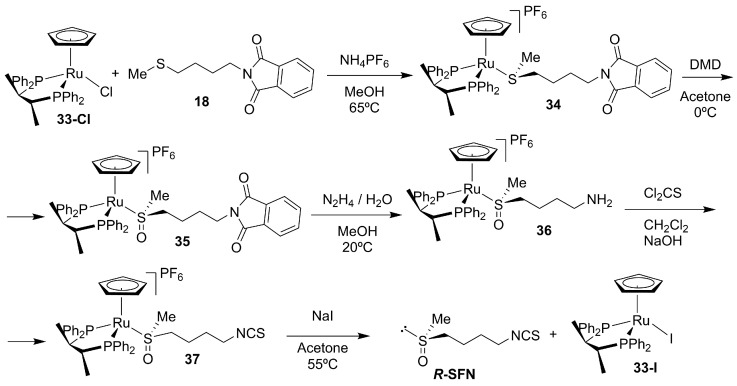 Figure 20
Figure 20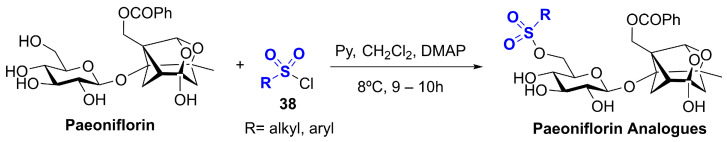 Figure 21
Figure 21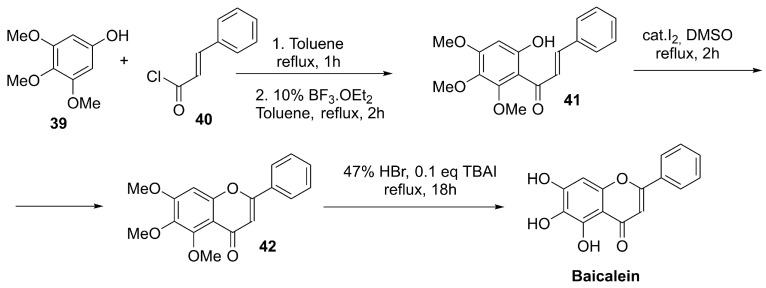 Figure 22
Figure 22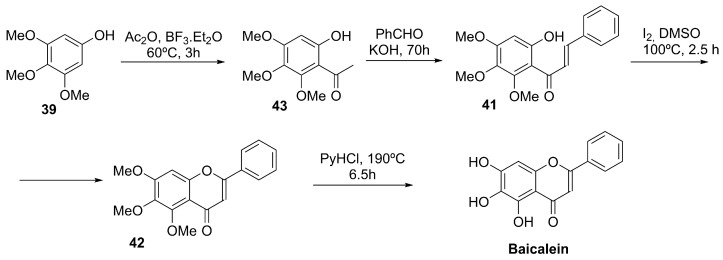 Figure 23
Figure 23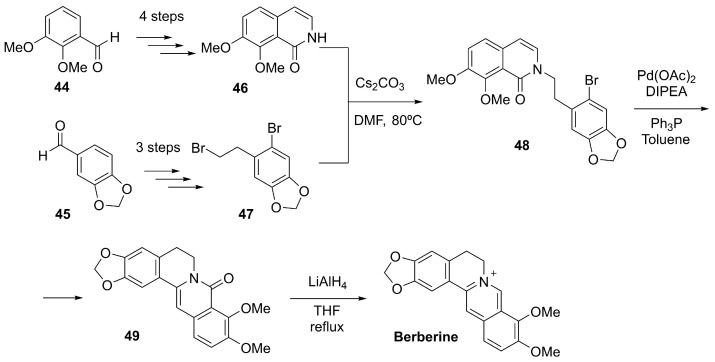 Figure 24
Figure 24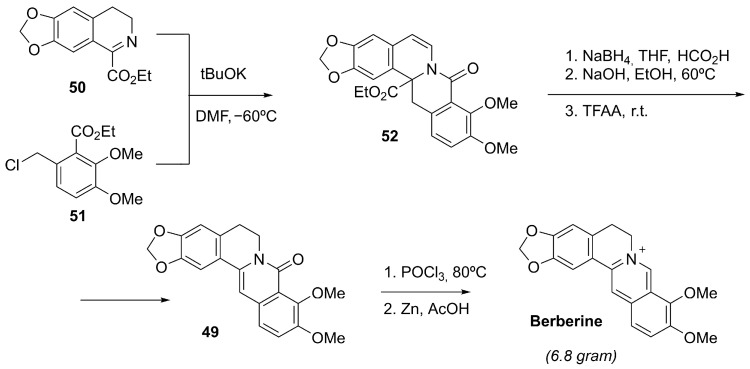 Figure 25
Figure 25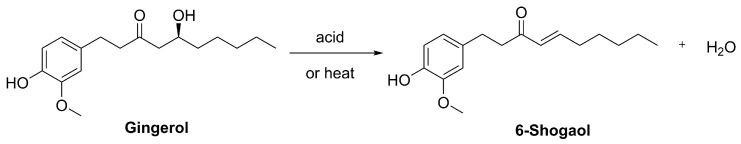 Figure 26
Figure 26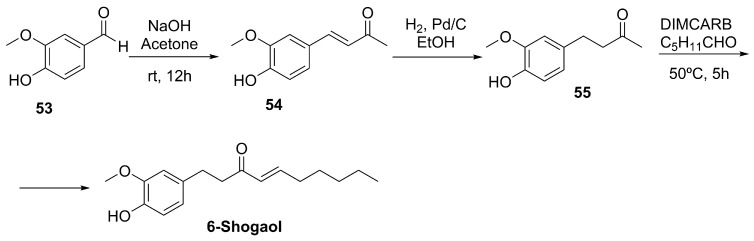 Figure 27
Figure 27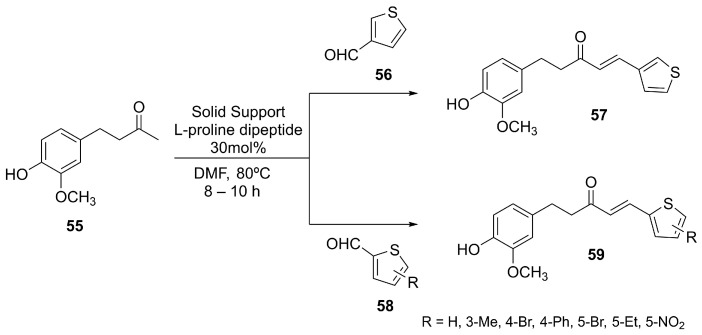 Figure 28
Figure 28- —MICIU/AEI/10.13039/501100011033
- —ERDF/EU
- —US (University of Seville)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics, phytochemicals, and oxidative stress · melanin and skin pigmentation · Berberine and alkaloids research
1. Introduction
The transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) has emerged as a pivotal regulator of cellular antioxidant and detoxification responses. Increasing evidence links NRF2 dysregulation to the pathogenesis of various dermatological disorders, including inflammatory conditions, pigmentary disorders, and photoaging [1,2,3,4]. Accordingly, the NRF2 signaling pathway is now recognized as a promising therapeutic target in the management of skin diseases [5,6,7]. This review examines the activation mechanisms of NRF2, associated skin diseases, and candidate molecules developed to modulate this pathway.
Beyond its role as a generic antioxidant switch, the KEAP1–NRF2–ARE axis is increasingly regarded as a regulatory hub that integrates the cutaneous exposome with barrier biology and immune signaling [5,6]. In the epidermis, NRF2 activity influences keratinocyte differentiation, redox buffering, xenobiotic metabolism, and stress-adaptive programs that shape inflammatory tone and tissue repair [6,7]. This translational interest is accompanied by an important nuance: while moderate and time-limited NRF2 activation is typically cytoprotective, sustained pathway hyperactivation in chronically inflamed environments may contribute to maladaptive outcomes such as aberrant differentiation or hyperproliferation [8] (Figure 1).
NRF2 activation, therefore, requires an appropriate magnitude, duration, and tissue localization of pathway engagement, often achieved through topical delivery and chemically defined small molecules. In this context, we first outline the principal molecular mechanisms governing NRF2 regulation and then critically review candidate NRF2 modulators for skin disorders, emphasizing their chemical features, available synthetic information, and the strength of preclinical and clinical evidence.
2. NRF2: Structure and Activation Pathways
2.1. NRF2 Protein Structure
NRF2 (nuclear factor erythroid 2–related factor 2), encoded by the NFE2L2 gene, is a stress-responsive transcription factor that orchestrates cellular defense programs against oxidative and electrophilic insults. NRF2 comprises seven NRF2-ECH homology domains (Neh1–Neh7) that coordinate its DNA binding, transcriptional activity, and regulated turnover (Figure 2).
The Neh2 domain harbors two KEAP1-binding motifs, ETGE (high affinity) and DLG (lower affinity), which enable KEAP1-dependent ubiquitination and proteasomal degradation under basal conditions [2]. The Neh1 domain contains the CNC-bZIP region required for heterodimerization with small Maf (sMAF) proteins and binding to antioxidant response elements (AREs). Transactivation is mediated largely by Neh4 and Neh5, which recruit co-activators such as CBP/p300, while Neh3 contributes to C-terminal transcriptional activation. The Neh6 domain provides a KEAP1-independent degradation route via degron motifs (including DSGIS and DSAPGS) that can be recognized by β-TrCP within the SCF (CUL1-based) E3 ligase complex. Finally, Neh7 has been described as a negative regulatory module through interactions with RXRα in specific cellular contexts [1,9].
2.2. Canonical Activation Pathway (KEAP1–CUL3 Axis)
Under homeostatic conditions, KEAP1 functions as a substrate adaptor for a CUL3-dependent E3 ubiquitin ligase complex, thereby maintaining low levels of NRF2 through continuous ubiquitin–proteasome mediated turnover [3,4] (Figure 1, “Basal state”). During oxidative or electrophilic stress, modification of key KEAP1 cysteine residues can impair the ability of the KEAP1–CUL3 ligase system to ubiquitinate NRF2 [10]. Consequently, NRF2 becomes stabilized, accumulates, and translocates to the nucleus, where it binds AREs and drives a broad cytoprotective transcriptional program involved in antioxidant defense, detoxification, and metabolic adaptation (Figure 1, “Activated state”). This KEAP1-centered mechanism accounts for the rapid and typically transient activation of NRF2 in response to redox imbalance [1].
2.3. Non-Canonical Activation Pathway (P62/Sqstm1–Autophagy Axis)
Beyond canonical KEAP1 cysteine sensing, NRF2 can also be activated through non-canonical mechanisms that alter KEAP1 availability. A major route involves p62/SQSTM1**,** an autophagy adaptor protein. Upon phosphorylation (notably at Ser349 within its KEAP1-interacting region), p62 binds KEAP1 and promotes its sequestration and/or turnover, thereby reducing the pool of functional KEAP1 and allowing NRF2 to accumulate and activate nuclear ARE-driven transcription. This pathway is particularly relevant in settings where p62 accumulates due to impaired or overloaded autophagy [11].
2.4. Kinase-Mediated Regulation and KEAP1-Independent Turnover
NRF2 signaling is further tuned by kinase-dependent events that influence NRF2 release, nuclear localization, and turnover. For example, PKC-dependent phosphorylation of NRF2 (e.g., at Ser40) has been associated with reduced KEAP1-mediated restraint in some experimental settings. In parallel, the GSK-3β/β-TrCP pathway provides a KEAP1-independent mechanism for limiting NRF2: GSK-3β promotes phosphorylation-dependent recognition of NRF2 Neh6 degrons by β-TrCP, facilitating SCF-mediated ubiquitination and degradation. In addition, GSK-3β–Fyn signaling has been linked to NRF2 nuclear export in certain contexts, adding a further layer of spatial control over NRF2 activity. Collectively, these pathways enable context-dependent fine-tuning of NRF2 signaling amplitude and duration beyond the KEAP1–CUL3 system [3,12].
2.5. Transcriptional/Epigenetic Modulation and Pathway Feedback
In addition to post-translational regulation, NRF2 signaling output can be influenced by changes in NFE2L2 and KEAP1 expression, including transcriptional and epigenetic mechanisms (e.g., promoter methylation and chromatin-associated regulation) that have been reported in chronic disease contexts. Importantly, NRF2 also participates in feedback control of the pathway by regulating components of its own regulatory network, including KEAP1, thereby contributing to the restoration of homeostasis once the stress response subsides. Dysregulated or persistent NRF2 activity may be associated with maladaptive consequences in specific pathological settings, emphasizing the importance of controlled and context-appropriate pathway engagement [1,4,13,14].
3. NRF2 and Skin Pathophysiology
The skin is constantly exposed to ultraviolet (UV) radiation, xenobiotics, microbes, and mechanical injury, and therefore relies on inducible cytoprotective systems to maintain homeostasis. Among these, the KEAP1–NRF2–ARE axis represents a central regulator of cutaneous defense. Under basal conditions, KEAP1 targets NRF2 for ubiquitin–proteasome degradation, thereby maintaining low basal NRF2 activity. When activated through any of the mechanisms described above, NRF2 escapes degradation, accumulates in the nucleus, and binds antioxidant response elements (AREs), thereby initiating a broad cytoprotective transcriptional program. Collectively, available data indicate that NRF2 activity contributes to keratinocyte viability, epidermal barrier integrity, and immune homeostasis [5,6,7] (Figure 1). Insufficient pathway activation may render the skin more susceptible to oxidative injury and chronic inflammation; by contrast, sustained or excessive NRF2 signaling may favor hyperproliferation, aberrant differentiation or even oncogenic progression. Accordingly, dysregulation of KEAP1–NRF2–ARE signaling has been implicated in psoriasis, atopic and allergic dermatitis, vitiligo, photodamage and photoaging, impaired wound healing, skin fibrosis, and both melanoma and non-melanoma skin cancer [5,6,7].
In the following subsections, we summarize how NRF2 contributes to the pathophysiology of major dermatological entities, leaving a detailed discussion of pharmacological NRF2 activators for the subsequent section.
3.1. Psoriasis
Psoriasis is a chronic immune-mediated dermatosis characterized by keratinocyte hyperproliferation, altered differentiation, and IL-23/IL-17-driven inflammation. Elevated oxidative stress and reduced antioxidant capacity have been consistently reported in affected patients, pointing to a relevant contribution of redox imbalance to disease pathogenesis [6].
Accumulating experimental and translational evidence suggests that NRF2 exerts context-dependent effects in psoriasis. Moderate activation of NRF2 enhances antioxidant defenses and suppresses NF-κB-dependent pro-inflammatory cytokines, which can attenuate the IL-23/IL-17 axis and reduce the severity of experimental psoriasiform dermatitis [6,15]. At the same time, NRF2 also induces so-called “stress keratins” such as K6, K16, and K17 as well as other genes that promote keratinocyte proliferation and acanthosis, features that are typical of psoriatic plaques [6]. Consistently, mouse models with epidermal NRF2 overactivity show psoriasiform lesions with strong expression of these keratins, illustrating how a pathway that is fundamentally cytoprotective can contribute to pathological hyperplasia under chronic stimulation [6]. Taken together, available data support the hypothesis that transient or moderate NRF2 activation is protective, whereas sustained activation within a chronically inflamed environment may promote epidermal hyperproliferation and plaque persistence [6,7]. This nuance is highly relevant for therapeutic strategies that aim to modulate NRF2 in psoriasis.
3.2. Atopic and Allergic Dermatitis
Atopic dermatitis (AD) arises from a combination of genetic barrier defects, type-2-skewed immunity, and heightened susceptibility to environmental triggers. Oxidative stress markers are elevated in AD, and recent proteomic and transcriptomic studies suggest that epidermal NRF2 activity is reduced in lesional skin compared with healthy controls [16,17].
NRF2 intersects with AD pathophysiology at multiple levels. First, it contributes to the regulation of cornified envelope proteins such as filaggrin, loricrin, and involucrin. These proteins are influenced not only by cytokines (IL-4, IL-13, IL-17A, IL-22) but also by AhR–NRF2 crosstalk; adequate NRF2 activity appears to support terminal differentiation and barrier formation, whereas dysregulation may exacerbate the filaggrin–loricrin–involucrin deficiency characteristic of AD [16]. Second, by strengthening antioxidant defenses and negatively regulating NF-κB and JAK–STAT pathways, NRF2 activation has been shown in experimental models to limit type-2 cytokine production and pruritogenic mediators, thereby attenuating AD-like inflammation [6,17].
The KEAP1–NRF2 system also functions as a sensor of electrophilic allergens and irritants. Contact sensitizers rapidly activate NRF2 in keratinocytes and dendritic cells; this early response facilitates adaptation to oxidative stress but also influences antigen presentation and the development of contact hypersensitivity [17]. Depending on the magnitude and duration of activation, NRF2 may either promote the resolution of inflammation or contribute to the persistence of chronic eczematous disease [6,17].
3.3. Vitiligo and Melanocyte Survival
Vitiligo is a depigmenting disorder in which melanocytes are lost through the combined effects of oxidative stress, metabolic disturbance and autoimmune attack. Lesional melanocytes and keratinocytes exhibit increased ROS, impaired mitochondrial function, and reduced activity of antioxidant enzymes such as catalase [18]. Oxidative stress in this setting promotes the release of danger signals, alters melanocyte antigens, and facilitates cytotoxic T-cell responses, thereby linking redox imbalance to autoimmunity [18].
NRF2 functions as a major transcriptional regulator of antioxidant responses in melanocytes in the face of this oxidative burden. When activated, NRF2 induces enzymes such as HO-1, NQO1, and GCLC that restore redox balance and support mitochondrial homeostasis, favoring melanocyte survival [18]. Genetic and metabolomic studies suggest that variants in NFE2L2 and several NRF2-regulated antioxidant genes may influence vitiligo susceptibility and disease course [19].
NRF2 also interacts with pathways involved in melanogenesis and melanocyte stem-cell maintenance, including Wnt/β-catenin signaling [20]. Reduced NRF2 activity could therefore contribute not only to melanocyte loss but also to impaired repigmentation. Conversely, in melanoma, constitutive NRF2 activation supports tumor survival under oxidative and therapeutic stress, highlighting the need for disease-specific NRF2 modulation [7].
3.4. Photoaging, Photodamage and Radiation Dermatitis
The skin is uniquely exposed to solar UV and visible radiation, which generate ROS, DNA lesions, and chronic low-grade inflammation. The KEAP1–NRF2 axis coordinates a substantial component of the intrinsic defense against these insults. In preclinical models, enhancement of NRF2 activity in the epidermis has been shown to increase antioxidant and phase-II detoxifying enzymes and to reduce UV-induced oxidative damage, inflammation, and apoptosis in keratinocytes and fibroblasts [5,20,21,22]. These NRF2-dependent responses translate into reduced photodamage and delayed photocarcinogenesis in hairless mice repeatedly exposed to solar-simulated radiation [5,21,22,23].
NRF2 has also been linked to premature and chronological skin aging. Work in Hutchinson–Gilford progeria syndrome demonstrated that progerin can sequester NRF2 at the nuclear lamina, thereby impairing its transcriptional activity and exacerbating oxidative stress; related mechanisms have been proposed to contribute to intrinsic photoaging in chronically sun-exposed skin [24].
Ionizing radiation used in cancer therapy produces an even more intense oxidative burden. Experimental data suggest that intact NRF2 signaling mitigates dermal and epidermal damage and reduces the severity of radiation-induced dermatitis, whereas insufficient NRF2 responses are associated with more severe acute lesions and impaired recovery [25,26]. Together, these observations support a central role for NRF2 in protecting the skin against both solar and therapeutic radiation, while also highlighting the need to avoid excessive pathway activation.
3.5. Wound Healing and Diabetic Ulcers
Cutaneous wound repair requires a tightly coordinated sequence of hemostatic, inflammatory, proliferative, and remodeling phases, all of which are sensitive to the redox environment. Experimental data indicate that NRF2 contributes to this coordination by limiting excessive oxidative stress, supporting keratinocyte and fibroblast survival, and modulating inflammatory gene expression [27].
In diabetic settings, chronic hyperglycemia and microangiopathy lead to sustained oxidative stress and a relative inadequacy of NRF2 activation. Experimental work shows that suppression of NRF2 signaling delays wound closure, prolongs inflammation, and impairs granulation tissue formation, whereas restoration of NRF2 activity normalizes antioxidant responses, improves re-epithelialization, and enhances collagen deposition [28,29,30]. These findings place NRF2 at the center of the impaired healing characteristic of diabetic foot ulcers and other chronic wounds.
3.6. Skin Fibrosis and Sclerosing Dermatoses
Fibrosing skin disorders, including systemic sclerosis, chronic radiation fibrosis, and hypertrophic scars, are driven by persistent myofibroblast activation and excessive extracellular matrix deposition. Oxidative stress and TGF-β signaling are important contributors to this process [31].
Although data are more limited than in other fields, available experimental evidence suggests that NRF2 may exert anti-fibrotic effects. Reduced NRF2 activity in keratinocytes and dermal fibroblasts is associated with increased ROS, enhanced expression of pro-inflammatory chemokines, and exaggerated matrix production [31]. In animal models, restoration of NRF2 signaling can attenuate dermal thickening and collagen accumulation, indicating that adequate NRF2 activity in the epidermis and dermis helps to restrain pro-fibrotic cascades [32]. Further work is needed to clarify how these findings translate to human conditions such as systemic sclerosis or chronic graft-versus-host disease.
3.7. Skin Cancer
The relationship between NRF2 and cutaneous carcinogenesis is highly stage dependent. During tumor initiation, NRF2 appears to behave predominantly as a tumor suppressor: by enhancing antioxidant capacity and detoxification, it reduces ROS-induced DNA damage and mutagenesis in keratinocytes [5,7]. In several chemical and UV-induced carcinogenesis models, loss of Nrf2 is associated with increased numbers and size of papillomas and squamous cell carcinomas, whereas genetic enhancement of NRF2 activity reduces tumor burden [33,34]. Constitutive NRF2 activation in established tumors promotes metabolic reprogramming that supports redox homeostasis and anabolic growth. NRF2 transcriptionally regulates enzymes involved in NADPH regeneration (e.g., glucose-6-phosphate dehydrogenase, malic enzyme 1) and glutathione biosynthesis (e.g., GCLC, GCLM), thereby enhancing intracellular reducing capacity and resistance to oxidative stress. This metabolic rewiring contributes to tumor survival, chemoresistance, and adaptation to hostile microenvironments [35,36].
Once tumors are established, however, many acquire somatic mutations in NFE2L2 or in components of the KEAP1–CUL3 E3 ligase complex, resulting in chronic NRF2 activation. This constitutive activity supports anabolic metabolism, protects malignant cells from therapy-induced stress, and contributes to immune evasion [37]. Recent work in mouse skin has shown that different modes of NRF2 gain-of-function (e.g., activating mutations versus Keap1 loss) can drive distinct squamous cell fates and influence the spectrum of cooperating oncogenic mutations [35]. Activating mutations in NFE2L2 typically disrupt KEAP1-binding motifs within the Neh2 domain, leading to impaired ubiquitination and constitutive nuclear accumulation of NRF2. In contrast, loss-of-function alterations in KEAP1 reduce its ability to target NRF2 for proteasomal degradation. Although both alterations result in persistent NRF2 activation, differences in mutational context and co-occurring genomic events may influence tumor phenotype and metabolic dependency [37].
A similar pattern is emerging in melanoma and other malignancies, where high NRF2 activity correlates with treatment resistance and poor prognosis. NRF2 can thus be viewed as a paradoxical regulator during early carcinogenesis that may confer a selective advantage when locked in a hyperactive state in established tumors [7,35,36]. From a translational perspective, these observations underscore the importance of exposure control and duration when considering NRF2-activating therapies. Although topical application generally results in localized and transient pathway engagement, the long-term safety of sustained NRF2 activation in chronically UV-exposed or premalignant skin warrants careful evaluation.
3.8. Other Inflammatory Dermatoses and Emerging Connections
Oxidative stress and redox imbalance have been described in several other inflammatory dermatoses. Increased markers of oxidative damage have been reported in lichen planus, chronic urticaria, and hidradenitis suppurativa, among others [38,39,40]. Although direct data on KEAP1–NRF2 signaling in these conditions are still scarce, it is plausible that inadequate NRF2 activation contributes to the persistent inflammation and tissue damage seen in these diseases.
NRF2 also intersects with pathways controlling keratinocyte differentiation, sebaceous gland function, and hair-follicle cycling, raising the possibility that dysregulated NRF2 contributes to disorders such as seborrheic dermatitis or certain alopecias, although this remains speculative [6,7]. As mechanistic and clinical data accumulate, it is likely that the NRF2 axis will be linked to an even broader spectrum of cutaneous pathology.
3.9. Conceptual Framework: Therapeutic Windows and Context-Dependent NRF2 Modulation in Dermatology
NRF2 activation in the skin cannot be considered uniformly beneficial or detrimental; rather, its biological effects are highly context-dependent and disease-specific. A translationally relevant framework requires distinguishing between insufficient, balanced, and excessive pathway activation.
Under physiological conditions, low basal NRF2 activity maintains redox homeostasis while allowing normal keratinocyte differentiation and immune surveillance [1,41]. In several dermatological disorders, including vitiligo, atopic dermatitis, chronic wounds, and radiation dermatitis, evidence suggests a relative NRF2 insufficiency or impaired stress-adaptive responses. Reduced NRF2 signaling has been associated with increased oxidative stress, mitochondrial dysfunction, and impaired barrier resilience in inflammatory skin disease [16]. In these contexts, pharmacological enhancement of NRF2 can restore antioxidant defenses, limits NF-κB–driven inflammatory amplification, and promote tissue protection [5,7]. Here, NRF2 activation functions as a mechanistically corrective intervention.
However, the magnitude and duration of activation critically determine the biological outcome. In psoriasis, transient NRF2 activation may attenuate oxidative stress and inflammatory cytokine production [6,15], whereas sustained or epidermis-restricted hyperactivation has been shown to promote stress keratin expression (K6, K16, K17), acanthosis, and psoriasiform phenotypes in experimental models [6,42]. Thus, therapeutic strategies in psoriasis likely require controlled, localized, and time-limited pathway activation.
The dual nature of NRF2 is most evident in skin carcinogenesis. During tumor initiation, NRF2 behaves predominantly as a tumor suppressor by limiting ROS-induced DNA damage and mutagenesis [7]. In contrast, established tumors frequently exploit constitutive NRF2 activation, often via KEAP1 or NFE2L2 mutations, to enhance metabolic reprogramming, cytoprotection, immune evasion, and therapeutic resistance [8,36,43]. In this setting, chronic pharmacological activation may be undesirable, particularly in patients with premalignant lesions or established non-melanoma skin cancer.
Beyond keratinocytes, NRF2 signaling also exhibits cell-type–specific effects in melanocytes, dermal fibroblasts, and cutaneous immune cells, further reinforcing the context-dependent nature of therapeutic modulation [44,45].
Collectively, these observations support a disease-stratified “therapeutic window” model for NRF2 modulation in dermatology, in which insufficient NRF2 activity increases oxidative vulnerability and inflammatory amplification, moderate and time-limited activation promotes cytoprotection and barrier reinforcement, and sustained or excessive activation may favor hyperproliferation, altered differentiation, or tumor support. Accordingly, NRF2-targeted strategies should be tailored to the disease context and aim for controlled pathway engagement [1,7].
The key distinctions between acute physiological activation and chronic hyperactivation across relevant cutaneous cell types are summarized in Table 1.
4. NRF2-Activating Compounds for Skin Disorders
A wide range of natural, semisynthetic, and synthetic compounds have been identified as modulators of the KEAP1–NRF2–ARE signaling axis, offering therapeutic potential for oxidative stress-mediated skin disorders. These molecules differ in origin, chemical structure, and their mechanisms of NRF2 activation. In the following section, we provide a detailed overview of selected NRF2-activating compounds, including their chemical structure, synthesis (where available), mechanism of action, dermatological applications, and current development status. Although some phytochemicals have shown potential in dermatology [46,47,48,49,50], this review focuses on pure bioactive compounds with rigorously defined chemical structures. However, the strength of evidence supporting direct KEAP1-dependent NRF2 activation varies among compounds, and in several cases NRF2 engagement appears secondary to broader redox modulation rather than a primary cysteine-sensing mechanism.
To facilitate interpretation, the compounds discussed below are organized by translational maturity in dermatology (from highest to lowest): (i) agents approved for dermatologic indications (tapinarof; dimethyl fumarate), (ii) approved drugs/supplements with clinical dermatology use or repurposing interest (indomethacin; simvastatin; folic acid), (iii) clinical-stage candidates evaluated in dermatologic settings (omaveloxolone; sulforaphane; molecular hydrogen), and (iv) preclinical candidates supported by skin-relevant models (bixin; 6-shogaol/thiophene analogs; paeoniflorin; baicalein; berberine; apigenin; afzelin).
4.1. Approved NRF2-Related Small Molecules in Dermatology
This section summarizes chemically defined small molecules (Figure 3) with dermatology regulatory approval and mechanistic links to oxidative-stress pathways relevant to NRF2 signaling, representing the highest level of translational maturity for skin indications.
4.1.1. Tapinarof (Vtama®, Benvitimod)
Tapinarof, also known as benvitimod, (E)-3,5-dihydroxy-4-isopropylstilbene (DHPS), is a chemically defined stilbenoid built on a trans-stilbene scaffold bearing a resorcinol-type phenolic motif (Figure 3) [51,52].
Although tapinarof was initially identified as a bacterial metabolite (reported in Photorhabdus spp. and related symbiotic bacteria) [52], its clinical and commercial development depends on scalable, well-controlled synthetic routes and robust impurity profiling. Accordingly, process patents describe industrially relevant preparations of tapinarof and the isolation of defined solid forms suitable for pharmaceutical use [53]. In parallel, several academic syntheses have been reported, including a practical late-stage demethylation of the (E)-3,5-dimethoxy precursor 1 (Scheme 1) under microwave irradiation using AlCl_3_/N,N-dimethylaniline or pyridinium chloride-based demethylating systems to furnish tapinarof (Scheme 1) [53].
Tapinarof engages NRF2 signaling indirectly, predominantly via AhR-driven pathways and AhR–NRF2 crosstalk in keratinocytes, thereby converging on NRF2/ARE cytoprotective programs (e.g., HO-1 and related stress-response enzymes) rather than through direct electrophilic modification of KEAP1 [54]. To date, direct covalent modification of KEAP1 cysteine residues by tapinarof has not been demonstrated. As a first-in-class topical AhR agonist, tapinarof induces transcriptional programs linked to epidermal differentiation and barrier reinforcement and modulates inflammatory signaling [2,4]. Consistent with an NRF2-focused framework, dual engagement of AhR- and NRF2-linked protective responses has been demonstrated in stress-relevant skin models [54,55], with NRF2 activation interpreted as secondary to AhR-driven transcriptional reprogramming. Clinically, tapinarof has demonstrated robust efficacy and favorable tolerability in pivotal Phase III trials in adults with plaque psoriasis and in both adult and pediatric patients with atopic dermatitis, supporting its approval for these indications. An additional Phase III clinical trial is currently ongoing to evaluate its efficacy and safety in the pediatric population with plaque psoriasis [56,57].
4.1.2. Dimethyl Fumarate (DMF)
Dimethyl fumarate (DMF) is the dimethyl ester of fumaric acid (Figure 3) and represents one of the simplest fumaric acid esters used therapeutically.
It is synthesized by esterification of fumaric acid with methanol under acidic catalysis (commonly sulfuric acid), yielding the dimethyl ester in high purity and yield. This process is well established and scalable for pharmaceutical production. Continuous-flow synthesis approaches have recently been developed to improve process efficiency and sustainability, further supporting industrial manufacture [58,59].
DMF is one of the best-characterized electrophilic activators of NRF2. It reacts covalently with cysteine residues of the cytosolic NRF2 repressor KEAP1, particularly Cys151, as demonstrated in biochemical and mutational studies, causing conformational changes that inhibit NRF2 ubiquitination and degradation [60]. This mechanism explains the dual antioxidant and anti-inflammatory effects observed in keratinocytes, neurons, and immune cells. This canonical electrophilic mechanism positions DMF among the most mechanistically validated direct KEAP1-dependent NRF2 activators currently used in clinical practice.
DMF has a long clinical history in the systemic treatment of moderate-to-severe plaque psoriasis, particularly in Europe under the brand Fumaderm^®^. Its clinical efficacy is attributed to immune modulation, reducing Th1/Th17 cytokines, and NRF2-driven antioxidant defense, thereby normalizing keratinocyte proliferation and inflammatory pathways [61]. Preclinical evidence also suggests potential benefits in other oxidative stress-driven skin diseases such as atopic dermatitis and vitiligo, although robust clinical data are still lacking [62]. DMF is approved for systemic use in psoriasis and relapsing–remitting multiple sclerosis (Tecfidera^®^). Ongoing clinical and translational research is exploring its repurposing for other inflammatory and pigmentary skin diseases, and topical DMF formulations are being developed for localized skin application [62].
4.2. Approved Drugs and Supplements with Clinical Dermatology Use or Repurposing Potential
Compounds represented in Figure 4 are approved for other indications (or used as supplements) but have been evaluated clinically in dermatology and show NRF2-associated antioxidant or cytoprotective effects, supporting their consideration as repurposing/adjunct strategies.
4.2.1. Indomethacin
Indomethacin is a well-established non-steroidal anti-inflammatory drug (NSAID) belonging to the indole acetic acid class. Its molecular structure consists of a substituted indole core linked to a chlorobenzoyl group (Figure 4), which confers potent cyclooxygenase (COX) inhibitory activity.
The most common synthetic route involves N-acylation of the appropriate arylhydrazine precursor 4 with 4-chlorobenzoyl chloride to generate the amide 5, prior to ring construction through a Fisher indole synthesis [63] (Scheme 2).
Beyond COX inhibition, indomethacin has been reported to modulate redox-sensitive signaling pathways, including activation of the KEAP1–NRF2 axis in experimental models. Mechanistically, indomethacin has been suggested to induce oxidative/electrophilic modifications of cysteine residues on KEAP1, which may inhibit KEAP1-mediated ubiquitination and proteasomal degradation of NRF2. This allows NRF2 to accumulate and translocate to the nucleus, where it binds antioxidant response elements (AREs) and drives the transcription of cytoprotective genes, including Hmox1 (heme oxygenase-1) and Nqo1 (NAD(P)H quinone dehydrogenase 1). This COX-independent mechanism was confirmed in murine myeloid cells using Nrf2 knockout models, demonstrating that indomethacin’s anti-inflammatory effects partly rely on NRF2-dependent gene expression [64]. However, direct biochemical evidence of covalent KEAP1 modification by indomethacin remains limited, and NRF2 activation may represent a secondary consequence of broader redox modulation.
Topical formulations of indomethacin have been investigated in dermatology for their ability to attenuate UVB-induced erythema and inflammation. In controlled human and animal studies, topical indomethacin (2.5–5%) significantly reduced erythema following UVB exposure, likely through inhibition of prostaglandin synthesis and modulation of local inflammatory mediators [65]. These findings support its potential utility in photodamage, actinic keratosis, and other inflammatory dermatoses where prostaglandins and oxidative stress play a key role.
Indomethacin is widely approved for systemic use and is also available as topical formulations in several countries for localized inflammatory pain conditions. Although NRF2 pathway engagement has been reported for a subset of NSAIDs, the NRF2-modulating effects of indomethacin remain primarily supported by preclinical evidence [64]. To our knowledge, no registered clinical trials have specifically evaluated indomethacin as an NRF2-targeting intervention for oxidative stress–driven dermatological disorders.
4.2.2. Simvastatin
Simvastatin is a semisynthetic derivative of lovastatin featuring a lactone ring essential for HMG-CoA reductase inhibition (Figure 4), which underlies its widespread use as a lipid-lowering agent in clinical practice [66]. At present, all commercially available simvastatin is derived either from this semisynthetic process or from biocatalytic transformation of fermentation-derived lovastatin or monacolin J (Figure 5).
Simvastatin is manufactured industrially using a semisynthetic pathway that begins with lovastatin, a metabolite produced by the fermentation of Aspergillus terreus. The conventional process involves hydrolysis of lovastatin to obtain monacolin J, followed by selective esterification at the C8 hydroxyl position (Figure 5). This approach streamlines production by eliminating the need for multiple protection and deprotection steps, and it remains the cornerstone of simvastatin manufacturing due to its efficiency and high yields [66,67]. Recent advances have focused on biocatalytic strategies, particularly those employing the acyltransferase LovD, which can be heterologously expressed in Escherichia coli. This enzyme facilitates the direct acylation of monacolin J with a 2,2-dimethylbutyryl donor in aqueous media, producing simvastatin in a single step. Ongoing efforts in directed evolution and protein engineering have enhanced the properties of LovD, improving its solubility, stability, and catalytic performance, thereby supporting sustainable and large-scale biocatalytic production of simvastatin [68].
Beyond its cholesterol-lowering effects, simvastatin has been reported to enhance the NRF2 pathway in melanocytes. The proposed mechanism includes inhibition of isoprenoid synthesis, leading to reduced prenylation of small GTPases and subsequent modulation of redox-sensitive signaling cascades that favor NRF2 nuclear translocation [69]. This results in increased expression of antioxidant enzymes, enhancing cellular resilience against oxidative stress [70]. In this context, NRF2 activation appears to be secondary to alterations in redox-sensitive signaling cascades rather than the result of direct electrophilic KEAP1 modification.
In dermatology, simvastatin has been explored as a repurposing candidate for vitiligo based on its NRF2-related antioxidant effects and anti-inflammatory activity. In vitro, it promotes NRF2 nuclear translocation in human melanocytes, increases antioxidant defenses, and may reduce oxidative stress–induced melanocyte loss [70]. However, a Phase II double-blind, placebo-controlled trial of oral simvastatin in vitiligo did not show a significant benefit versus placebo and reported adverse events (e.g., myalgia, elevated liver enzymes) [71]. Thus, despite encouraging mechanistic data, its clinical efficacy in vitiligo remains unproven.
4.2.3. Folic Acid
Folic acid (pteroylmonoglutamic acid) is a synthetic, oxidized form of folate (vitamin B9), a water-soluble micronutrient required for one-carbon metabolism and therefore essential for DNA synthesis, repair, and methylation. Structurally, it comprises a pteridine ring linked to p-aminobenzoic acid (PABA) and conjugated to a glutamic acid residue (Figure 4).
Industrial supply is based on multistep chemical synthesis, and patent literature describes optimized manufacturing routes. An improved process employs the diimine intermediate 6 that is converted to folic acid in the presence of sulfite over pH 3–8 (Scheme 3, method A), enabling substantially higher yields than earlier approaches [72]. Alternative patented methods have also been disclosed using halogenated pyruvaldehyde oxime intermediates 9 and aminopyrimidine salts under milder and potentially more environmentally friendly conditions (Scheme 3, method B) [73]. Although chemical synthesis remains the industrial standard, fermentation-based production has been explored as a greener option; for example, metabolic engineering of Ashbya gossypii through overexpression of folate-pathway genes has enabled folic acid production at mg/L–g/L titers [74].
Beyond its nutritional role, folic acid has attracted interest for antioxidant and cytoprotective effects in pigmentary disorders. In melanocyte models, folic acid has been reported to enhance resistance to oxidative stress, with mechanistic data consistent with NRF2 pathway engagement in cell-based experimental systems (including reduced KEAP1 expression and increased NRF2 nuclear translocation) and downstream cytoprotective responses [75]. To our knowledge, the precise molecular mechanism linking folic acid to NRF2 activation remains incompletely defined, and direct biochemical evidence of KEAP1 cysteine modification has not been demonstrated. Clinically, folic acid is widely used as an oral supplement with a favorable safety profile and is often included in combination regimens for vitiligo. A clinical study reported improved repigmentation outcomes when oral vitamin B12 and folic acid were combined with sun exposure compared with either intervention alone; however, larger randomized controlled trials are still needed to establish efficacy and to define optimal dosing and treatment protocols [76].
4.3. Dermatology Clinical-Stage Candidates Targeting Oxidative Stress/NRF2-Related Pathways
Compounds represented in Figure 6 have entered early clinical testing for dermatologic indications, providing proof-of-concept that pharmacologic engagement of oxidative-stress responses, including NRF2-linked programs, may translate into measurable skin benefit.
4.3.1. Omaveloxolone (RTA 408)
Omaveloxolone (Figure 6) is a semisynthetic derivative of oleanolic acid, a naturally occurring pentacyclic triterpenoid found in various medicinal plants such as Ligustrum lucidum and Olea europaea (Figure 7).
The semisynthetic route to omaveloxolone is based on late-stage functionalization of the oleanane scaffold starting from RTA 401 (11, Scheme 4), a pre-functionalized oleanolic acid derivative, to introduce substituents that tune physicochemical and ADME properties. These modifications increase overall lipophilicity and metabolic stability while enhancing NRF2-activating potency, thereby improving suitability for topical and/or systemic administration relative to the parent natural product [77]. From a synthetic standpoint, the sequence proceeds through carboxylic acid activation of 11 using diphenylphosphoryl azide (DPPA) and triethylamine in toluene, generating the corresponding acyl azide 12, which, upon heating, undergoes a Curtius rearrangement to the key isocyanate intermediate 13. Subsequent transformation of 13 (via hydrolytic conversion) affords the primary amine 14. Finally, 14 is derivatized by fluorinated acylation to deliver omaveloxolone [77].
The presence of activated alkenes as electrophilic Michael acceptor moieties in the structure facilitates covalent binding to cysteine residues on KEAP1, as demonstrated for related synthetic triterpenoids in biochemical and mutational studies, thereby promoting the NRF2 nuclear translocation and transcriptional activation of antioxidant and cytoprotective genes.
Preclinical studies demonstrated that topical application of omaveloxolone markedly attenuates radiation-induced dermatitis in murine models, reducing skin inflammation, ulceration, and epidermal thickening while enhancing expression of NRF2-dependent genes [78]. Early-phase clinical trials confirmed the safety and bioactivity of omaveloxolone in human skin, positioning it as a promising candidate for topical treatment of cutaneous radiation injury and other inflammatory skin conditions [79]. While its dermatological application is currently in Phase II with encouraging results, its systemic use has advanced further, receiving regulatory approval and continuing to be evaluated in pediatric development programs [80].
4.3.2. Sulforaphane (SFN)
Sulforaphane (SFN), shown in Figure 6, [(R)-1-isothiocyanato-4-(methylsulfinyl)butane] is a structurally simple chiral sulfoxide first isolated in 1992 from broccoli extract [81]. Like other naturally occurring isothiocyanates, it is formed through the enzymatic hydrolysis of glucosinolates present in cruciferous vegetables, a process catalyzed by the enzyme myrosinase upon plant tissue disruption (Scheme 5) [82]. Among these vegetables, broccoli is especially rich in glucoraphanin, the stable aliphatic glucosinolate that serves as the direct biochemical precursor of SFN [81,83]. The molecule is structurally simple yet notable for containing a chiral sulfinyl group. As in other naturally occurring isothiocyanates, the sulfinyl sulfur in SFN exhibits an R-configuration, which results from a stereospecific oxidation catalyzed by flavin-containing monooxygenases [81].
Various synthetic and semisynthetic methods have been developed for SFN production, ranging from enzymatic hydrolysis to total chemical and asymmetric syntheses. In the context of semisynthesis, the biotransformation that naturally occurs in cruciferous vegetables, namely, the enzymatic hydrolysis of glucosinolates to isothiocyanates, can be reproduced in the laboratory to obtain high-yield production. A gram-scale, highly enantioselective method employs purified myrosinase from Tuscan black kale in a biphasic system to hydrolyze glucoraphanin, yielding enantiopure R-sulforaphane (>99% yield) (1.09 g of pure R-SFN from 150 g of defatted seed meal) [84].
Although SFN is defined by its chiral sulfoxide center, many syntheses have focused on producing it in racemic form. Racemic synthesis is simpler, more cost-effective, and enables the preparation of larger amounts of SFN compared to asymmetric routes that require chiral catalysts or auxiliaries. As an example of racemic synthesis of SFN, a practical multi-step approach starts from commercially available phthalimide salt 16 and 1,4-dibromobutane 15 (Scheme 6) [85,86]. The sequence begins with a Gabriel synthesis to form N-(4-bromobutyl) phthalimide 17, followed by substitution with sodium methanethiolate to yield N-(4-(methylthio)butyl) phthalimide 18. Subsequent hydrazinolysis releases 4-(methylthio)butylamine 19, which undergoes thiophosgene-mediated transformation into the corresponding isothiocyanate 20. Finally, oxidation of the thioether with m-CPBA furnishes racemic SFN in reliable yield.
With the aim of improving previously described methodologies, Chen et al. developed an alternative route to racemic SFN starting from 4-aminobutan-1-ol, 21 [87]. This method avoids the use of toxic reagents such as thiophosgene, employs a mild one-pot conversion of the amine 19 to the isothiocyanate 20 using O-phenyl chlorothioformate, and achieves a higher overall yield (64%), making it suitable for multigram synthesis (Scheme 7).
Regarding stereoselective synthesis, Whitesell and Wong reported a classic, versatile synthesis of both enantiomers of SFN via the chiral sulfinate ester derived from trans-2-phenylcyclohexanol [88]. Starting from the trans-(1R,2S) alcohol, 24, through thionyl chloride and dimethyl zinc, they achieved the (R)-methane sulfinate 25 with a high stereochemical induction (92% de), which delivered R-sulforaphane in good yield (Scheme 8).
Building upon the diacetone-D-glucofuranose (DAG) chiral auxiliary strategy [89], our group synthesized the chiral sulfinate ester S-32 as precursor and achieved stereoinversion via a Grignard-mediated pathway. Subsequent Staudinger/aza-Wittig transformation delivered R-SFN (Scheme 9) [90,91]. This methodology demonstrated excellent versatility, enabling the stereoselective preparation of structurally diverse SFN derivatives [92,93,94].
Schenk and Dürr developed one of the first enantioselective syntheses of R-SFN using the ruthenium complex 33-Cl, with the chiral bisphosphine ligand (R,R)-CHIRAPHOS as a stereodirecting auxiliary (Scheme 10) [95]. The sulfonium salt 34 was asymmetrically oxidized to the chiral sulfoxonium salt 35 with ~80% ee, which was subsequently converted into the isothiocyanate 37 through thiophosgene treatment, affording R-SFN after deprotection (Scheme 10). Although this method provided one of the first examples of catalytic stereocontrol in sulforaphane synthesis, its relatively modest enantioselectivity and multistep nature have limited its use compared to auxiliary-based or biocatalytic approaches.
Given the relative instability and liquid state of SFN, various research groups have focused on developing structurally related analogs with improved physicochemical properties [96,97,98,99,100,101,102,103]. These efforts aim to enhance stability, bioavailability, and potency, often favoring solid compounds more suitable for pharmaceutical development. The following figure illustrates the chemical structures of some of the most representative SFN analogs (Figure 8).
SFN activates the NRF2 pathway primarily through covalent modification of cysteine residues on KEAP1. This canonical electrophilic mechanism has positioned SFN as one of the most extensively characterized direct KEAP1-dependent NRF2 activators. SFN contains an electrophilic isothiocyanate group, which reacts with specific cysteines, particularly Cys151, Cys273, and Cys288, on KEAP1, as confirmed in biochemical and mutational studies using cysteine-targeted approaches.
SFN is recognized for its strong antioxidant and cytoprotective properties, which have attracted considerable interest in dermatological use, particularly in addressing photoaging and inflammatory skin diseases. Notably, SFN has demonstrated significant efficacy in shielding the skin from UV-induced damage and mitigating the effects of cutaneous aging [19,33].
The clinical development of SFN in dermatology is currently at an early stage, with promising results from both clinical and preclinical studies. A Phase I clinical trial (NCT03730649) at Johns Hopkins University is investigating the effects of topical SFN lotion on markers of skin aging and UV/visible light exposure in healthy adults; although the sample size is small, this represents an important step toward clinical application [104]. Nevertheless, dermatology-specific clinical evidence remains limited, and most mechanistic data derive from preclinical or systemic studies. In addition, a completed Phase II study (NCT00982319) assessed oral broccoli sprout extract, rich in SFN, for its systemic antioxidant effects, providing further rationale for its use in skin health, even though this trial was not dermatology-specific [105]. Preclinical research supports these findings, as numerous in vitro and animal studies have shown that SFN can protect against UV-induced skin damage, reduce inflammation and oxidative stress, and ameliorate features of atopic dermatitis by activating the NRF2/HO-1 axis and reducing immune cell infiltration and IgE levels [106]. These data together highlight the growing interest and therapeutic potential of SFN for dermatological applications. It remains one of the most advanced natural NRF2 activators being explored for dermatological indications.
4.3.3. Molecular Hydrogen (H2)
It is worth noting that molecular hydrogen (H_2_, Figure 6), although not synthesized in the conventional chemical sense for therapeutic purposes, can be administered through various delivery systems, including hydrogen-rich water, hydrogen gas inhalation, and topical hydrogen-enriched solutions [107]. These methods have been developed to introduce molecular hydrogen into biological systems, where it can exert selective antioxidant and cytoprotective effects.
Molecular hydrogen (H_2_) has been reported to modulate NRF2 indirectly by reducing oxidative stress, thereby favoring NRF2 stabilization and nuclear accumulation. In dermatology-relevant models, hydrogen-rich media protect keratinocytes and melanocytes under oxidative challenge by lowering intracellular ROS and limiting apoptosis, consistent with reinforcement of endogenous antioxidant defenses. Importantly, molecular hydrogen does not function as an electrophilic KEAP1 modifier, and direct cysteine modification has not been demonstrated. NRF2 activation in this context is therefore interpreted as secondary to redox modulation rather than canonical KEAP1-dependent sensing. In human melanocytes, H_2_ has been reported to enhance NRF2 signaling in cellular models; however, covalent KEAP1 modification has not been demonstrated [108,109].
Molecular hydrogen is currently being explored in both preclinical and early-phase clinical studies for a range of oxidative and inflammatory diseases [110]. In dermatology, its use remains experimental, with most data coming from in vitro and animal models. However, its favorable safety profile and antioxidant properties make it a promising candidate for adjunctive therapy in conditions like vitiligo. Current evidence remains largely preclinical or exploratory, and controlled dermatology-specific clinical validation is still limited.
4.4. Preclinical Dermatology Candidates with Defined Structures and NRF2-Linked Mechanisms
In this section, we describe several chemically defined compounds (Figure 9) that have shown compelling preclinical efficacy in skin-relevant models and activate NRF2 either directly (e.g., via KEAP1 modulation) or indirectly through upstream signaling pathways; together, they constitute a promising pipeline for future topical and/or systemic development.
For most of the compounds discussed in this section, NRF2 pathway engagement has been demonstrated primarily in cell-based or animal models. In several cases, direct biochemical validation of KEAP1 cysteine modification is lacking; therefore, NRF2 activation should be interpreted as indirect or context-dependent.
4.4.1. Bixin
Bixin is a structurally defined apocarotenoid (C25) (Figure 9) and the major lipophilic pigment of annatto (Bixa orellana), widely used as a natural colorant. In practice, bixin is commonly obtained by extraction and purification from annatto preparations rather than by de novo total synthesis [111].
Beyond its pigmentary applications, bixin has emerged as a compound with robust NRF2 pathway engagement and strong in vivo dermatological relevance. In murine models of solar UV exposure, systemic administration of bixin attenuated acute photodamage, including oxidative DNA damage and inflammatory responses, and genetic validation showed that protection is NRF2-dependent [112]. Mechanistically, bixin has been reported to activate NRF2 via the canonical KEAP1 sensor mechanism involving Cys151, consistent with an electrophile-driven mode of NRF2 stabilization and ARE gene induction [22,113]. Although NRF2 dependence has been genetically validated in vivo, direct biochemical demonstration of covalent KEAP1 cysteine adduct formation by bixin remains limited in skin-specific systems. Complementing systemic studies, topical bixin has also been shown to confer NRF2-dependent protection against UV-induced photodamage and to mitigate stress-induced hair graying in a PUVA-related model, supporting its potential as a topical cytoprotective agent [5,23,114]. Collectively, bixin exemplifies an NRF2-activating small molecule with dermatological in vivo validation and provides a rationale for photoprotective strategies that enhance endogenous antioxidant defenses beyond conventional UV filtering [112,114].
4.4.2. Paeoniflorin
Paeoniflorin (PF) is a monoterpene glycoside primarily isolated from the roots of Paeonia species, especially Paeonia lactiflora and Paeonia suffruticosa. It is biosynthesized in plants via the terpenoid pathway and consists of a pinane skeleton glycosylated at the C-1 position with a β-D-glucopyranosyl moiety (Figure 9) [115].
The traditional method for obtaining PF involves solvent-based extraction from dried Paeonia lactiflora roots, followed by chromatographic quantification, although concerns about solvent toxicity and yield variability persist. To address these limitations, green extraction strategies such as the use of deep eutectic solvents (DESs) have been proposed, achieving improved selectivity and sustainability [116]. Comparative transcriptomic analyses across distinct P. lactiflora genotypes have identified candidate genes associated with terpenoid backbone and monoterpenoid biosynthesis, revealing genotype- and organ-dependent differences in upstream precursor pathways and monoterpene synthase expression, and providing a resource for future metabolic engineering efforts [113]. MicroRNA (miRNA) regulatory networks have been implicated in the tissue-specific regulation of PF accumulation and monoterpenoid-related pathways in P. lactiflora, highlighting an additional post-transcriptional layer of control within secondary metabolism [117]. In parallel, microbial biotransformation has emerged as a complementary approach: a paeoniflorin-converting enzyme from Cunninghamella blakesleeana has been identified and functionally characterized, supporting the feasibility of enzyme-based conversion routes that could be leveraged for scalable biocatalytic processing [118]. Recent advances have enabled the semisynthetic modification of PF, with several novel derivatives synthesized to enhance its antioxidant and pharmacological potential through targeted structural modifications, including reacting PF with various sulfonyl chlorides 38 (Scheme 11) [119].
PF activates the NRF2 signaling pathway primarily through modulation of redox-sensitive upstream targets. Experimental evidence suggests that PF may disrupt the KEAP1–NRF2 complex by inducing oxidative or electrophilic modifications of KEAP1, thereby inhibiting NRF2 degradation. Additionally, PF may engage kinase-mediated mechanisms, such as PI3K/Akt or MAPK pathways, that influence the phosphorylation status of NRF2, facilitating its stabilization and activation. In a murine model of ulcerative colitis, PF treatment markedly increased NRF2 and HO-1 expression while reducing oxidative stress markers, supporting NRF2 pathway engagement consistent with upstream redox- and kinase-mediated mechanisms [120]. Direct biochemical evidence of covalent KEAP1 cysteine modification by paeoniflorin has not been demonstrated, and NRF2 activation is therefore best interpreted as indirect and context-dependent.
Although direct clinical development of PF for dermatologic indications is still limited, mechanistic studies support its relevance in redox- and inflammation-driven epithelial injury. PF attenuated UVA-associated oxidative stress through NRF2 signaling in skin-relevant cellular settings [121] and inhibited NLRP3 inflammasome activation via NRF2/HO-1 in a Sjögren’s syndrome model, pointing to a broader anti-inflammatory stress-adaptive mechanism [122]. In addition, PF reduced NF-κB/NLRP3–linked inflammation and improved outcomes in diabetic foot ulcer models, including keratinocyte-relevant in vitro systems [123]. Clinically, evidence is available mainly for Total Glucosides of Peony preparations (with paeoniflorin as a major constituent), which showed efficacy and safety in a multicenter, randomized, double-blind, placebo-controlled trial in primary Sjögren’s syndrome [124] and reduced hepatotoxicity when co-administered with methotrexate and leflunomide in rheumatoid arthritis studies [125,126].
4.4.3. Baicalein
Baicalein (5,6,7-trihydroxy-2-phenyl-4H-1-benzopyran-4-one; Figure 9) is a naturally occurring flavone with a planar 2-phenylchromen-4-one scaffold bearing hydroxyl groups at C-5, C-6, and C-7, a substitution pattern associated with strong polyphenolic antioxidant activity. It is mainly obtained by extraction from Scutellaria species (Lamiaceae), particularly Scutellaria baicalensis and Scutellaria lateriflora, where it occurs as a major bioactive constituent together with its glycoside baicalin (Figure 10) [127,128]. Baicalein has also been reported in other botanical sources, including Oroxylum indicum [129], and has been isolated from Thymus vulgaris (thyme) [130].
In plants, baicalein is produced through the flavonoid pathway. In the 4-deoxyflavone branch characteristic of Scutellaria roots, chrysin (5,7-dihydroxyflavone) is generated by flavone synthase and can subsequently undergo enzymatic 6-hydroxylation (flavone 6-hydroxylase) to yield baicalein. Although extraction from Scutellaria species remains the main source for experimental use, synthetic approaches have been developed to secure larger quantities. For multigram supply, Kim et al. fine-tuned a reported route by lowering the loading of a strong Lewis acid in the key Fries rearrangement and using tetrabutylammonium iodide (TBAI) as an additive to accelerate demethylation of precursor 42 in refluxing HBr, enabling preparation of >10 g of baicalein (Scheme 12) [131].
Using also trimethoxyphenol 39 as a starting material, Chen et al. described a total synthesis involving the condensation of the ketone derivative 43 with benzaldehyde, followed by intramolecular cyclization (I_2_/DMSO) of the α,β-unsaturated aromatic ketone 41 and high-temperature demethylation of chromenone 42, affording baicalein in 59% overall yield (Scheme 13) [132].
In vitiligo-relevant human melanocytes exposed to H_2_O_2_, baicalein has been reported to activate NRF2, promoting nuclear translocation and antioxidant defenses, which lower ROS and apoptosis; these effects are abolished by NRF2 silencing and appear independent of ERK or PI3K/Akt signaling [133]. Although NRF2 dependence is supported by silencing experiments, direct biochemical evidence of covalent KEAP1 cysteine modification in skin-relevant systems remains limited. More recent evidence suggests baicalein may also weaken KEAP1–NRF2 binding via direct interaction with KEAP1, consistent with Michael-acceptor reactivity [134].
Topical baicalein reduced disease severity and inflammatory mediators in an NC/Nga mouse model of atopic dermatitis induced by Dermatophagoides pteronyssinus [135], and oral administration limited acute UVB-induced skin thickening while suppressing MMP-9/VEGF and COX-2/NF-κB signaling in hairless mice, consistent with photoprotective activity [124]. Overall, dermatology-focused evidence remains preclinical and derives from in vitro studies and animal models (e.g., vitiligo-relevant oxidative stress, atopic dermatitis, and UV injury) [133,134,135,136]. In humans, oral baicalein tablets have shown acceptable safety and predictable pharmacokinetics in a Phase I multiple-ascending-dose study in healthy volunteers [137], and a Phase II randomized, placebo-controlled trial has been registered (NCT03830684) for a non-dermatologic indication, with no results posted to date [138].
4.4.4. Berberine
Berberine is a natural isoquinoline alkaloid characterized by a planar, quaternary ammonium structure (Figure 9), and it is primarily extracted from the roots, rhizomes, and bark of plants such as Berberis vulgaris and other species of the Berberidaceae family [139].
While berberine has traditionally been obtained from medicinal plants, several efficient laboratory synthesis methods have also been established. Tajiri et al. achieved the total chemical synthesis of berberine (and related analogs) using an intramolecular Heck coupling of 48 as the central step to construct the isoquinolinium core. After final reduction in the amide 49, this approach enabled the generation of fully synthetic berberine (Scheme 14) [140].
Subsequently, Yan et al. patented an alternative scalable, convergent synthesis of berberine hydrochloride employing a trifluoroacetic anhydride–promoted decarbonylative elimination of the intermediate 52, facilitating gram-scale production without requiring complex purification procedures (Scheme 15) [141].
Berberine has been reported to enhance NRF2 pathway activity, at least in part, by alleviating intracellular oxidative stress; accordingly, NRF2 silencing in human melanocytes markedly reduces berberine-mediated protection against H_2_O_2_-induced damage [142]. In vitro, berberine has shown relevance for pigmentary disorders such as vitiligo by engaging both the NRF2/ARE and melanogenic programs. In PIG1 melanocytes exposed to H_2_O_2_, berberine increased NRF2 nuclear translocation, boosted antioxidant defenses (e.g., HO-1 and SOD), and lowered ROS levels and apoptosis, while also enhancing melanogenesis through upregulation of MITF and its target genes in an NRF2-dependent manner [142]. NRF2 activation in this context appears to result from modulation of upstream redox-sensitive signaling rather than direct electrophilic KEAP1 modification. Although clinical evidence is still lacking, topical delivery systems (e.g., berberine-loaded hyalurosomes) have shown activity in preclinical vitiligo models, improving biochemical readouts and pigmentation-related gene expression [143].
4.4.5. Apigenin
Apigenin (4′,5,7-trihydroxyflavone) (Figure 9) is a naturally occurring flavonoid widely found in plants such as parsley, chamomile, and celery, as well as various other fruits and vegetables. In plants, it is biosynthesized via the phenylpropanoid pathway, starting from phenylalanine, which is transformed through a series of enzymatic steps involving phenylalanine ammonia-lyase (PAL), chalcone synthase (CHS), and flavone synthase (FNS) to yield the core flavone structure [144].
In addition to its natural production, apigenin can also be synthesized through chemical strategies, such as the cyclization of chalcone intermediates or targeted hydroxylation of flavone precursors, approaches that are useful in pharmaceutical development [145]. Nevertheless, the most widely used method remains plant-based extraction, employing both classical solvent techniques (e.g., ethanol, methanol) and more sustainable technologies such as ultrasound-assisted extraction, supercritical fluid extraction, and microwave-assisted extraction [146,147].
Apigenin has been reported to enhance NRF2 signaling activity in experimental models primarily by interfering with its cytoplasmic repressor KEAP1, leading to NRF2 stabilization and release [148]. In addition, apigenin has been shown to activate upstream kinases such as ERK and PI3K/Akt, which in turn phosphorylate NRF2, further promoting its dissociation from KEAP1 [149]. It has also been demonstrated that apigenin increases intracellular reactive oxygen species (ROS) in a controlled manner, serving as a redox signal to initiate KEAP1-NRF2 uncoupling without causing cytotoxicity [150,151]. While NRF2 nuclear translocation and downstream antioxidant gene induction have been observed, a canonical KEAP1 cysteine-sensing mechanism has not been formally established.
These multifaceted interactions enable apigenin to act as a robust NRF2 activator across different tissues, particularly in settings of oxidative stress [152]. In dermatology, apigenin is therefore of interest for conditions in which redox imbalance and melanocyte dysfunction contribute to disease pathogenesis. In vitiligo-relevant models, apigenin has been reported to engage the NRF2 pathway and reinforce antioxidant defenses, thereby limiting ROS-driven melanocyte apoptosis. In addition, it may help preserve pigment cell homeostasis by modulating redox-sensitive signaling within the skin microenvironment, including pathways implicated in the redox-sensitive regulation of melanocyte homeostasis in this niche [153].
At present, apigenin remains at a preclinical stage for dermatological indications. While multiple in vitro and animal studies support its bioactivity and tolerability, to date no dermatology-specific clinical trials have been registered. Nevertheless, apigenin is already available in cosmetic products and dietary supplements, reflecting its favorable safety profile and broad biological activity [154].
4.4.6. Afzelin
Afzelin, 4′,5,7-trihydroxy-3-(α-D-rhamnopyranosyloxy) flavone, is a naturally occurring flavonol glycoside, specifically the 3-O-rhamnoside of kaempferol (Figure 9). It is commonly found in various plant species, particularly those rich in kaempferol derivatives, such as Prunus persica, Fagopyrum esculentum, and Elaeagnus umbellata. These natural matrices constitute the primary source of afzelin for pharmacological studies, with the compound occurring as a minor constituent in polar flavonoid-enriched fractions [155,156,157].
Despite its natural abundance, the increasing demand for standardized and reproducible sources has prompted the exploration of alternative production methods. Biotechnological approaches, particularly those involving glycosyltransferase-mediated glycosylation, offer a viable route for the biosynthesis of afzelin and structurally related flavonol glycosides. Plant UDP-glycosyltransferases (UGTs), which catalyze the regioselective transfer of sugars to specific hydroxyl groups of flavonoids, have been studied extensively in this context. Kaempferol-specific UGTs capable of catalyzing 3-O-rhamnosylation represent promising tools for the generation of afzelin analogs under controlled conditions [158].
Although afzelin has not yet been synthesized via total chemical synthesis in an industrial context, chemoenzymatic strategies and recombinant UGT platforms are under active investigation. These systems aim to enable scalable, environmentally benign production routes, especially for applications requiring consistent bioactive profiles in pharmaceutical and cosmeceutical formulations [159].
Afzelin has been shown to indirectly activate the NRF2 pathway through inhibition of glycogen synthase kinase 3 beta (GSK-3β), a negative regulator of NRF2. By suppressing GSK-3β, afzelin prevents the phosphorylation and subsequent nuclear export of NRF2, thereby increasing its nuclear accumulation and activation of target antioxidant genes. This mechanism is consistent with KEAP1-independent stabilization of NRF2 rather than canonical cysteine-sensing modification of KEAP1. In pigment cell models related to vitiligo, afzelin has demonstrated protective effects against oxidative stress. Its capacity to enhance NRF2 signaling contributes to the maintenance of redox balance and preservation of melanocyte function [160].
Currently, the application of afzelin in dermatology remains at the preclinical stage. Its effects have been demonstrated in vitro, but there are no clinical trials or commercially available dermatological products containing afzelin to date [160]. Further studies are needed to validate its pharmacokinetics, formulation, and efficacy in vivo.
4.4.7. 6-Shogaol
6-Shogaol, (4E)-1-(4-Hydroxy-3-methoxyphenyl)dec-4-en-3-one (Figure 9), is a bioactive compound derived from gingerol, the principal pungent component of fresh ginger (Scheme 16).
6-Shogaol can be obtained either through semisynthesis or through total chemical synthesis starting from simpler aromatic precursors. Both pathways are relevant for pharmacological development and structural modification. In semisynthesis, 6-shogaol is typically obtained through acid- or heat-induced dehydration of 6-gingerol, a reaction that naturally occurs during processing and involves the removal of a water molecule from the β-hydroxy ketone moiety to form the α,β-unsaturated ketone (Scheme 16) [161,162]. A more sophisticated and environmentally friendly approach has been developed, employing the acidic ionic liquid 1-butyl-3-methylimidazolium hydrosulfate ([Bmim]HSO_4_) under ultrasound irradiation to achieve the dehydration more efficiently and sustainably [163].
Alternatively, 6-shogaol can be synthesized de novo starting from vanillin 53. The typical pathway involves a Claisen–Schmidt condensation between vanillin and acetone to produce dehydrozingerone 54, followed by reduction, aldol condensation with hexanal, and dehydration steps to yield 6-shogaol (Scheme 17) [164].
To improve stability and pharmacological performance, synthetic analogs of 6-shogaol have been developed via thiophene derivatization (Scheme 18), considering that the vanillyl and the α,β-unsaturated carbonyl groups present in 6-shogaol are considered essential pharmacophores responsible for its biological activity [165]. The prepared shogaol thiophene compounds (STCs) (57 and 59, Scheme 18) showed anti-inflammatory effects via NRF2-dependent and -independent mechanisms. The STCs exhibited lower reactivity with thiols than 6-shogaol, suggesting they may cause fewer side effects and display greater metabolic stability.
Mechanistically, 6-shogaol, as an electrophilic molecule, is proposed to modify cysteine residues on KEAP1 via covalent bond formation, thereby releasing NRF2 and promoting the transcription of antioxidant response genes. Although its electrophilic structure is consistent with KEAP1 cysteine targeting, direct biochemical demonstration of covalent KEAP1 adduct formation in dermatologic models remains limited.
6-Shogaol is currently in preclinical development, with no registered clinical trials for dermatological use to date. In vitro studies using human epidermal melanocytes (HEMn-MPs) have demonstrated that 6-Shogaol activates the NRF2/ARE pathway, upregulates antioxidant enzymes (such as HO-1 and NQO1), and protects against oxidative stress–induced cell damage, including challenges from H_2_O_2_ and rhododendrol. These findings underscore its potential for preventing melanocyte loss in vitiligo [166]. While these results are promising, future work is necessary, including in vivo efficacy studies and eventual clinical trials, to fully establish its role in the treatment of pigmentary disorders.
To facilitate translational interpretation and comparative evaluation of NRF2-modulating compounds discussed above, a summary of their chemical class, mechanism of NRF2 engagement, dermatologic indications, level of evidence, and regulatory status is provided in Table 2.
5. Conclusions
Pharmacological modulation of the KEAP1–NRF2–ARE pathway represents a promising, mechanism-based strategy for dermatological conditions in which oxidative stress and chronic inflammation contribute to disease initiation or persistence. The evidence discussed in this review indicates that diverse natural, semisynthetic, and synthetic compounds can reinforce endogenous cytoprotective programs, support epidermal barrier resilience, and improve the stress tolerance of key cutaneous cell types, including keratinocytes and melanocytes. Importantly, the translational landscape is heterogeneous: while some NRF2 modulators have reached advanced clinical development and established therapeutic use, many others remain supported primarily by preclinical data despite encouraging in vitro and in vivo findings. Moving forward, rigorous, well-controlled clinical studies will be essential to define clinical efficacy, long-term safety, and the optimal magnitude, duration, and tissue localization of NRF2 engagement—particularly for topical delivery, where maximizing local benefit while minimizing systemic exposure is more readily achievable. Overall, targeting NRF2 emerges as a versatile approach with potential value across a spectrum of oxidative stress–related skin disorders, including photoaging, pigmentary alterations, inflammatory dermatoses, and cutaneous injury states, provided that pathway activation is appropriately tuned to disease context and treatment goals.
6. Future Directions
Despite substantial advances in understanding the KEAP1–NRF2–ARE axis in skin biology, important translational gaps remain before NRF2-targeted approaches can be broadly and safely implemented in dermatology. A major priority is to better resolve cell-type-specific NRF2 programs across keratinocytes, melanocytes, fibroblasts, and cutaneous immune cells, as these cell types likely differ in redox thresholds and downstream transcriptional outputs under inflammatory, pigmentary, fibrotic, and neoplastic conditions. In this context, combining disease-relevant models with high-resolution molecular profiling may help clarify when NRF2 activation is truly corrective versus potentially maladaptive.
Defining disease-specific therapeutic windows remains equally important. As emphasized in this review, insufficient NRF2 signaling can contribute to oxidative vulnerability and persistent inflammation, whereas prolonged or excessive activation may favor hyperproliferation, altered differentiation, or tumor-supportive stress adaptation in certain settings. Future pharmacological development should therefore prioritize control over the magnitude, duration, and tissue localization of pathway engagement. Topical delivery and formulation strategies that promote localized, time-limited NRF2 activation are particularly attractive for maximizing benefit while limiting systemic exposure.
Progress in clinical translation will also depend on the development of robust biomarkers of NRF2 pathway engagement. Practical pharmacodynamic readouts, such as ARE-driven gene expression signatures (e.g., HO-1, NQO1 and related targets) in skin samples, could support patient stratification, dose optimization, and early detection of excessive pathway activation. Incorporating such biomarkers into early clinical studies would strengthen mechanistic attribution and improve interpretability of clinical outcomes.
Long-term safety evaluation remains essential, especially for chronic dermatoses that may require prolonged treatment and for individuals with extensive UV exposure or premalignant lesions. Given the well-recognized context dependence of NRF2 in carcinogenesis, future studies should explicitly consider exposure duration, cumulative dose, and patient risk factors when evaluating NRF2-activating therapies.
Finally, rational combination strategies may expand therapeutic utility while improving tolerability. NRF2 modulation could be explored alongside barrier-repair agents, targeted anti-inflammatory therapies, phototherapy, or other redox-modulating interventions, with the goal of achieving efficacy at lower individual drug exposures. Overall, future research should move beyond generalized antioxidant paradigms toward disease-stratified, biomarker-guided NRF2 modulation that aligns pathway engagement with the specific biology and treatment goals of each dermatologic indication.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yamamoto M. Kensler T.W. Motohashi H. The KEAP 1–NRF 2 system: A thiol-based sensor–effector apparatus for maintaining redox homeostasis Physiol. Rev.2018981169120310.1152/physrev.00023.201729717933 PMC 9762786 · doi ↗ · pubmed ↗
- 2Itoh K. Wakabayashi N. Katoh Y. Ishii T. Igarashi K. Engel J.D. Yamamoto M. KEAP 1 represses nuclear activation of antioxidant responsive elements by NRF 2 through binding to the amino-terminal Neh 2 domain Genes Dev.199913768610.1101/gad.13.1.769887101 PMC 316370 · doi ↗ · pubmed ↗
- 3Kobayashi A. Kang M.-I. Okawa H. Ohtsuji M. Zenke Y. Chiba T. Igarashi K. Yamamoto M. Oxidative stress sensor KEAP 1 functions as an adaptor for CUL 3-based e 3 ligase to regulate proteasomal degradation of NRF 2Mol. Cell. Biol.2004247130713910.1128/mcb.24.16.7130-7139.200415282312 PMC 479737 · doi ↗ · pubmed ↗
- 4Zhang D.D. Lo S.C. Cross J.V. Templeton D.J. Hanninke M. KEAP 1 is a redox-regulated substrate adaptor protein for a CUL 3-dependent ubiquitin ligase complex Mol. Cell. Biol.200424109411095310.1128/MCB.24.24.10941-10953.200415572695 PMC 533977 · doi ↗ · pubmed ↗
- 5Kahremany S. Hofmann L. Gruzman A. Dinkova-Kostova A.T. Cohen G. NRF 2 in dermatological disorders: Pharmacological activation for protection against cutaneous photodamage and photodermatosis Free Radic. Biol. Med.202218826227610.1016/j.freeradbiomed.2022.06.23835753587 PMC 9350913 · doi ↗ · pubmed ↗
- 6Kobayashi E.H. Suzuki T. Funayama R. Nagashima T. Hayashi M. Sekine H. Tanaka N. Moriguchi T. Motohashi H. Nakayama K. NRF 2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription Nat. Commun.201671162410.1038/ncomms 1162427211851 PMC 4879264 · doi ↗ · pubmed ↗
- 7Dinkova-Kostova A.T. Copple I.M. Advances and challenges in therapeutic targeting of NRF 2Trends Pharmacol. Sci.20234413714910.1016/j.tips.2022.12.00336628798 · doi ↗ · pubmed ↗
- 8De Nicola G.M. Karreth F.A. Humpton T.J. Gopinathan A. Wei C. Frese K. Mangal D. Yu K.H. Yeo C.J. Calhoun E.S. Oncogene-induced NRF 2 transcription promotes ROS detoxification and tumorigenesis Nature 201147510610910.1038/nature 1018921734707 PMC 3404470 · doi ↗ · pubmed ↗