Glnk Mediates Carbapenem Resistance Through the NtrB/NtrC-OprD Regulatory Pathway in Pseudomonas aeruginosa
Xiaomeng Sun, Yiming Li, Xuetao Gong, Qitong Du, Yongxin Jin, Zhihui Cheng, Shouguang Jin, Weihui Wu

TL;DR
This study reveals how a gene mutation in Pseudomonas aeruginosa affects carbapenem resistance through a regulatory pathway involving nitrogen metabolism.
Contribution
The discovery of a new regulatory pathway linking GlnK, NtrB/NtrC, and OprD to carbapenem resistance in P. aeruginosa.
Findings
A mutation in the glnK gene decreases carbapenem resistance in P. aeruginosa.
The NtrB/NtrC system regulates oprD, which is crucial for carbapenem uptake.
Deleting ntrB, ntrC, or oprD in the glnK mutant restores carbapenem resistance.
Abstract
Pseudomonas aeruginosa is a major causative agent of nosocomial infections worldwide. Carbapenems are the first-line agents for combating severe P. aeruginosa infections. However, the increasing prevalence of carbapenem-resistant P. aeruginosa (CRPA) has developed as a critical threat to global healthcare systems. In this study, we demonstrated that a mutation in the core nitrogen metabolism regulatory gene glnK decreases carbapenem resistance in P. aeruginosa. OprD, the major porin for carbapenem uptake, is upregulated in the glnK mutant, resulting in decreased resistance. We further found that the NtrB/NtrC two-component regulatory system is upregulated in the glnK mutant. An electrophoretic mobility shift assay (EMSA) and genetic studies revealed a direct regulatory role of NtrC on the expression of oprD. Deletion of ntrB, ntrC, or oprD in the glnK mutant restored the bacterial…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
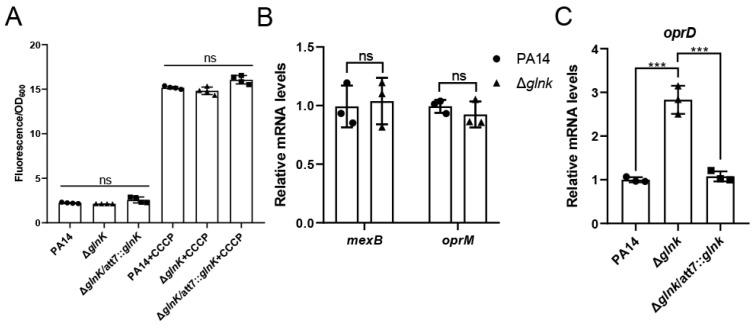 Figure 1
Figure 1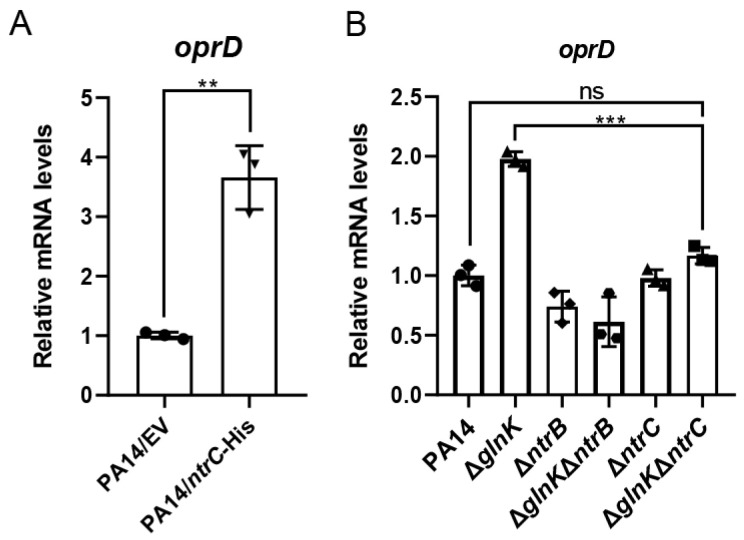 Figure 2
Figure 2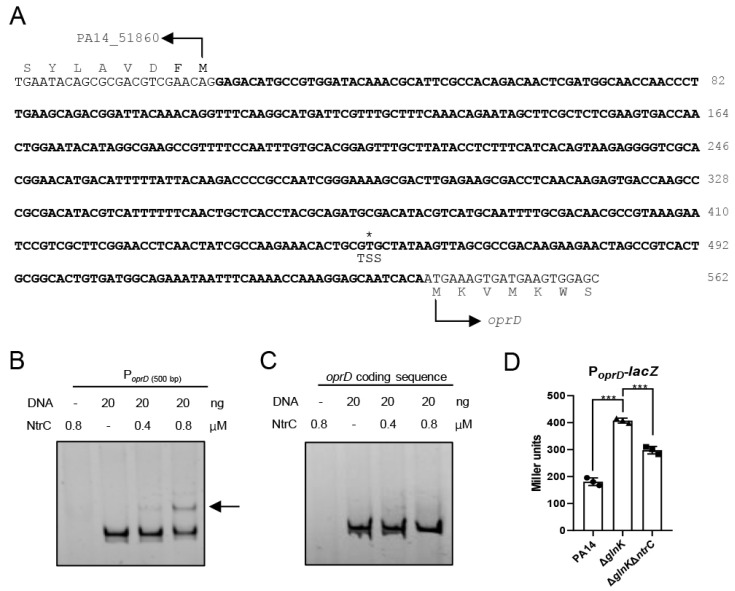 Figure 3
Figure 3- —National Natural Science Foundation of China
- —Nankai University Tianjin Applied and Fundamental Research Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Bacterial Genetics and Biotechnology · Bacterial biofilms and quorum sensing
1. Introduction
Antimicrobial resistance (AMR) poses a tremendous threat to global public health. It compromises the clinical efficacy of antibiotics in combating bacterial infections, imposing overwhelming burdens on healthcare systems and elevating mortality rates of infectious diseases [1,2]. A global burden study revealed that 4.71 million deaths were associated with bacterial AMR between 1990 and 2021, and 1.14 million deaths were attributable to bacterial AMR in 2021. It was predicted that these figures would surge to 8.22 million AMR-associated deaths globally by 2050 [3]. AMR-related environmental risks are increasing sharply. The inappropriate use and overprescription of antibiotics in clinical and agricultural settings have exacerbated global AMR prevalence [4,5]. Among the multidrug-resistant pathogens driving this crisis, Pseudomonas aeruginosa stands out as one of the most problematic pathogens [6].
P. aeruginosa is an opportunistic pathogen notorious for its robust antibiotic resistance and ability to colonize and persist in diverse microenvironments, such as nosocomial settings and the lungs of cystic fibrosis patients [7]. It has a relatively large genome, which arms the bacterium with a repertoire of antibiotic resistance determinants [8,9]. These determinants enable the pathogen to evade the inhibitory or killing effects of antibiotics by preventing the entry or actively extruding antibiotics, enzymatically degrading or modifying antibiotics, and modifying cellular targets [10,11,12]. Additionally, spontaneous chromosomal mutations and the acquisition of genes conferring antibiotic resistance via horizontal transfer (HGT) further augment its resistance [13,14,15].
β-lactams are one of the most widely used classes of antimicrobial agents in clinical practice against bacterial infections. Characterized by a conserved core β-lactam ring, these antibiotics exert their activities by targeting penicillin-binding proteins (PBPs), the enzymes catalyzing peptidoglycan cross-linking [16]. Major subgroups of β-lactam antibiotics include penems, penicillins, carbapenems, cephalosporins, and monobactams, among which carbapenems are considered reliable antibiotics for severe P. aeruginosa infections [17]. However, P. aeruginosa has progressively developed resistance to carbapenems, primarily via mechanisms including overexpression of efflux pumps such as MexAB-OprM, mutations in their PBP genes, reduction of outer membrane permeability, and acquisition of carbapenemases [18,19,20,21,22]. The outer membrane protein OprD is a substrate-specific porin for the uptake of basic amino acids. It is also a major porin for the entry of carbapenems. Mutation of oprD has been found to lead to carbapenem-resistance. Among 382 carbapenem-resistant P. aeruginosa (CRPA) isolates collected from 78 hospitals across Japan from 2019 to 2020, 87.1% (333/382) strains harbored mutations in oprD [23]. Another study identified the oprD mutation in 97.1% (68/70) CRPA strains [24].
GlnK, a homologue of the PII signal protein GlnB, functions as a core regulator of nitrogen metabolism in P. aeruginosa. Unlike many other bacteria that encode both GlnK and GlnB PII signal proteins, P. aeruginosa only harbors GlnK. In response to nitrogen fluctuations, the GlnK protein undergoes post-translational modification, altering its interaction with its cognate binding partners and thus modulating key metabolic pathways [25]. For instance, GlnK interacts with the ammonia transport protein AmtB to fine-tune ammonia transport [26]. Studies in E. coli demonstrate that GlnK regulates the activity of NtrB, the master regulator of nitrogen metabolism, to exert hierarchical control over nitrogen metabolic processes [27]. NtrB functions as both a kinase and a phosphatase, catalyzing the phosphorylation and dephosphorylation of NtrC. In E.coli, this dual enzymatic activity is governed by GlnK in a post-translational modification-dependent manner, in which unmodified GlnK interacts with NtrB, inhibiting its kinase activity and activating the phosphatase activity. In contrast, modified GlnK is unable to interact with NtrB, thereby permitting NtrB to phosphorylate NtrC [28]. Phosphorylated NtrC activates the nitrogen (ntr) regulon, such as the glutamine synthetase gene glnA, and coordinates with CbrB to regulate histidine utilization (hut) genes [27,29]. These regulatory mechanisms enable bacteria to dynamically optimize carbon-nitrogen metabolism in response to diverse nutrient conditions, which is critical for their colonization in the host.
It has been reported that nutritional conditions trigger global metabolic responses in bacteria, which affect antibiotic resistance [30]. Carbon sources have been shown to tune tobramycin tolerance in P. aeruginosa [31]. In addition, distinct nutrient cues shape the evolution of resistance of P. aeruginosa [32,33]. These results demonstrate a functional link between nutrient metabolism and antibiotic resistance. However, the role of carbon-nitrogen metabolic balance in bacterial antibiotic resistance remains largely unknown.
Here, we demonstrate that GlnK modulates P. aeruginosa’s carbapenem susceptibility by regulating the NtrB*/N*trC two-component system and subsequent OprD. Our findings reveal a regulatory link between metabolism and carbapenem resistance.
2. Materials and Methods
2.1. Plasmids, Primers, and Bacterial Strains
The plasmids, primers, and bacterial strains used in this study are summarized in Table 1. Bacteria were grown aerobically in LB broth (5 g/L yeast extract, 10 g/L tryptone, and 5 g/L NaCl, pH 7.4) at 37 °C and 200 rpm. When needed, antibiotics were added at the following concentrations: for E. coli, 10 μg/mL tetracycline, 100 μg/mL ampicillin, and 10 μg/mL gentamicin; for P. aeruginosa, 50 μg/mL tetracycline, 150 μg/mL carbenicillin, and 50 μg/mL gentamicin.
2.2. Construction of the glnK Mutant
The glnK mutant was constructed as described earlier, with slight modifications [38]. Briefly, the glnK deletion mutant was constructed using the suicide vector pEX18Tc via homologous recombination. Approximately 1 kb DNA fragments upstream and downstream of the glnK gene were amplified from wild-type P. aeruginosa PA14 genomic DNA using primer pairs glnK-LF/glnK-LR and glnK-RF/glnK-RR, respectively, and fused by overlap PCR. The resulting PCR product fragment was digested with Sac I and BamH I, and cloned into the pEX18Tc. The resulting recombinant plasmid was transformed into E.coli S17-1 by electroporation and then mobilized into P. aeruginosa PA14 by conjugation. Transconjugants were selected on LB agar containing tetracycline (50 μg/mL, to counterselect wild-type PA14) and kanamycin (25 μg/mL, to counterselect E. coli S17-1). These single-crossover integrants were then streaked onto LB agar supplemented with 7.5% sucrose. Sucrose-resistant colonies were screened for the glnK deletion mutant using colony PCR with primers glnK-LF and glnK-RR. Other deletion mutants were constructed using the same strategy.
2.3. Minimum Inhibitory Concentration (MIC) Assay
For the MIC determination assay, the standard broth microdilution approach was carried out in triplicate, in accordance with the Clinical and Laboratory Standards Institute (CLSI) guidelines. Briefly, antibiotics were two-fold serially diluted in cation-adjusted Mueller–Hinton broth (CAMHB, 3.0 g/L beef extract, 17.5 g/L casamino acid, 1.5 g/L soluble starch, 0.5 mM CaCl_2_, 0.5 mM MgCl_2_, and pH 7.3 ± 0.1), followed by mixing with an equal volume of bacterial suspension in a 96-well microplate. The final bacterial concentration was adjusted to 5 × 10^5^ CFU/mL. MIC values were defined as the lowest concentration of antibiotics yielding no visible bacterial growth after incubation at 37 °C for 20 h. The assays were performed with three biological repeats. Since the antibiotics were two-fold serially diluted, the standard deviations were calculated with log_2_-transformed MICs (log_2_MIC).
2.4. Ethidium Bromide Influx Assay
The assay was performed as previously described with slight modifications [39,40]. Bacteria were cultured in LB medium until OD_600_ reached 0.7, and then washed and resuspended in PBS supplemented with 0.5% (m/v) glucose. The cells were incubated with 100 μM of carbonyl cyanide m-chlorophenylhydrazone (CCCP) or not, and 1 μg/mL of ethidium bromide, followed by incubation for 30 min at 37 °C. The accumulation of ethidium bromide was measured using Varioskan™ Flash (ThermoFisher, Waltham, MA, USA) with an excitation wavelength of 530 nm and an emission wavelength of 585 nm. The relative uptake of ethidium bromide was normalized to the OD_600_ of each sample.
2.5. RNA Extraction and Real-Time Quantitative PCR
Overnight cultured cells were diluted 1:50 in LB and incubated at 37 °C to an OD_600_ of 1.0. Total RNA was isolated using the TransZol Up Plus RNA Kit (TransGen Biotech, Beijing, China) according to the manufacturer’s instructions. Complementary DNA (cDNA) was then synthesized from respective RNA samples utilizing HiScript III RT SuperMix for qPCR (Vazyme, Nanjing, China), followed by amplification with Super Multiple Probe qPCR PreMix (Vazyme, Nanjing, China). The 30S ribosomal protein gene rpsL, a well-validated housekeeping gene for internal controls in P. aeruginosa, was utilized for normalization of gene expression levels [41].
2.6. β-Galactosidase Activity Assay
The β-galactosidase activity assay was conducted according to a previous protocol, with slight modifications [42]. Bacteria were cultured in LB at 37 °C with shaking at 200 rpm until the OD_600_ reached 1.0, and then cells were harvested and resuspended in an equal volume of LacZ-buffer (0.04 M NaH_2_PO_4_, 0.06 M Na_2_HPO_4_, 0.001 M MgSO_4_, 0.01 M KCl, and 0.05 M β-ME). A 500 μL aliquot of the suspension was mixed with 10 μL of 0.1% (m/v) SDS and 10 μL of chloroform, followed by vortexing for 10 s to lyse the cells. Subsequently, orthonitrophenyl—galactopyranoside (ONPG, 4 mg/mL, BBI Life Sciences, Shanghai, China), at a volume of 100 μL, was added and incubated at 37 °C. The reaction was stopped by adding 500 μL of 1 M Na_2_CO_3_ solution once the reaction mixture turned yellow. The calculation of β-galactosidase activity was as previously described [43].
2.7. Electrophoretic Mobility Shift Assay (EMSA)
The electrophoretic mobility shift assay (EMSA) was performed with slight procedural modifications based on a previous protocol [44]. Briefly, DNA fragments amplified by PCR from the P. aeruginosa PA14 genome were incubated with purified recombinant NtrC protein in a 20 μL reaction buffer (100 mM Tris–HCl, pH 7.4, 5 mM EDTA, 500 mM KCl, 35 mM MgCl_2_, and 5 mM DTT) for 30 min at 25 °C. Electrophoresis was carried out at 10 mA for 60 min in 0.5 × TBE buffer after loading the samples onto an 8% native polyacrylamide gel. The entire process was carried out on ice.
2.8. Statistical Analysis
All analyses were performed using GraphPad Prism 8.0 (San Diego, CA, USA), and data are presented as the mean ± SD. Comparisons between groups were conducted using Student’s t test or one-way ANOVA followed by Dunnett’s multiple comparison test. A p-value less than 0.05 was considered statistically significant.
3. Results
3.1. Susceptibility of the glnK Mutant to Carbapenem Antibiotics
To investigate the role of glnK in β-lactam resistance, we measured the MICs of various subclasses of β-lactam antibiotics, including carbapenems, cephalosporins, monobactam, penicillin, and cephamycin for wild-type PA14, the glnK mutant (ΔglnK), and the complemented strain (ΔglnK/att7::glnK). The ΔglnK mutant exhibited a two- to four-fold increased susceptibility to carbapenem antibiotics, including meropenem (MEM), imipenem (IMP), and doripenem (DRPM), compared with the wild-type strain PA14, and this susceptibility was restored by complementation with the glnK gene (ΔglnK/att7::glnK) (Table 2). In contrast, no significant changes in MICs were observed for other β-lactam subclasses. These results indicate a role of GlnK in carbapenem resistance.
3.2. Upregulation of OprD in the glnK Mutant Contributes to the Increased Carbapenem Susceptibility
To explore the mechanism of glnK-mediated carbapenem resistance, we first measured the bacterial efflux activity by measuring intracellular accumulation of ethidium bromide (EB) [45]. The EB influx assay is a well-established method for the functional assessment of efflux pumps [46,47], in which reduced efflux activity results in increased intracellular EB accumulation, thereby enabling reliable comparison of efflux pump function among isogenic strains. In this assay, PA14, ΔglnK, and ΔglnK/att7::glnK treated with carbonyl cyanide m-chlorophenylhydrazine (CCCP), an efflux pump inhibitor, served as the control. In the absence of CCCP, intracellular EB accumulation was comparable among the strains of PA14, ΔglnK, and ΔglnK/att7::glnK, while CCCP addition elevated the intracellular EB levels in all strains to a similar level (Figure 1A), indicating that efflux pump activity was comparable among these strains. Given that MexAB-OprM is the major efflux system for the extrusion of carbapenems from the cell interior [48]. We further examined the gene expression levels of mexB and oprM by RT-qPCR. Consistent with the efflux activity result, no significant difference in mexB and oprM expression was observed between PA14 and the ΔglnK mutant (Figure 1B). In addition, the expression levels of other efflux pump genes (mexC, mexE, and mexY) were similar between PA14 and the ΔglnK mutant (Supplementary Figure S1). Together, these results suggest that the efflux activity is not responsible for the increased carbapenem susceptibility of the ΔglnK mutant. In P. aeruginosa, the basic amino acid porin OprD is a major channel for the diffusion of the carbapenem antibiotics [49,50]. Differential gene expression (DGE) analysis revealed that the oprD gene was upregulated in the ΔglnK mutant (Supplementary Table S1B). RT-qPCR results confirmed the upregulation of oprD in the ΔglnK mutant. Complementation with the glnK gene restored the expression level of oprD (Figure 1C). To examine the role of OprD in the increased carbapenem susceptibility of the ΔglnK mutant, we deleted the oprD gene. In both wild-type PA14 and ΔglnK mutant, deletion of oprD increased the MICs of the carbapenem antibiotics by 8- to 256-fold, resulting in the same MICs in the ΔoprD and ΔglnKoprD mutants (Table 3). These results demonstrate that the upregulated oprD contributes to the increased carbapenem susceptibility in the ΔglnK mutant.
3.3. GlnK Regulates oprD Through the NtrB/NtrC Two-Component System
We previously found that the mutation of glnK resulted in upregulation of the ntrB/ntrC genes [35]. Since NtrB/NtrC regulates nitrogen metabolism genes, we hypothesized that GlnK might modulate oprD expression through this two-component regulatory system. To verify this hypothesis, we overexpressed the regulatory gene ntrC in PA14, which increased the expression of oprD (Figure 2A). Deletion of ntrB or ntrC in the ΔglnK mutant reduced the expression of oprD to a similar level as that in the wild-type PA14 (Figure 2). Furthermore, deletion of ntrB or ntrC in the ΔglnK mutant increased bacterial resistance to the carbapenem antibiotics (Table 4). Further deletion of oprD in the ΔglnKΔntrB mutant (ΔglnKΔntrBΔoprD) resulted in a MIC comparable to that of the ΔoprD single mutant (Supplementary Table S2), demonstrating a major role of OprD in carbapenem resistance. Collectively, these results demonstrate that GlnK regulates oprD by modulating the NtrB/NtrC two-component system.
3.4. NtrC Directly Regulates oprD
To determine whether NtrC directly activates the transcription of oprD, we performed an electrophoretic mobility shift assay (EMSA) using purified NtrC and a DNA fragment (500 bp) covering the oprD promoter region (Figure 3A). The DNA fragment was shifted by NtrC, whereas the fragment (500 bp) from the oprD encoding region was not shifted, indicating direct binding of NtrC to the oprD promoter region. Subsequently, we conducted an oprD promoter-lacZ transcriptional fusion. Compared to the wild-type PA14, LacZ expression was higher in the ΔglnK mutant, which was reduced by the deletion of the ntrC gene (Figure 3D). These findings indicate that NtrC directly regulates oprD in P. aeruginosa.
4. Discussion
In this study, we demonstrate that P. aeruginosa GlnK modulates the expression of oprD via the NtrB-NtrC two-component system, which affects bacterial resistance to carbapenem.
In proteobacteria, GlnK functions as a central integrator of nitrogen and energy signals that fine-tunes target-specific interactions to coordinate nitrogen metabolism. GlnK is post-translationally modified by the bifunctional uridylyltransferase/uridylyl-removing enzyme GlnD and interacts with various target proteins in response to cellular nitrogen status. GlnD catalyzes the uridylylation and deuridylylation of GlnK under nitrogen-limiting and replete conditions, respectively [52]. This reversible modification at the conserved tyrosine residue in GlnK’s T-loop dynamically changes its conformation and binding affinity for downstream targets [53]. Additionally, GlnK also senses cellular nitrogen and energy status by integrating with 2-oxoglutarate (2-OG), ATP, and ADP, which modulates its conformational state and subsequent interactions with target proteins [54]. In E. coli, GlnK interacts with ribonucleotide monophosphatase UmpH to inhibit its phosphatase activity toward uridine 5′-monophosphate [55]. The GlnK-UmpH complex is modulated by the GlnK uridylylation status, as well as the levels of its allosteric effectors ATP, ADP and 2-OG, with the interaction detectable in the presence of either ADP or ATP alone but abrogated when ATP is combined with 2-OG. Notably, uridylylated GlnK (GlnK∼UMP_3_) fails to interact with UmpH regardless of the presence of these molecules.
The two-component regulatory system (TCS) NtrB/NtrC is recognized as a core regulatory hub governing gene expression involved in nitrogen assimilation and utilization. Beyond its canonical function in nitrogen metabolism, increasing evidence has revealed the pleiotropic regulatory targets of NtrB/NtrC in P. aeruginosa, including factors involved in pathogenicity and antibiotic resistance. Alford et al. reported that double deletion of ntrB and ntrC led to a significant (~50%) reduction in abscess size relative to the wild-type LESB58 strain in a chronic abscess infection model [56]. The ΔntrBC mutant exhibits impaired swarming motility and biofilm formation, which are associated with chronic infections [56]. Furthermore, RNA-seq results revealed elevated expression of T3SS genes in ΔntrB and ΔntrC mutants [56]. This result is consistent with our previous finding that mutation of glnK results in upregulation of ntrB/ntrC, thereby repressing T3SS gene expression [35]. These results support a role of NtrB/NtrC in regulating the acute and chronic infection lifestyles in P. aeruginosa [57]. In our study, genes involved in nitrite assimilation, ammonium uptake, glutamine and glutamate synthesis, and urea detoxification and assimilation were upregulated in the ΔglnK mutant (Supplementary Table S1A). Alford et al. demonstrate that deletion of ntrB or ntrC reduced the expression of these genes [56]. For instance, the nitrite reductase small subunit gene nirD was downregulated in the ntrB and ntrC mutants, while upregulated in the ΔglnK strain. Since we observed that the mutation of glnK results in upregulation of ntrB and ntrC [35], these results indicate that the upregulation of the genes in the ΔglnK mutant might be mediated through the NtrB/NtrC two-component regulatory system.
Besides virulence factors, NtrB/NtrC has also been found to influence antibiotic resistance determinants. The previous RNA-seq analysis demonstrated the upregulation of muxABC and opmB in the ntrB and ntrC mutant strains. The MuxABC-OpmB is a resistance-nodulation-cell division (RND)-type multidrug efflux pump system, which contributes to the bacterial resistance to aztreonam, macrolides, novobiocin, and tetracycline [58]. In addition, NtrB/NtrC has been reported to directly affect ciprofloxacin resistance [59]. The MIC of the fluoroquinolone ciprofloxacin was reduced by 8-fold in ΔntrB and ΔntrC strains and by ≥16-fold in the ΔntrBC double mutant, whereas no significant differences in the MICs were observed for tobramycin, chloramphenicol, and tetracycline [59]. In this study, our findings revealed that the upregulation of ntrB/ntrC in the ΔglnK mutant increases bacterial susceptibility to carbapenems (Table 2 and Table 4), which is due to upregulation of OprD (Figure 1C and Figure 2).
OprD is a well-characterized outer-membrane porin in P. aeruginosa, which serves as a channel for basic amino acid transportation and plays a critical role in the entry of carbapenems. Mutations in the oprD gene confer carbapenem resistance, especially to imipenem [60]. One study performed genomic sequencing analyses on carbapenem-resistant P. aeruginosa (CRPA) strains, identified a high prevalence of oprD mutations with no significant variations in the expression of common carbapenemases or efflux pump activity, and demonstrated that the loss of OprD is sufficient for carbapenem resistance [61]. Consistent with these results, our results revealed that deletion of ntrB in the ΔglnK restored carbapenem resistance (Table 4). In clinical isolates, oprD missense or nonsense mutations are frequently identified. These mutations result in conformational changes in this porin, thereby conferring carbapenem resistance [49,62].
The expression of oprD is upregulated when arginine, histidine, glutamate, or alanine is used as the sole carbon source in a minimal medium supplemented with ammonium sulfate as the nitrogen source. Similarly, oprD is upregulated when glutamate or alanine is used as the nitrogen source [51]. Arginine-mediated oprD upregulation is attributed to the activation of the arginine-responsive repressor ArgR, which functions as a direct activator of the oprD gene [51]. Here, we identified the role of NtrB/NtrC in regulating the expression of oprD. Given the role of NtrB/NtrC in regulating nitrogen metabolism, it is likely that P. aeruginosa may exhibit different levels of carbapenem susceptibility under various nutritional conditions.
These findings highlight the critical relevance of the nitrogen metabolism regulator to bacterial carbapenem susceptibility, as specifically evidenced by an approximately four-fold reduction in the MIC of meropenem in the glnK mutant (Table 2). As a common empirical antibiotic, meropenem resistance is associated with increased treatment failure and mortality in cystic fibrosis (CF) patients with P. aeruginosa infections [63,64]. Of note, elevated amino acid levels are present in the airways of CF patients [65], which may modulate bacterial susceptibility to meropenem. In fact, it has been reported that sequential P. aeruginosa isolates collected over 3 years from a CF patient exhibited a striking shift in meropenem MIC, from 0.25 μg/mL to 4 μg/mL and finally to 64 μg/mL, which was accompanied by a significant deterioration in the patient’s pulmonary function [66]. Mechanistically, this altered carbapenem susceptibility was mainly due to the progressive downregulation of the outer membrane porin OprD. Thus, it is likely that under the CF patients’ lung environment, mutation of oprD does not affect bacterial growth, presumably due to the high amino acid levels.
5. Conclusions
This study identifies a regulatory pathway by which GlnK mediates carbapenem susceptibility in P. aeruginosa. We demonstrate that mutation of glnK leads to upregulation of ntrB/ntrC, which activates oprD expression, enhancing bacterial susceptibility to carbapenems. These findings establish a functional link between nitrogen metabolism and antibiotic resistance, expanding the understanding of the complex regulatory network governing oprD expression. Collectively, this GlnK-mediated regulatory pathway provides a clue for the development of strategies to enhance the efficacy of carbapenems for combating P. aeruginosa infections.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Carmeli Y. Cisneros J.M. Paul M. Daikos G.L. Wang M. Torre-Cisneros J. Singer G. Titov I. Gumenchuk I. Zhao Y. Aztreonam-avibactam versus meropenem for the treatment of serious infections caused by Gram-negative bacteria (REVISIT): A descriptive, multinational, open-label, phase 3, randomised trial Lancet Infect. Dis.20252521823010.1016/S 1473-3099(24)00499-739389071 · doi ↗ · pubmed ↗
- 2Jones K.E. Patel N.G. Levy M.A. Storeygard A. Balk D. Gittleman J.L. Daszak P. Global trends in emerging infectious diseases Nature 200845199099310.1038/nature 0653618288193 PMC 5960580 · doi ↗ · pubmed ↗
- 3GBD 2021 Antimicrobial Resistance Collaborators Global burden of bacterial antimicrobial resistance 1990–2021: A systematic analysis with forecasts to 2050 Lancet 20244041199122610.1016/S 0140-6736(24)01867-139299261 PMC 11718157 · doi ↗ · pubmed ↗
- 4Hotor P. Kotey F.C.N. Donkor E.S. Antibiotic resistance in hospital wastewater in West Africa: A systematic review and meta-analysis BMC Public Health 202525136410.1186/s 12889-025-22513-w 40217451 PMC 11987346 · doi ↗ · pubmed ↗
- 5Zhao X. Qi G. Feng Y. Du C. Application of nematicide avermectin enriched antibiotic-resistant bacteria and antibiotic resistance genes in farmland soil Environ. Res.202322711580210.1016/j.envres.2023.11580237003554 · doi ↗ · pubmed ↗
- 6Macesic N. Uhlemann A.C. Peleg A.Y. Multidrug-resistant Gram-negative bacterial infections Lancet 202540525727210.1016/S 0140-6736(24)02081-639826970 · doi ↗ · pubmed ↗
- 7Letizia M.A. Diggle S.A. Whiteley M.A. Pseudomonas aeruginosa: Ecology, evolution, pathogenesis and antimicrobial susceptibility Nat. Rev. Microbiol.20252370171710.1038/s 41579-025-01193-840442328 PMC 13064840 · doi ↗ · pubmed ↗
- 8Stover C.K. Pham X.Q. Erwin A.L. Mizoguchi S.D. Warrener P. Hickey M.J. Brinkman F.S. Hufnagle W.O. Kowalik D.J. Lagrou M. Complete genome sequence of Pseudomonas aeruginosa PAO 1, an opportunistic pathogen Nature 200040695996410.1038/3502307910984043 · doi ↗ · pubmed ↗