Identification and Drought-Responsive Expression Analysis of the ZmSPS Gene Family in Maize and Preliminary Investigation of the ZmSPS3 Regulatory Network
Minghao Sun, Wei Zhao, Shuai Hou, Haoxin Meng, Luyao Wang, Erna Wu, Enhao Zhou, Yuyang Duan, Yue Wang, Quan Cai, Baitao Guo, Tao Yu, Jianguo Zhang

TL;DR
This study identifies and analyzes the ZmSPS gene family in maize, focusing on ZmSPS3's role in drought response and its regulatory interactions.
Contribution
The study introduces ZmSPS3 as a key drought-responsive gene and reveals its physical interactions with proteins involved in stress regulation.
Findings
Seven ZmSPS genes were identified in maize, showing high conservation among monocots.
ZmSPS3 expression significantly increased under drought stress.
ZmSPS3 interacts with protein kinases and F-box proteins, suggesting roles in stress regulation.
Abstract
Sucrose phosphate synthase (SPS) is a key rate-limiting enzyme that regulates carbon partitioning and stress tolerance in plants. In this study, we systematically characterized the SPS gene family in maize (Zea mays L.) and identified key members and their interaction networks involved in drought responses. A total of seven ZmSPS genes were identified through genome-wide bioinformatics analyses. Motif composition, gene structure, phylogenetic relationships, and synteny analyses indicated that the ZmSPS gene family is highly conserved among monocot species. Promoter analysis revealed that the upstream regions of ZmSPS genes are enriched with multiple stress responsive cis-acting elements. Drought stress treatments combined with quantitative real-time PCR (RT-qPCR) analyses showed that the expression of ZmSPS3 was significantly upregulated with increasing stress duration. Furthermore,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
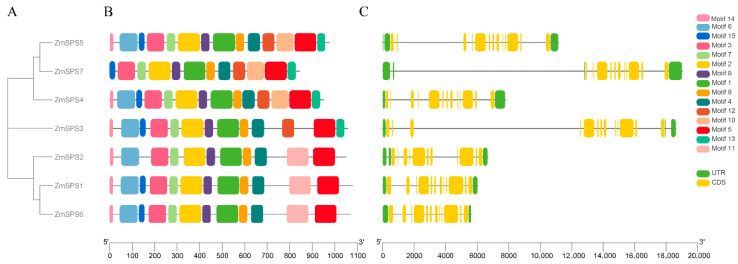 Figure 1
Figure 1 Figure 2
Figure 2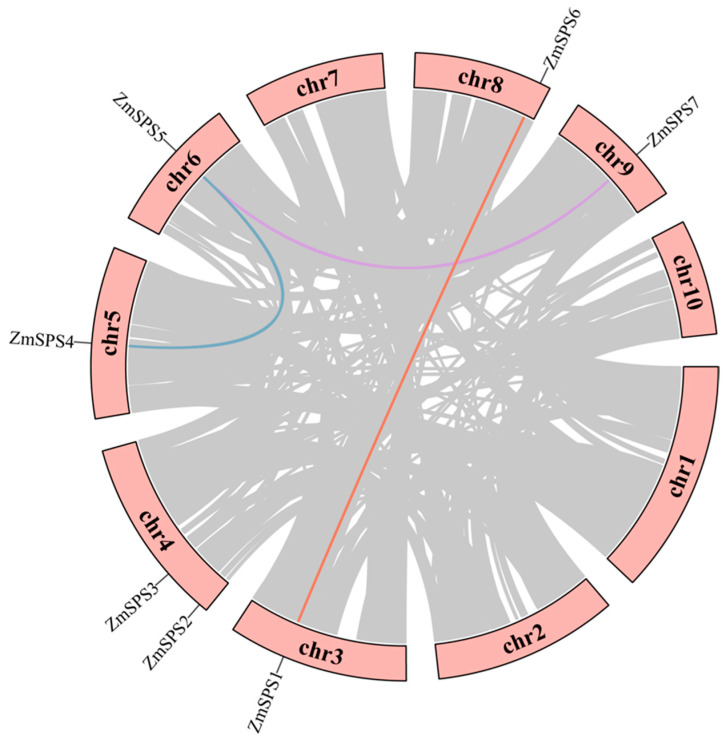 Figure 3
Figure 3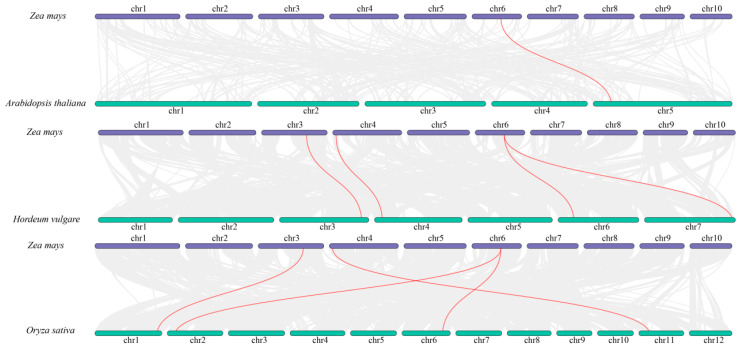 Figure 4
Figure 4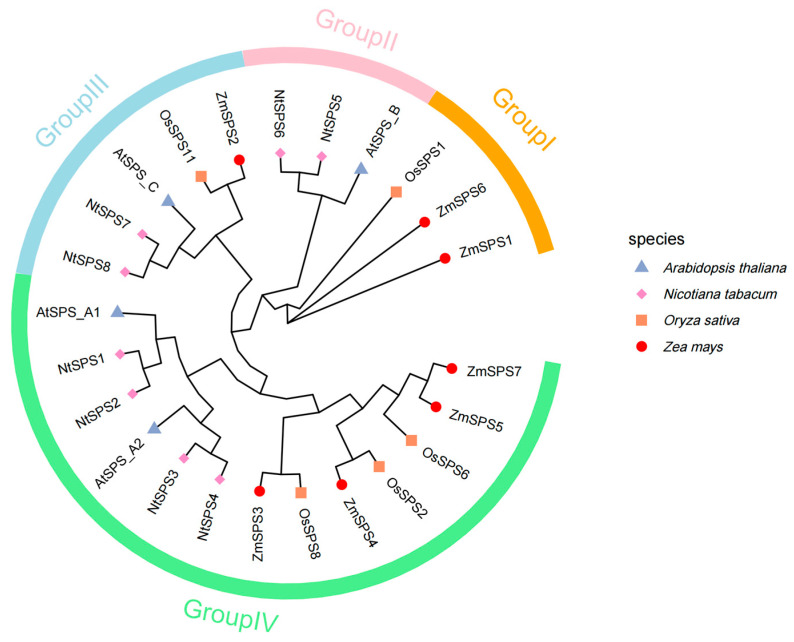 Figure 5
Figure 5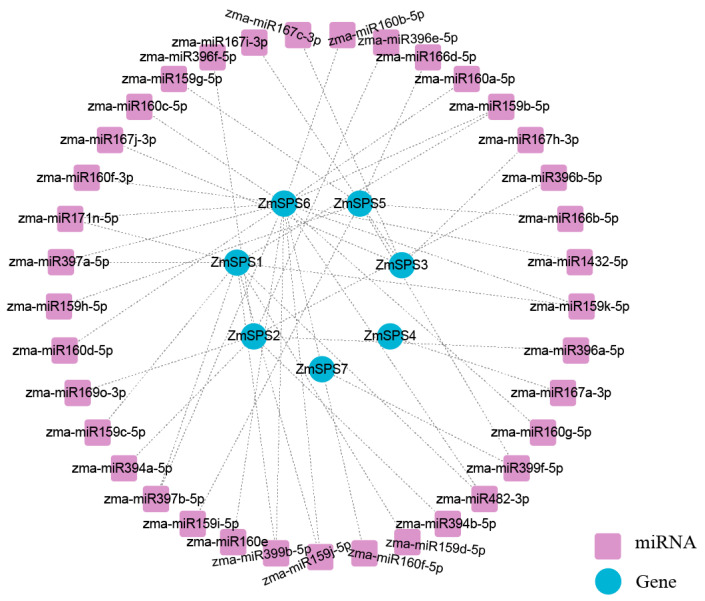 Figure 6
Figure 6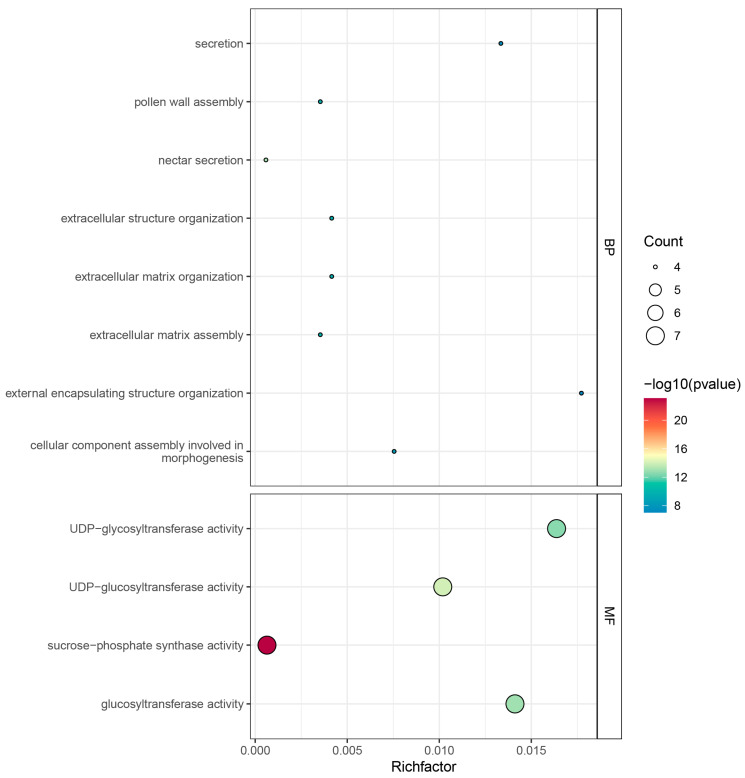 Figure 7
Figure 7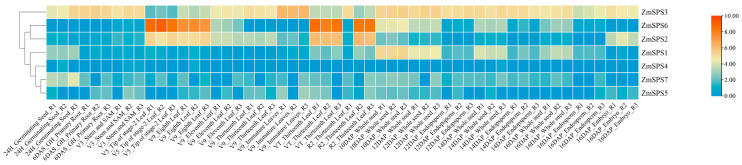 Figure 8
Figure 8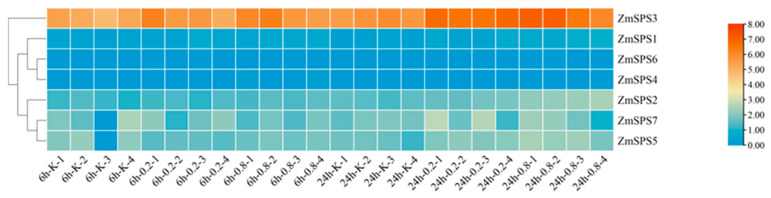 Figure 9
Figure 9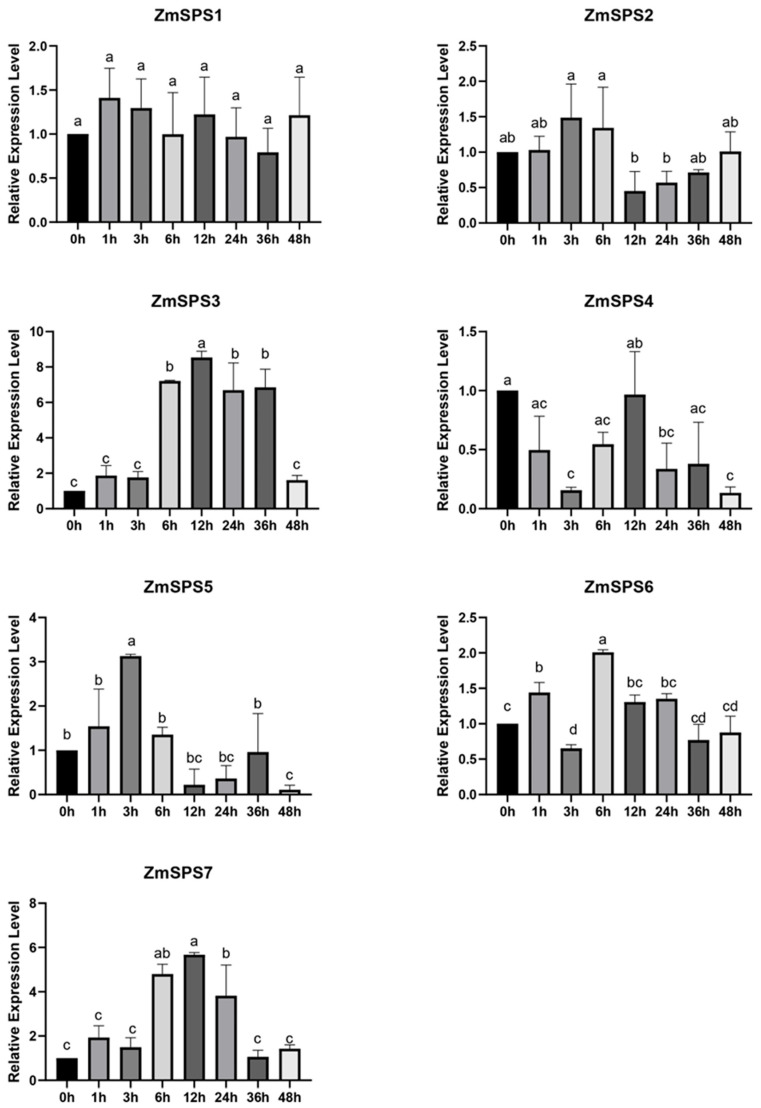 Figure 10
Figure 10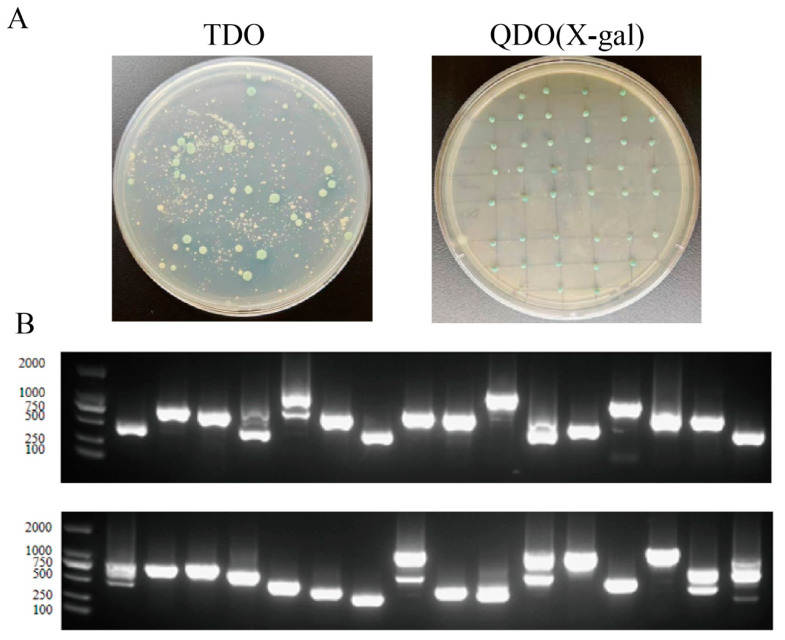 Figure 11
Figure 11 Figure 12
Figure 12- —Research Business Fee Project of Provincial Research Institutes
- —Finance Department of Heilongjiang Province
- —Heilongjiang Province Science and Technology Innovation Base Incentive Program
- —The Innovation Project of Heilongjiang Academy of Agricultural Sciences
- —Postdoctoral Innovation Practice Base, Maize Research Institute, Heilongjiang Academy of Agricultural Sciences
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant nutrient uptake and metabolism · Plant Stress Responses and Tolerance · Enzyme Structure and Function
1. Introduction
Maize (Zea mays L.) is one of the most widely produced food crops worldwide, ranking first in both planting area and total production, and it also serves as an important source of animal feed and industrial raw materials [1]. As a major food resource for humans, maize provides essential carbohydrates and proteins and is extensively used in the production of biofuels, starch, and other industrial products [2,3]. The growth, development, and yield of maize are highly sensitive to environmental factors such as water availability, light conditions, and nutrient supply. Among these factors, drought stress is a major abiotic constraint that severely limits maize growth and productivity [4,5,6]. Drought stress suppresses photosynthesis and metabolic activities and disrupts cellular osmotic balance, thereby directly affecting the synthesis and allocation of photosynthetic assimilates and ultimately reducing crop yield [7,8]. During this process, sucrose, as the primary product of photosynthesis, functions not only as an energy source but also as a key molecule involved in osmotic regulation and signal transduction. Sucrose therefore plays a crucial role in maintaining cellular homeostasis and enhancing drought tolerance in maize [9].
In higher plants, sucrose is primarily produced through photosynthesis. The dynamic balance between its synthesis in source organs, transport via the phloem to sink organs, and utilization in sink tissues collectively determines plant growth, development, and final crop yield [10]. Analyzing crop yield improvement from the perspective of source-sink regulation has become a mainstream approach in recent years [11]. In addition to serving as a primary carbon and energy source, sucrose also functions as a signaling molecule, widely participating in the regulation of plant growth, development, and various physiological processes, thereby profoundly affecting crop yield [12,13]. The source-to-sink transport of photosynthetic products in plants involves several key steps: (1) CO_2_ fixation in chloroplasts; (2) transport of carbon assimilation products to the cytoplasm; (3) sucrose synthesis; and (4) sucrose transport to sink tissues and its utilization therein [14,15].
During this process, intermediates generated from CO_2_ fixation via carbon assimilation pathways are ultimately converted into sucrose through the coordinated actions of sucrose-phosphate synthase (SPS) and sucrose-6-phosphate phosphatase (SPP) [16]. Specifically, SPS catalyzes the transfer of a glucose moiety from uridine-5′-diphosphate glucose (UDPG) to fructose-6-phosphate (F-6-P), producing sucrose-6-phosphate (Suc-6-P). Subsequently, Suc-6-P is dephosphorylated by SPP to form sucrose, which is transported from source organs to sink tissues via the phloem to meet the carbon and energy demands required for plant growth, development, and stress adaptation [17,18].
Sucrose-phosphate synthase (SPS) is a soluble enzyme localized in the cytoplasm, with a molecular weight of approximately 120 kDa and an optimal reaction pH of around 7.0. It plays a central catalytic role in plant sucrose biosynthesis [19,20]. Based on phylogenetic analysis, the plant SPS gene family can be classified into four subfamilies: A, B, C, and D. Notably, the D subfamily has so far been identified only in monocotyledonous grasses, indicating a certain degree of species specificity [20,21]. Protein structure analysis shows that SPS proteins typically consist of multiple highly conserved functional domains, including the sucrose synthase domain (sucrose-synth), the glycosyltransferase domain (glycos-transf-1), and the sucrose-6-phosphate phosphatase domain (S6PP) [22,23]. These conserved domains collectively form the structural basis of SPS and are critical for its catalytic activity and physiological functions.
Previous studies have demonstrated that SPS plays a crucial role in plant growth, development, and responses to abiotic stress. In Arabidopsis thaliana (Arabidopsis), genetic functional analyses of the AtSPS gene family revealed that the atspsa1/atspsc double mutant and the atspsa1/atspsa2/atspsc triple mutant exhibited pronounced dwarf phenotypes during both vegetative and reproductive stages, with severely impaired development of shoots, floral organs, and siliques. Further studies showed that the atspsa1/atspsb/atspsc triple mutant and the quadruple mutant caused defective seed germination [24]. Moreover, overexpression of AtSPSA1 in Arabidopsis significantly increased sucrose content in stems and was accompanied by enhanced stem height, fiber length, and overall plant height [25].
In maize, comparative studies of varieties with different growth rates have shown a significant positive correlation between SPS activity and plant growth rate. This suggests that the capacity to convert photosynthetically produced carbohydrates into sucrose, regulated by SPS, may be an important limiting factor determining early growth in maize [21]. Moreover, under high-temperature stress, inhibition of SPS and other enzymes involved in sucrose metabolism and starch synthesis markedly reduces starch accumulation and kernel weight, further highlighting the critical role of SPS in maize grain filling and yield formation. In Manihot esculenta, MeSPS genes are highly expressed in source tissues, such as leaves and stems, and their expression levels are significantly positively correlated with root starch content. High expression of MeSPS accelerates starch accumulation in cassava tuberous roots [26]. In Solanum lycopersicum, overexpression of SlSPS significantly increases SPS activity and sucrose content, thereby promoting plant growth and enhancing tolerance to high-temperature stress. Conversely, knockout of SlSPS suppresses growth, reduces sucrose metabolism, and increases sensitivity to heat stress [27]. Similarly, in Nicotiana tabacum, NtSPS5 and NtSPS6 have been identified as positive regulators in drought stress responses, contributing to enhanced drought tolerance [28]. Collectively, studies across different species consistently indicate that SPS plays a conserved and crucial role in plant growth and development, yield formation, and abiotic stress adaptation by regulating sucrose synthesis and the allocation of photosynthetic assimilates.
To date, studies on maize SPS have primarily focused on the identification of gene numbers, and systematic investigations of their functions and regulatory mechanisms remain lacking [20,29]. In this study, we systematically identified members of the maize SPS gene family using bioinformatics approaches and conducted comprehensive analyses of their physicochemical properties, promoter elements, evolutionary relationships, and synteny. Additionally, potential functions of ZmSPS genes were preliminarily explored through miRNA target prediction and Gene Ontology (GO) annotation. Combined with transcriptome data under drought stress and validation by RT-qPCR, we further characterized the transcriptional response patterns of ZmSPS genes. Moreover, yeast two-hybrid library screening and pairwise interaction assays were employed to investigate the potential regulatory pathways of ZmSPS under drought stress. This study provides important insights into the role of sucrose metabolism in maize drought tolerance and has significant implications for improving water-use efficiency and enhancing crop yield.
2. Results
2.1. Identification and Physicochemical Properties of the Maize ZmSPS Gene Family
The ZmSPS gene family members in maize were identified using two complementary approaches. First, previously reported studies were combined with the SPS HMM profiles (PF00534, PF00862, PF05116) to search the maize whole genome and obtain candidate genes. Second, known SPS members in rice were used to query maize protein sequences, yielding additional candidate genes. The candidate genes obtained from the two methods were then merged. The merged candidates were manually screened for the presence of SPS core domains using multiple databases, including CDD, Phmmer, Pfam, and SMART. Ultimately, seven ZmSPS genes were confirmed and named ZmSPS1-7 according to their physical positions on the chromosomes. Analysis of their encoded proteins revealed that the CDS lengths ranged from 2532 bp (ZmSPS7) to 3237 bp (ZmSPS1), corresponding to protein lengths of 844–1079 amino acids (Table 1). The predicted molecular weights ranged from 94.50 to 119.42 kDa, with minor differences among family members, indicating high structural conservation. The theoretical isoelectric points (pI) of ZmSPS proteins mainly ranged from 6.04 to 7.38, with ZmSPS4 (pI = 7.38) and ZmSPS5 (pI = 7.03) being neutral or slightly basic, while the other 5 members were slightly acidic. Hydropathicity analysis showed that all ZmSPS proteins had negative GRAVY values, ranging from −0.20 to −0.42, suggesting that the family members are overall hydrophilic. Subcellular localization prediction indicated that all seven ZmSPS proteins are cytoplasmic, consistent with the physiological role of sucrose synthesis occurring primarily in the cytoplasm, further supporting the critical role of ZmSPS genes in maize carbon metabolism.
2.2. Gene Structure and Conserved Motif Analysis of the ZmSPS Gene Family
To further investigate the evolutionary relationships and structural features of the maize SPS gene family, a phylogenetic tree of ZmSPS proteins was constructed, and conserved motifs and gene structures were comprehensively analyzed (Figure 1). Phylogenetic analysis revealed that the seven ZmSPS members could be clearly divided into three major evolutionary clades (Figure 1A). Clade I included ZmSPS5, ZmSPS7, and ZmSPS4; Clade II contained only ZmSPS3, showing a relatively independent evolutionary position; and Clade III comprised ZmSPS2, ZmSPS1, and ZmSPS6. Members within the same clade were more closely related, suggesting that they may have similar or related biological functions. Conserved motif analysis indicated that ZmSPS family members generally possess highly conserved structural features (Figure 1B). All ZmSPS proteins contained most of the same motifs (motifs 1, 2, 3, 4, 5, 7, 8, 9), and the order of these motifs was highly consistent across different members, reflecting strong structural stability during evolution. However, certain motif differences were observed among the evolutionary clades, showing branch-specific patterns. For example, members of Clade III (ZmSPS1, ZmSPS2, and ZmSPS6) uniquely contained motif 11 at the C-terminal, which was absent in Clades I and II. Additionally, ZmSPS7 lacked motif 14 at the N-terminal, and the position of motif 15 differed from other members, suggesting potential functional divergence or specialization.
Gene structure analysis showed that all ZmSPS genes contained multiple exons and introns (Figure 1C, Table S2), but the numbers varied. ZmSPS1, ZmSPS2, and ZmSPS6 had 12 exons and 11 introns; ZmSPS4 and ZmSPS7 had 13 exons and 12 introns, while ZmSPS3 and ZmSPS5 contained 14 exons and 13 introns. Overall, genes within the same clade displayed similar structures, reflecting evolutionary conservation of the family, while structural variations among different clades may contribute to functional divergence.
2.3. Cis-Acting Element Analysis of ZmSPS Promoters
To explore the transcriptional regulatory mechanisms of the maize ZmSPS gene family, the 2000 bp upstream sequences of all seven ZmSPS genes were extracted and analyzed for cis-acting elements (Figure 2). The results showed that the promoter regions were enriched with elements related to light responsiveness, hormone regulation, stress response, and growth and development. Notably, light-responsive elements (G-Box), hormone-responsive elements (ABRE), and stress-related elements (MYB, STRE, MYC, GC-motif) were present in all members. Among them, ABRE elements were most abundant in ZmSPS1, ZmSPS2, ZmSPS3, and ZmSPS5, with 9, 9, 7, and 8 copies, respectively, suggesting that these four genes may play central roles in ABA-mediated abiotic stress responses. The light-responsive core element G-Box was highly represented in ZmSPS1 and ZmSPS2 promoters, with 8 and 9 copies, respectively, consistent with the role of SPS in photosynthate metabolism. ZmSPS1 also contained a relatively high number of STRE elements. Overall, the high enrichment of ABRE and G-Box elements in the ZmSPS promoters indicates that transcriptional regulation of this gene family is primarily driven by light and ABA signaling.
2.4. Chromosomal Distribution and Synteny Analysis of ZmSPS Genes
To elucidate the physical locations and expansion mechanisms of the maize ZmSPS gene family, chromosomal mapping and intraspecies synteny analysis of all seven ZmSPS genes were performed using TBtools v2.376 software. The results showed that ZmSPS genes were unevenly distributed on six chromosomes (Figure 3), located on chr3, chr4, chr5, chr6, chr8, and chr9. Notably, chr4 contained two members (ZmSPS2 and ZmSPS3), while the other chromosomes each carried a single member. This broad, cross-chromosomal distribution suggests that the SPS family has undergone considerable spatial diversification during maize evolution.
Intraspecies synteny analysis indicated that the expansion of the ZmSPS gene family primarily relied on segmental duplications across chromosomes. Three clear pairs of duplicated genes were identified and marked with colored lines in the Figure 3: ZmSPS4-ZmSPS5 (chr5-chr6, blue), ZmSPS1-ZmSPS6 (chr3-chr8, red), and ZmSPS5-ZmSPS7 (chr6-chr9, purple). Interestingly, ZmSPS5 was syntenic with both ZmSPS4 and ZmSPS7. All three genes belong to Clade I in the phylogenetic tree, reflecting high evolutionary conservation. These results indicate that the expansion of the ZmSPS family is closely associated with whole-genome duplication (WGD) events in maize.
To investigate the evolutionary driving forces and potential functional divergence of the maize ZmSPS gene family, the nonsynonymous substitution rate (Ka), synonymous substitution rate (Ks), and their ratio (Ka/Ks) were calculated for the syntenic gene pairs. The results (Table S3) showed that all identified gene pairs had Ka/Ks < 1.0, ranging from 0.1133 to 0.2798. Among them, ZmSPS4/7 exhibited the lowest value (0.1133), whereas ZmSPS1/2 showed a relatively higher value (0.2798). These findings indicate that the ZmSPS gene family has undergone strong purifying selection during evolution, suggesting that family members tend to retain their original amino acid sequences to maintain functional stability as key enzymes in sucrose biosynthesis.
2.5. InterSpecies Synteny Analysis of the SPS Gene Family
To explore the evolutionary conservation and interspecies homology of the maize ZmSPS gene family, synteny analysis was performed with Arabidopsis, rice, and barley (Figure 4). The results revealed that ZmSPS genes are highly conserved across both monocots and dicots, while more complex expansion patterns are observed within the Poaceae family. In comparison with Arabidopsis, only one significant orthologous pair was detected: ZmSPS5 on maize chr6 exhibited synteny with AT5G11110.1 on Arabidopsis chr5 (Table S4). In contrast, broader syntenic relationships were observed between maize and rice or barley. Four orthologous gene pairs were identified in rice, with genes on maize chr3, chr4, and chr6 corresponding to their respective rice chromosomes. Notably, the ZmSPS genes in poaceae displayed clear one-to-many expansion patterns. For example, ZmSPS5 on maize chr6 corresponded to two transcripts in rice (Os02t0184400-01 and Os06t0643800-01) and to chr6 and chr7 in barley, whereas genes on chr3 and chr4 corresponded to single orthologs in rice and barley. This distribution pattern reflects both expansion and conservation within the Poaceae. ZmSPS5 on chr6 is the only member syntenic across all compared species, suggesting it represents a core ancestral gene of the ZmSPS family. In contrast, SPS members on chr3 and chr4 exhibit synteny only with rice and barley but are absent in Arabidopsis, indicating that these genes may have arisen through segmental duplication during the early evolution of monocots or poaceae and subsequently became stably inherited.
2.6. Phylogenetic Analysis of the Maize ZmSPS Gene Family
A phylogenetic tree was constructed based on the SPS protein sequences from maize, rice, Arabidopsis, and tobacco to clarify the evolutionary positions and relationships of ZmSPS family members (Figure 5). The analysis showed that SPS proteins from the four species were divided into four major groups (Group I–IV), exhibiting clear monocot–dicot differentiation patterns. Group I (yellow branch) included maize ZmSPS1 and ZmSPS6, as well as rice OsSPS1, indicating high conservation within monocots. Group II (pink branch) contained ZmSPS2, which clustered with AtSPS_B from Arabidopsis and corresponding proteins from tobacco and rice, suggesting potential functional diversification. Group III (blue branch) mainly comprised members from Arabidopsis, tobacco, and rice, with no maize ZmSPS member detected, implying possible loss or functional degeneration during maize evolution. Group IV (green branch) was the largest subgroup, including maize ZmSPS3, ZmSPS4, ZmSPS5, and ZmSPS7. Maize members in this group formed close sister relationships with rice OsSPS members (ZmSPS4 with OsSPS8, ZmSPS5/7 with OsSPS6), reflecting high homology within the poaceae family.
Furthermore, the phylogenetic tree revealed clear species clustering: monocots (maize and rice) and dicots (Arabidopsis and tobacco) tended to cluster separately within each subgroup. In all branches containing maize members, ZmSPS preferentially clustered with rice OsSPS, consistent with the previously observed inter-species synteny results. This finding further confirms the close evolutionary relationship and high conservation of SPS genes between maize and rice.
2.7. miRNA-Mediated Regulatory Network Analysis of the Maize ZmSPS Gene Family
To investigate the post-transcriptional regulation of the maize ZmSPS gene family, a miRNA-target regulatory network was predicted and constructed for the seven ZmSPS members. The results showed that this gene family is extensively regulated by multiple miRNAs (Figure 6), forming a highly interconnected many-to-many network, in which a single miRNA can target multiple ZmSPS genes, and each ZmSPS gene can be regulated by multiple distinct miRNAs. A total of 34 different miRNAs were identified, highlighting the complex post-transcriptional regulatory landscape of this family. Network analysis revealed differential regulation among the genes. ZmSPS6, ZmSPS5, and ZmSPS1 were positioned at the network core, interacting with multiple miRNAs, such as members of the zma-miR167, zma-miR159, and zma-miR160 families. In addition, several specific regulatory interactions were observed, including ZmSPS1 regulated by zma-miR397a-5p and ZmSPS3 regulated by zma-miR1432-5p, suggesting that these specific interactions may contribute to functional divergence in particular tissues or developmental stages.
2.8. GO Functional Enrichment Analysis of the Maize ZmSPS Gene Family
To elucidate the biological functions of the ZmSPS family members in maize growth, development, and metabolic regulation, GO enrichment analysis was performed on the seven identified members (Figure 7). At the molecular function (MF) level, the family genes were primarily associated with catalytic activities, with the most significantly enriched term being sucrose-phosphate synthase activity. Additionally, ZmSPS genes were significantly enriched in UDP-glycosyltransferase activity and glucosyltransferase activity, reflecting their biochemical role in catalyzing sucrose-phosphate synthesis using UDP-glucose.
At the biological process (BP) level, ZmSPS genes were involved in multiple processes ranging from fundamental metabolism to complex developmental events. They were significantly enriched in processes such as secretion and nectar secretion, highlighting their central role in carbohydrate allocation and nectar formation. Furthermore, ZmSPS genes participated in extracellular structure organization, extracellular matrix assembly, and pollen wall assembly. Some members were enriched in cellular component assembly involved in morphogenesis, indicating that sucrose not only serves as an energy source but also acts as a precursor for the construction of cell wall-related structures.
2.9. Maize ZmSPS Gene Family Tissue Expression Pattern Analysis
The expression patterns of the seven ZmSPS family members were analyzed across different tissues and developmental stages using maize transcriptome data (Figure 8). The results revealed significant functional differentiation among family members, suggesting that they coordinately regulate maize metabolism through both division of labor and collaboration. ZmSPS2 and ZmSPS6 exhibited highly coordinated expression during leaf development. Both genes maintained very high transcript levels in leaf tips at the V5 stage, at the V9 stage, mature leaves at the VT stage, and leaves at the R2 stage, indicating that they are the core genes for sucrose synthesis in leaves, supporting plant growth and kernel filling. ZmSPS3 displayed tissue-specific expression during immature tissue development, reaching its peak in V9 immature leaves. Similarly, ZmSPS5 generally showed low expression across most tissues but had relatively higher expression in V9 immature leaves, suggesting a specific regulatory role during early leaf development. During seed germination and early developmental stages, ZmSPS1 and ZmSPS7 exhibited notable specificity. ZmSPS1 showed the highest expression in 10 DAP whole seeds, primarily participating in sugar accumulation and allocation during early kernel development. ZmSPS7 was most highly expressed in 24H germinating seeds, indicating its role in nutrient mobilization and energy supply during seed germination. ZmSPS4 showed relatively low expression across most tissues.
Although all members belong to the same gene family, their expression varies markedly across tissues and developmental stages: ZmSPS2 and ZmSPS6 are highly expressed in leaves; ZmSPS3 is relatively enriched in immature leaves, while ZmSPS5 shows localized enrichment in young leaves; ZmSPS1 and ZmSPS7 are active during kernel development and germination; and ZmSPS4 generally exhibits low expression, possibly being induced only under specific conditions.
2.10. ZmSPS Gene Family Transcriptome Expression Analysis Under Drought Stress
To evaluate the responsiveness of the maize SPS gene family under drought stress, transcriptome data from drought-treated plants were analyzed, examining the expression patterns of seven ZmSPS members under mild (−0.2 MPa) and severe (−0.8 MPa) PEG treatments at 6 h and 24 h. The results revealed significant differential expressions among family members in response to drought (Figure 9): ZmSPS3 exhibited consistently high expression under drought stress. Its transcript levels were higher than those of other family members across all treatments and control, and expression increased significantly over time, reaching peak levels after 24 h under both mild and severe stress, indicating that ZmSPS3 serves as a key regulatory gene in drought response. The branch members ZmSPS2, ZmSPS5, and ZmSPS7 showed dynamic response patterns. ZmSPS2 remained relatively stable throughout the treatments, with a slight upregulation observed at 24 h under severe stress. ZmSPS5 and ZmSPS7 displayed increasing trends under mild stress at both 6 h and 24 h, particularly pronounced at 24 h. The low-sensitivity members ZmSPS1, ZmSPS4, and ZmSPS6 exhibited generally low expression under all treatments. These findings indicate considerable diversity among ZmSPS family members in spatial expression and in response to abiotic stress.
2.11. RT-qPCR Validation of the ZmSPS Gene Family Under PEG-Induced Osmotic Stress
To verify the expression patterns of ZmSPS genes under osmotic stress conditions, the relative expression levels of the seven ZmSPS family members were analyzed by RT-qPCR during 0–48 h of PEG treatment. The results revealed distinct temporal expression patterns among the ZmSPS genes (Figure 10).
Among them, ZmSPS3 exhibited the most pronounced response to stress treatment, with expression rapidly increasing at 6 h and peaking at 12 h (about 8.5-fold higher than the control), remaining relatively high at 24 h and 36 h. ZmSPS7 displayed a similar trend, reaching its highest expression (about 6-fold) at 12 h before gradually declining. ZmSPS5 showed early induction, peaking at 3 h (about 3-fold) and then gradually decreasing. ZmSPS6 exhibited a transient increase (about 2-fold) at 6 h, then returned to lower levels. In contrast, ZmSPS1 showed minimal change throughout the treatment period. ZmSPS2 slightly increased at 3–6 h but decreased significantly after 12 h. ZmSPS4 was initially suppressed under drought but showed partial recovery at 12 h. Overall, the RT-qPCR results were largely consistent with the transcriptome analysis, indicating that ZmSPS family members exhibit distinct dynamic expression patterns in response to osmotic stress.
2.12. ZmSPS3 Yeast Two-Hybrid (Y2H) Library Screening
Analysis based on transcriptome data showed that ZmSPS3 exhibited strong induction expression characteristics under drought stress. Then, we performed RT-qPCR analysis on maize B73 seedlings treated with PEG-induced osmotic stress. The results showed that both ZmSPS3 and ZmSPS7 exhibited an upward expression trend; the response of ZmSPS3 was the most significant, with its expression level at 12 h of treatment increasing by more than 8 times compared to 0 h, and its induction intensity ranked first among all family members. Considering that drought response is a complex process involving the synergistic action of multiple genes, this study integrated transcriptome and RT-qPCR and finally selected ZmSPS3 as the target gene for subsequent research.
Self-activation testing of the ZmSPS3 bait construct pGBKT7-ZmSPS3 indicated that it does not possess transcriptional self-activation activity (Figure S1), making it suitable for subsequent Y2H screening. The pGBKT7-ZmSPS3 bait was co-transformed with the maize Y2H library and screened on selective media. The DDO plates showed a sufficient number of single clones to meet the analysis requirements; positive clones from the primary screen were re-screened on TDO plates and grew normally on QDO/X plates while turning blue (Figure 11A), indicating reliable screening results.
PCR verification of the positive clones revealed insert sizes mainly ranging from 500 to 1000 bp, all exceeding 250 bp and meeting the library quality standards. Clones with clear bands were sequenced, yielding multiple valid sequences (Figure 11B). Through homology comparison and functional annotation, several candidate proteins potentially interacting with ZmSPS3 were identified (Table S5).
2.13. ZmSPS3 and Candidate Protein Yeast Two-Hybrid Validation
To further verify the reliability of the Y2H library screening results, a pairwise yeast two-hybrid assay was performed to examine the interactions between ZmSPS3 and the candidate proteins: a protein kinase (Zm00001eb129820), an F-box protein (Zm00001eb426160), and RABD2C (Zm00001eb356710). The bait vector ZmSPS3-BD was co-transformed with the AD vectors of the candidate proteins (Zm00001eb129820-AD, Zm00001eb426160-AD, and Zm00001eb356710-AD) into yeast cells (Figure 12). All co-transformants grew normally on DDO. On QDO, these co-transformants continued to grow robustly and formed clear colonies, indicating stable interactions between ZmSPS3 and the two candidate proteins. In contrast, all negative controls failed to grow on QDO medium, effectively ruling out autoactivation or nonspecific interactions. These results demonstrate that ZmSPS3 can specifically interact with the protein kinase and F-box protein in yeast cells.
3. Discussion
The number of SPS gene family members in maize has previously been reported as six or seven [20,29]. In this study, a total of seven members were systematically identified, which are mainly distributed across six chromosomes, and encode proteins of approximately 120 kDa. The Ka/Ks ratios for all gene pairs are well below 1, indicating that this gene family has undergone strong purifying selection during evolution. Phylogenetic analysis and cross-species synteny analysis revealed that maize SPS genes are closely related to those in other monocotyledons, such as rice and barley, whereas they are more distantly related to dicotyledons like Arabidopsis and tobacco, suggesting that maize SPS genes may have functions highly similar to those in rice and barley.
Sucrose metabolism is a central process in plant responses to abiotic stress [10,18]. In this study, although both ZmSPS3 and ZmSPS7 were induced by stress, the response of ZmSPS3 was the most prominent. Notably, the expression of ZmSPS3 increased by more than 8-fold at 12 h compared to 0 h under PEG treatment. Notably, ZmSPS3 showed a strong correlation between the enrichment of stress responsive cis-elements in its promoter region and its transcriptional response, suggesting that it may serve as a key rate-limiting gene in regulating carbon allocation and the accumulation of osmoprotectants in maize under drought conditions.
Analysis of cis-acting elements in the promoter regions of the ZmSPS gene family revealed that these genes not only contain drought-responsive MBS elements but are also enriched with various hormone-responsive motifs, such as the abscisic acid (ABA) responsive element ABRE, the methyl jasmonate (MeJA) responsive element TGACG-motif, and the auxin-responsive element AuxRR-core. The presence of ABRE elements in ZmSPS promoters provides a molecular basis for the coordination between sucrose and ABA signaling. For example, in grape, sucrose can synergistically induce the expression of the maturation-related gene ASR together with ABA [30,31]. ABA signaling not only directly activates ZmSPS transcription, but the resulting sucrose may further act as a secondary messenger to feedback and enhance the ABA signaling pathway [30]. As shown in fruit ripening studies, sucrose can increase ABA levels by inducing ABA biosynthesis genes (VvNCED2) and repressing ABA catabolic genes (VvCYP707A) [32]. Goren et al. found that suppressing sucrose synthase genes in tomato had little effect on total sucrose and other soluble sugars but increased the expression of the auxin transport-related gene PIN1 in shoot apices and leaves [33]. These findings suggest that the ZmSPS family may function in multiple signaling pathways, participating not only in sucrose metabolism but also in modulating plant growth, development, and stress responses through interactions with ABA, auxin, and other hormone signaling pathways.
In this study, yeast two-hybrid assays verified that ZmSPS3 physically interacts with a protein kinase and an F-box protein. This finding extends research on maize SPS from solely transcriptional regulation to the complex layer of post-translational regulation. Previous studies have shown that in rice, OsCPK17 can phosphorylate OsSPS4, participating in low-temperature stress responses [34]. In addition, multiple phosphorylation sites have been reported in SPS proteins, such as Ser^158^ [35], Ser^229^ [36], and Ser^424^ [37]. Based on the yeast two-hybrid library results for ZmSPS3 in this study, its interaction with the protein kinase suggests the possibility of phosphorylation modifications. In further studies, we will employ phosphorylation site prediction and protein experiments to validate the interaction between the protein kinase and ZmSPS3 and to elucidate its role in drought stress response.
F-box proteins are core components of the SCF E3 ubiquitin ligase complex. The interaction between ZmSPS3 and the F-box protein suggests that plants may finely regulate ZmSPS3 degradation via the ubiquitin-proteasome pathway. Moreover, sucrose can regulate the ethylene signaling negative regulator CONSTITUTIVE TRIPLE RESPONSE1 (CTR1) through the F-box protein ZEITLUPE, thereby stabilizing GIGANTEA (GI) and maintaining a normal circadian rhythm in plants [38]. This mechanism allows plants to rapidly reduce SPS protein abundance when drought is alleviated or environmental conditions change, thus preventing feedback inhibition or metabolic imbalance caused by excessive sucrose accumulation.
However, several limitations in the present study should be acknowledged. First, the osmotic stress simulated by PEG treatment, while informative, does not fully replicate the physiological complexity of soil-based drought stress in field conditions. Second, our functional insights into ZmSPS3 are currently limited to transcriptomic responses and protein–protein interactions, lacking direct evidence from physiological and biochemical measurements, such as SPS enzymatic activity and sucrose content, as well as photosynthetic phenotypic data. Consequently, further investigations involving stable genetic transformation and comprehensive phenotypic characterization under more realistic drought conditions are required. Future research will focus on evaluating the growth performance and water-use efficiency of transgenic maize lines to fully elucidate the biological role of ZmSPS3 in the complex drought-response regulatory networks.
4. Materials and Methods
4.1. Plant Material and Drought Stress Treatment
The maize inbred line B73 was used as the experimental material. Seeds were soaked in distilled water for 24 h and then placed in a dark incubator at 37 °C for germination. After germination, seedlings were transplanted into seedling pots containing a 1:1 (v/v) mixture of perlite and soil. Plants were grown in a controlled-environment growth chamber with day/night temperatures of 25 °C/22 °C and a 16 h/8 h light/dark photoperiod. When seedlings reached the three-leaf stage, osmotic stress was simulated using a 12% PEG6000 solution to mimic drought conditions (Coolaber, Beijing, China) [39]. Leaf samples were collected at 0, 1, 3, 6, 12, 24, 36, and 48 h after treatment. Each time point included three biological replicates, with each replicate weighing approximately 0.1 g, and three technical replicates were set for each biological sample. All collected leaf samples were immediately frozen in liquid nitrogen and then stored at −80 °C for subsequent analyses.
4.2. Identification of the ZmSPS Gene Family
The maize genome (Zm-B73-REFERENCE-NAM-5.0), protein sequences, and annotation files were downloaded from the Phytozome database (https://phytozome-next.jgi.doe.gov/, accessed on 4 October 2025). Members of the SPS gene family were initially identified using HMMER v3.3 based on Hidden Markov Models (HMMs) for the SPS family (PF00534, PF00862, PF05116) by searching against the maize protein sequences [40]. In addition, BLASTP version 2.15.0 searches were performed using Oryza sativa (rice) SPS protein sequences against the maize protein dataset, and the results were combined to generate a list of candidate gene IDs. The candidate sequences were further confirmed for the presence of conserved SPS domains using SMART (http://smart.embl.de/, accessed on 4 October 2025), the NCBI Conserved Domain Database (https://www.ncbi.nlm.nih.gov/cdd/, accessed on 4 October 2025), Pfam (http://pfam.xfam.org/, accessed on 4 October 2025), and Phmmer (https://www.ebi.ac.uk/Tools/hmmer/search/phmmer, accessed on 4 October 2025). Sequences lacking core SPS domains were discarded, and the remaining sequences were designated as the final set of ZmSPS genes.
4.3. Physicochemical Properties and Subcellular Localization Prediction of the ZmSPS Gene Family
The physicochemical properties of ZmSPS proteins were analyzed using the ProtParam online tool (https://web.expasy.org/protparam/, accessed on 5 October 2025). Subcellular localization of SPS proteins was predicted using DeepLoc-2.0 (https://services.healthtech.dtu.dk/services/DeepLoc-2.0/, accessed on 5 October 2025).
4.4. Conserved Domain and Gene Structure Analysis of ZmSPS Gene Family
The conservation of ZmSPS protein sequences was analyzed using the MEME Suite 5.59 online tool (https://meme-suite.org/meme/tools/meme, accessed on 5 October 2025). The number of motifs to be identified was set to 15, with motif widths constrained between 6 and 100 residues. Based on the maize genome GFF annotation file, exon and intron distribution information of the ZmSPS genes was extracted. Gene structures were subsequently visualized using appropriate visualization tools.
4.5. Prediction of Cis-Elements in the Promoters of the ZmSPS Gene Family
Based on the maize whole-genome sequence and corresponding annotation files, the 2000 bp upstream sequences from the start codon (ATG) of each ZmSPS gene were extracted as the putative promoter regions. These sequences were submitted to the PlantCARE online database (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 10 October 2025) for identification and functional annotation of cis-acting elements. The distribution of core cis-elements was integrated and visualized using the R programming language (version 4.3.1). Additionally, potential miRNA targets of ZmSPS genes were predicted using the psRNATarget online tool (https://www.zhaolab.org/psRNATarget/, accessed on 10 October 2025) with default parameters.
4.6. Chromosomal Localization and Evolutionary Synteny Analysis
Based on genome annotation information, the physical distribution of ZmSPS genes on the 10 maize chromosomes was visualized using TBtools v2.376 [41]. Multiple sequence alignment of SPS protein sequences from maize, rice, Arabidopsis, and tobacco (Nicotiana tabacum) was performed using MAFFT v7.526 [42]. Phylogenetic trees were constructed using IQ-TREE (v3) based on the Maximum Likelihood (ML) method [43]. The best-fit substitution model was automatically selected using ModelFinder. To assess the reliability of tree branches, ultrafast bootstrap (-bb) analysis was performed with 1000 replicates. Phylogenetic trees were visualized and beautified using Evolview3 (https://www.evolgenius.info/evolview-v3/, accessed on 10 October 2025). Synteny analysis was conducted using TBtools to assess both intra-genome collinearity within maize and inter-species collinearity with rice (Oryza sativa), barley (Hordeum vulgare), and Arabidopsis (Arabidopsis thaliana) genomes, which were downloaded for this purpose.
4.7. Transcriptome Analysis of the ZmSPS Gene Family
Transcriptome datasets with accession numbers SRP010680 and PRJNA226757 were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/, accessed on 10 October 2025). Gene expression of ZmSPS members at different developmental stages and under drought stress was quantified using TPM (Transcripts Per Million) values after alignment with Salmon V1.10.3 software [44]. Visualization and hierarchical clustering of gene expression patterns were performed using the heatmap module of TBtools.
4.8. Quantitative Real-Time PCR (RT-qPCR) and Data Analysis
To further validate the expression patterns of ZmSPS genes, total RNA was extracted from maize leaves using FreeZol Reagent (Vazyme, Nanjing, China). The RNA was then reverse transcribed into cDNA using HiScript III RT SuperMix for qPCR (+gDNA wiper) (Vazyme, Nanjing, China). Primers for RT-qPCR were designed using NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/, accessed on 20 October 2025), and their sequences are listed in Table S1. RT-qPCR reactions were performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) on a QuantStudio 3 real-time PCR system, following the protocol described by Mo et al. [45]. Relative gene expression levels were calculated using the LV method [46] and normalized against ZmActin as the internal reference gene (Table S1) [47]. Each sample included three biological replicates and three technical replicates. Statistical analyses were conducted using SPSS software (v26.0), and graphs were generated with GraphPad Prism 9.0.
4.9. GO Annotation and Functional Enrichment Analysis of ZmSPS Genes Family
The maize whole-genome protein sequences were functionally annotated using the EggNOG-mapper v5 online platform (http://eggnog5.embl.de/#/app/home, accessed on 25 October 2025) to obtain detailed Gene Ontology (GO) annotations for ZmSPS family members. Based on the annotation results, functional enrichment analysis was performed using the clusterProfiler R package (v4.0) [48]. The analysis covered three core GO categories: biological process (BP), cellular component (CC), and molecular function (MF). GO terms related to stress responses and resistance were further visualized using the ggplot2 (version 3.5.1) R (version 4.3.1) package, providing an intuitive representation of the biological functions of the ZmSPS gene family.
4.10. Yeast Two-Hybrid Screening of ZmSPS3
The full-length coding sequence (CDS) of ZmSPS3 was cloned into the pGBKT7 vector to construct the bait plasmid pGBKT7-ZmSPS3 (Table S1). Using the PEG/LiAc method, the bait plasmid was co-transformed with the empty pGADT7 vector into Y2HGold yeast competent cells [49]. Transformed yeast strains were plated on double dropout (SD/-Leu-Trp, DDO) and quadruple dropout (SD/-Leu-Trp-His-Ade, QDO) media to evaluate the transcriptional autoactivation activity and potential cytotoxicity of the bait protein. The pGBKT7-ZmSPS3 bait strain was subsequently co-transformed with a maize whole-tissue cDNA library. Transformants were first plated on TDO medium for primary screening, and colony-forming units (CFUs) were used to assess library coverage. Positive clones from the primary screen were transferred to QDO plates containing X-Gal for stringent secondary screening. Clones showing healthy growth and blue coloration were selected as potential interacting candidates. PCR amplification of positive clones from the secondary screen was performed using vector-specific primers Y2H-F/Y2H-R (Table S1). PCR products with clear and single bands were recovered and sequenced. The resulting sequences were analyzed for homology and functional annotation using NCBI BLAST and the UniProt database. Additionally, sequencing results were compared against the MaizeGDB database (https://maizegdb.org/, accessed on 30 October 2025) to identify candidate proteins potentially interacting with ZmSPS3. The list of aligned genes and functional annotation results are presented in the corresponding Table S5.
4.11. Yeast Two-Hybrid Validation
To validate the physical interactions between ZmSPS3 and candidate proteins, the full-length CDS of Zm00001eb426160, Zm00001eb129820, and Zm00001eb356710 were individually cloned into the pGADT7 vector (Table S1). The resulting prey plasmids were co-transformed with the pGBKT7-ZmSPS3 bait plasmid into Y2H yeast competent cells. Transformants were plated on double dropout (SD/-Leu-Trp, DDO) medium and quadruple dropout (SD/-Leu-Trp-His-Ade, QDO) stringent selection medium. Direct interactions between ZmSPS3 and each candidate protein were confirmed based on colony growth on QDO medium.
5. Conclusions
In this study, the SPS gene family in maize was systematically identified using bioinformatics approaches, resulting in the characterization of seven ZmSPS members. Their gene structures, conserved motifs, promoter elements, evolutionary relationships, and synteny were comprehensively analyzed. GO annotation and miRNA target prediction further revealed the potential transcriptional regulatory network of the SPS genes. Combined with transcriptome analysis under PEG-induced osmotic stress and validation by RT-qPCR, the results showed that ZmSPS3 was significantly upregulated under drought, with expression levels continuously increasing over time, indicating its key role in maize drought response. Furthermore, yeast two-hybrid experiments suggested interactions of ZmSPS3 with proteins, including a protein kinase and an F-box protein, pointing toward possible regulatory mechanisms involving post-translational modification and protein stability. Overall, these findings enrich the functional annotation of the maize SPS gene family and provide preliminary experimental evidence for constructing its molecular regulatory network under drought stress conditions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jubily A.H. El-Hashash E.F. Salem K.F.M. Breeding and Biotechnology Approaches for Genetic Enhancement in Maize (Zea mays L.)Breeding and Biotechnology of Grass and Bast Fiber Crops Salem K.F.M. Al-Khayri J.M. Jain S.M. Advances in Plant Breeding Strategies Springer Nature Cham, Switzerland 2025 Volume 12107173978-3-032-00402-4
- 2Azadi M.S. Shokoohfar A. Mojdam M. Lak S. Fazel M.A. Investigation of Changes in Corn Hybrids Grain Protein, Proline, and Micronutrient Content under the Influence of Drought and Fertilizers Turk. J. Agric. For.20224641342310.55730/1300-011X.3014 · doi ↗
- 3Azam M.G. Sarker U. Uddin M.S. Screening Maize (Zea mays L.) Genotypes for Phosphorus Deficiency at the Seedling Stage Turk. J. Agric. For.20224680282110.55730/1300-011X.3044 · doi ↗
- 4Noman A. Ali S. Naheed F. Ali Q. Farid M. Rizwan M. Irshad M.K. Foliar Application of Ascorbate Enhances the Physiological and Biochemical Attributes of Maize (Zea mays L.) Cultivars under Drought Stress Arch. Agron. Soil Sci.2015611659167210.1080/03650340.2015.1028379 · doi ↗
- 5Chávez-Arias C.C. Ligarreto-Moreno G.A. Ramírez-Godoy A. Restrepo-Díaz H. Maize Responses Challenged by Drought, Elevated Daytime Temperature and Arthropod Herbivory Stresses: A Physiological, Biochemical and Molecular View Front. Plant Sci.20211270284110.3389/fpls.2021.70284134367221 PMC 8341156 · doi ↗ · pubmed ↗
- 6Zhang L. Gao M. Hu J. Zhang X. Wang K. Ashraf M. Modulation Role of Abscisic Acid (ABA) on Growth, Water Relations and Glycinebetaine Metabolism in Two Maize (Zea mays L.) Cultivars under Drought Stress Int. J. Mol. Sci.2012133189320210.3390/ijms 1303318922489148 PMC 3317709 · doi ↗ · pubmed ↗
- 7Qiao M. Hong C. Jiao Y. Hou S. Gao H. Impacts of Drought on Photosynthesis in Major Food Crops and the Related Mechanisms of Plant Responses to Drought Plants 202413180810.3390/plants 1313180838999648 PMC 11243883 · doi ↗ · pubmed ↗
- 8Seleiman M.F. Al-Suhaibani N. Ali N. Akmal M. Alotaibi M. Refay Y. Dindaroglu T. Abdul-Wajid H.H. Battaglia M.L. Drought Stress Impacts on Plants and Different Approaches to Alleviate Its Adverse Effects Plants 20211025910.3390/plants 1002025933525688 PMC 7911879 · doi ↗ · pubmed ↗