Novel 8-trifluoromethylquinobenzothiazines—Synthesis and Evaluation for Antiproliferative and Antibacterial Activity
Daria Klimoszek, Anna Majewska, Małgorzata Jeleń, Marta Struga, Beata Morak-Młodawska, Małgorzata Dołowy

TL;DR
This study develops new 8-trifluoromethylquinobenzothiazine compounds with potential anticancer and antibacterial properties.
Contribution
The synthesis and biological evaluation of novel 8-trifluoromethylquinobenzothiazines with antiproliferative and antibacterial activity is presented.
Findings
Derivatives 8 and 12 showed micromolar cytotoxicity against lung and pancreatic cancer cells with moderate selectivity.
Compound 3 exhibited antibacterial activity against Gram-positive bacteria, including MRSA, with bactericidal effects confirmed.
Abstract
Background: Phenothiazine derivatives bearing trifluoromethyl substituents have attracted increasing interest as multifunctional scaffolds in drug repositioning strategies, particularly in cancer and infectious diseases. Structural modification of classical phenothiazines by incorporation of a quinoline moiety has previously been shown to enhance biological activity. Objectives: The present study aimed to develop an efficient synthesis of 8-trifluoromethylquinobenzothiazines and to evaluate the anticancer and antibacterial potential of their N-substituted analogues inspired by triflupromazine, trifluoperazine, and fluphenazine. Methods: 6H-8-Trifluoromethylquinobenzothiazine was synthesized by cyclization of 2-amino-4-trifluoromethylbenzenethiol and 3-bromo-2-chloroquinoline. The resulting quinobenzothiazine, unsubstituted at the nitrogen atom, was subjected to N-alkylation reactions to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
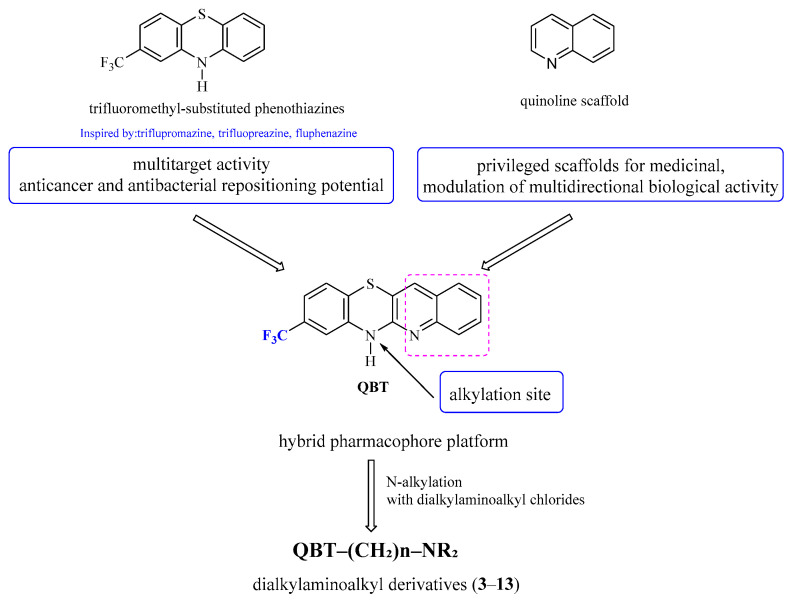 Figure 1
Figure 1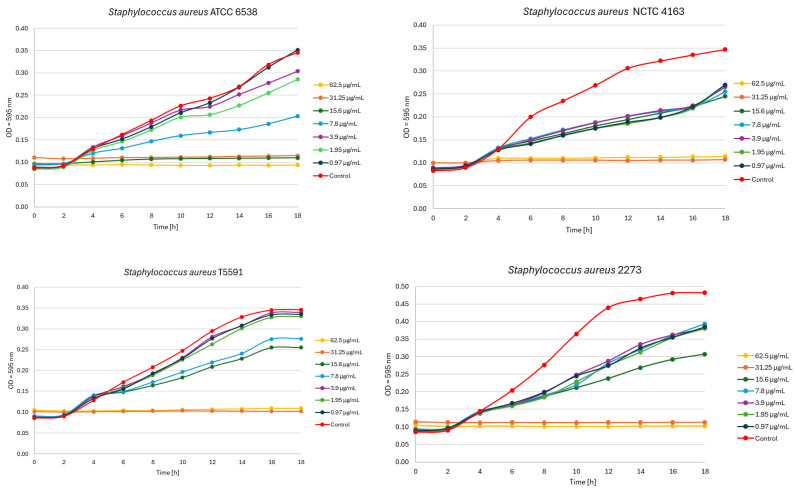 Figure 2
Figure 2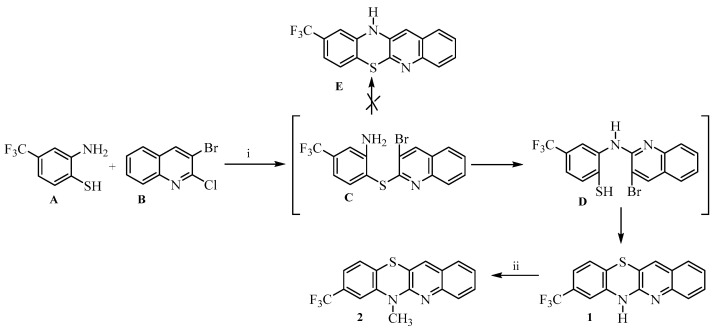 Figure 3
Figure 3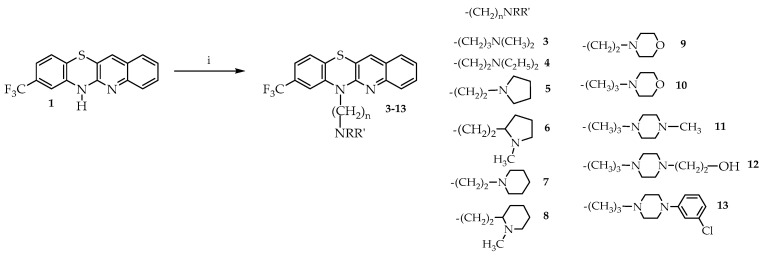 Figure 4
Figure 4- —Medical University of Silesia in Katowice
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhenothiazines and Benzothiazines Synthesis and Activities · Synthesis and Reactivity of Heterocycles · Quinazolinone synthesis and applications
1. Introduction
Cancer and bacterial infections remain leading global health challenges. According to the World Health Organization, cancer accounts for nearly 10 million deaths annually, with lung and pancreatic cancers ranking among the most lethal malignancies worldwide. Data from GLOBOCAN indicate a continuous increase in cancer incidence, emphasizing the urgent need for novel therapeutics with improved selectivity profiles [1]. Simultaneously, antimicrobial resistance (AMR) constitutes a major threat to global public health. Methicillin-resistant Staphylococcus aureus (MRSA) is classified among high-priority pathogens due to limited treatment options and high mortality rates [2].
Phenothiazine (PTZ) is a tricyclic heterocyclic compound containing nitrogen and sulfur atoms, first described by Heinrich A. Bernthesen at the end of the 19th century. Compounds containing a phenothiazine skeleton, initially used as dyes (e.g., methylene blue), quickly found applications in medicine, including in the treatment of malaria and as antiseptics [3,4,5]. In the mid-20th century, phenothiazines became the basis of modern psychiatry—chlorpromazine, the first antipsychotic drug, paved the way for the widespread use of PTZ as a privileged structure in pharmaceutical design [6]. At present, phenothiazines are a group of important compounds used not only in psychiatry, but also in the search for new therapeutics for cancer and bacterial diseases, including tuberculosis, as well as in materials chemistry as photoredox components [7,8,9].
They have become an important pillar of drug repositioning strategies due to their multidirectional effects on cancer cells and microorganisms, as well as their favorable pharmacological properties, which may accelerate clinical translation compared to the development of new molecular entities [10,11]. Their potential stems from their easy functionalization—various substitutions on benzene rings and nitrogen atoms allow for the modulation of both biological and physicochemical properties [7].
Compounds containing a trifluoromethyl group (–CF_3_) also occupy an important place within this class, as the CF_3_ moiety is widely used in drug design due to its ability to simultaneously modulate the electronic and lipophilic properties of molecules. Such properties translate into increased binding strength to the protein target, improved selectivity, and a favorable pharmacokinetic profile, including bioavailability and potential penetration into the CNS (central nervous system) [12,13,14,15]. In turn, pharmacological studies have shown that 2-CF_3_-PTZ derivatives may exhibit increased anticancer activity, including against oral cancer—in in vitro and in vivo studies, the efficacy of selected analogues was greater than that of classic phenothiazine drugs such as trifluoperazine [16]. In addition, synthetic reports indicate the possibility of obtaining stable and easily modifiable analogues of 2-CF_3_-PTZ, which opens up new perspectives in medicinal chemistry [15].
Triflupromazine, trifluoperazine, and fluphenazine—antipsychotic drugs containing a trifluoromethyl substituent at the second carbon of the phenothiazine ring—are increasingly becoming the subject of research into their potential repositioning because these drugs are multi-targeted, clinically well-known and often penetrate the CNS, which shortens the path to applications in new oncological and neurological indications and in infectious diseases. Their common theme is the modulation of cancer cell signaling pathways and the tumor microenvironment, as well as antibacterial activity, which may support therapies for resistant infections [10,11,17,18,19].
Triflupromazine, originally introduced as a sedative and antiemetic, effectively reduces anxiety and postoperative nausea while enhancing opioid and barbiturate effects without significantly affecting respiratory or hemodynamic function [20,21]. Although its clinical use has declined with the advent of second-generation neuroleptics, triflupromazine remains of scientific interest due to its diverse biological activity beyond central nervous system effects [22,23]. It exhibits potent antibacterial properties against numerous Gram-positive and Gram-negative strains, including Staphylococcus aureus, Shigella spp., and vibrios, with MIC values of 2–100 µg/mL, and increases survival in Salmonella typhimurium-infected mice [24]. Preclinical studies demonstrate that triflupromazine inhibits lactate dehydrogenase A (LDHA), suppressing glycolysis and enhancing cisplatin efficacy in lung cancer models, while also exerting cytotoxic effects on glioblastoma stem cells independent of dopamine receptor blockade [25]. Moreover, copper (II) complexes of triflupromazine with glycine or histidine display strong anticancer activity against liver (HepG2) and breast (MCF-7) cells, with low toxicity to normal cells [26].
Trifluoperazine (TFP) exhibits broad anticancer activity against various tumor types, including nasopharyngeal carcinoma (NPC) [27,28,29]. Thanks to its established safety profile and ability to cross the blood–brain barrier, trifluoperazine (TFP) represents a promising therapeutic option for central nervous system tumors and brain metastases [30,31]. In glioblastoma multiforme (GBM), TFP increases intracellular calcium levels by releasing Ca^2+^ from the endoplasmic reticulum through calmodulin (CaM) binding, thereby suppressing proliferation, migration, and invasion [30,31]. This mechanism also involves COX-2 upregulation, prostaglandin production, and PPARγ activation, leading to apoptosis in glioblastoma cells [32,33]. Additionally, TFP suppresses proliferation, migration, and clonogenicity of non-small cell lung cancer (NSCLC) cells, while its synthetic analog 3dc shows even stronger pro-apoptotic effects via caspase activation, DNA fragmentation, and inhibition of PI3K/AKT and ERK pathways. Both compounds significantly reduce tumor growth in vivo, supporting their potential in NSCLC therapy [34]. Furthermore, TFP exhibits neuroprotective effects against cytotoxic drug-induced neurotoxicity [35]. It may also modulate multidrug resistance by inhibiting calmodulin, P-glycoprotein, and topoisomerase II activity [36]. Finally, TFP demonstrates antiviral properties, effectively inhibiting dengue and Zika virus replication at the post-entry stage [37].
Fluphenazine, beyond its neuropsychiatric applications, exhibits therapeutic potential in oncology, infectious diseases, and as a tool for investigating drug resistance and membrane transport [38,39]. It shows cytotoxicity across multiple cancer cell types, including lung, breast, colon, liver, brain, leukemia, oral, ovarian, and skin cancers [38]. In triple-negative breast cancer (TNBC) models, fluphenazine inhibits proliferation, induces G0/G1 arrest, activates mitochondrial apoptosis, and suppresses migration and invasion by reducing ERK1/2 and AKT phosphorylation [40]. Molecular docking studies revealed that fluphenazine derivatives can inhibit caspase-3 by binding to its active site, forming stabilizing hydrogen bonds with Ser219 and Arg269 residues [41]. Phenothiazines, including fluphenazine, also modulate P-glycoprotein and signaling pathways while exhibiting antimicrobial, antioxidant, and neuroprotective effects [42]. In liver and breast cancer, fluphenazine induces cuproptosis—a copper-dependent cell death—and disrupts autophagy, limiting tumor growth [43,44]. It may also overcome drug resistance by sensitizing cells to antimitotic agents such as eribulin and inducing G2 arrest, DNA damage, and apoptosis via P-gp inhibition [45]. Additionally, fluphenazine shows antimicrobial activity against Gram-positive and Gram-negative bacteria, protecting mice from S. typhimurium infections as a “non-antibiotic” antimicrobial [46]. Structural optimization has yielded derivatives like CWHM-974 with enhanced antifungal potency and reduced resistance induction in C. albicans [39,47].
Our previous studies demonstrated that modifying the structure of phenothiazines by introducing one or two quinoline rings, leading to the formation of quinobenzothiazines and diquinothiazines, allowed for the creation of derivatives with multifaceted biological activity. Previous studies have demonstrated that selected quinoline-modified phenothiazines exhibit significant anticancer activity against numerous cancer cell lines, as well as antioxidant and immunomodulatory effects. These compounds inhibit mitogen-induced proliferation of peripheral blood mononuclear cells, limit TNFα production, and influence the activity of enzymes such as butyrylcholinesterase [48,49,50,51,52,53,54]. In vivo models, their anti-inflammatory and immunosuppressive effects have been observed, including inhibition of delayed-type hypersensitivity reactions, experimental psoriasis, and colitis, as well as an effect on the expression of IFNβ and genes dependent on this pathway [55,56]. The accumulated data indicate that phenothiazines modified with a quinoline ring constitute a promising class of compounds with potential use in the therapy of cancer and autoimmune diseases, which justifies their further modifications and studies on structure–activity relationships.
Despite the well-documented biological activity of trifluoromethyl-substituted phenothiazines and their growing interest in drug repositioning strategies, structural modifications involving hybridization with quinoline frameworks remain insufficiently explored (Figure 1). In particular, quinobenzothiazine systems bearing a trifluoromethyl substituent have not been systematically investigated in the context of anticancer and antibacterial activity.
2. Results
2.1. Synthesis and Spectral Data
In this study, a series of novel dialkylaminoalkyl derivatives of 8-trifluoromethylquinobenzothiazine were designed, synthesized, and evaluated for their anticancer and antibacterial activity. 6H-8-Trifluoromethylquinobenzothiazine 1 was prepared by the reaction of 2-amino-4-trifluoromethylbenzenethiol A and 3-bromo-2-chloroquinoline B (Scheme 1). The reaction was carried out in DMF under reflux for 1 h.
We then carried out alkylation reactions with methyl iodide towards derivative 2 (Scheme 1), and with dialkylaminoalkyl chloride hydrochlorides towards the target derivatives 3–13 (Scheme 2).
2.2. Biological Evaluationof Newly Synthesized Compounds
2.2.1. Cytotoxic Activity
The cytotoxic potential of newly synthesized compounds 1–13 (Scheme 1 and Scheme 2) was evaluated for their possible application in cancer therapy. Their effects were assessed on three human carcinoma cell lines: breast adenocarcinoma (MDA-MB-231), pancreatic cancer (Mia-PaCa-2), lung carcinoma (A-549), as well as on normal HaCaT keratinocytes, with IC_50_ values determined using the MTT assay [57,58]. For reference, the results were compared to the IC_50_ of the widely used chemotherapeutic agents doxorubicin and cisplatin (Table 1).
2.2.2. In Vitro Antibacterial Activity
Next, the antibacterial efficacy of the newly synthesized 8-trifluoromethylquinobenzothiazines 1–13 was assessed by pre-screening them for minimum inhibitory concentrations (MICs). Most of the tested 8-trifluoromethylquinobenzothiazines inhibited the growth of bacteria used in the study at a concentration of ≥125 µg/mL. The most promising substances from the presented series were substances 3, 5, and 8 (Table 2).
The antibacterial activity of the investigated derivatives was evaluated against reference Gram-positive bacteria, including several Staphylococcus aureus strains (ATCC 6538, NCTC 4163, 2273 [MSSA], and T5591 [MRSA]) and Enterococcus faecalis (ATCC 29212), as well as against Gram-negative bacilli such as Escherichia coli (ATCC 25922) and Pseudomonas aeruginosa (ATCC 15442). All strains are susceptible to ciprofloxacin, a fluorophinolone antibiotic.
An OD-based growth kinetics study was also performed for the most active compound 3. Figure 2 shows the growth inhibition of S. aureus strains based on OD exposed to compound 3.
3. Discussion
The present study is a continuation of our ongoing efforts focused on the synthesis and biological evaluation of quinobenzothiazine derivatives [48]. Here, we aimed to develop an efficient and selective synthetic route to 6H-8-trifluoromethylquinobenzothiazine as a versatile core scaffold enabling the preparation of novel analogues of clinically established phenothiazines, namely triflupromazine, trifluoperazine, and fluphenazine. In the newly designed structures, the benzene ring of the phenothiazine system was replaced with a quinoline moiety, while diverse dialkylaminoalkyl substituents were introduced at the thiazine nitrogen atom (Figure 1). This structural modification was motivated by previously reported in vitro and in vivo activities of quinoline-modified phenothiazines, as well as by the well-documented anticancer and antibacterial potential of trifluoromethyl-substituted phenothiazine drugs [48]. The applied design strategy aimed to combine the favorable pharmacological features of trifluoromethyl phenothiazines with the biological relevance of the quinoline scaffold. The study was initiated by developing an efficient synthetic route to 6H-8-trifluoromethylquinobenzothiazine 1. We used 2-amino-4-trifluoromethylbenzenethiol A and 3-bromo-2-chloroquinoline B as substrates (Scheme 1). The intermediate product in this type of syntheses of the phenothiazine system is phenylquinoline sulfide C. The initially formed phenylquinoline sulfide intermediate C may undergo Ullmann-type cyclization toward quinobenzothiazine E or proceed via a Smiles rearrangement (S→N type), in which the quinolinyl moiety migrates from sulfur to nitrogen, forming intermediate D that subsequently cyclizes to quinobenzothiazine 1. According to literature data, the reaction progress of this type of the synthesis of phenothiazine systems in most cases depends on the conditions used [59,60]. If the products of Ullmann cyclization and Smiles rearrangement are the same, it is impossible to determine which path will lead to the closure of the phenothiazine system (this occurs when substrates not substituted with additional substituents are used). When the substrate in the synthesis of the quinobenzothiazine system is substituted o-aminobenzenethiol as a substrate, as in the reaction we performed, the possibility of formation of two phenothiazines 1 and E should be considered. We had already synthesized 6H-8-trifluoroquinobenzothiazine 1 in our earlier studies, but in those studies we used 2,2′-dichloro-3,3′-diquinolyl disulfide and m-trifluoromethylaniline [61]. This reaction resulted in the formation of 6H-8-trifluoromethylquinobenzothiazine and 6H-10-trifluoromethylquinobenzothiazine in yields of 28% and 22%, respectively, which required chromatographic separation. The separation was hampered by the small difference in R_f_ coefficients. In further studies, we confirmed the formation of the 8-trifluoromethylquinobenzothiazine system using single-crystal X-ray analysis [62]. Comparison of the substances obtained in these reactions allowed us to tentatively assume that the reaction of 2-amino-4-trifluoromethylbenzenethiol A and 3-bromo-2-chloroquinoline B involves a Smiles rearrangement. We unequivocally confirmed the assumed structure of the quinobenzothiazine system by measuring ^1^H NMR spectra and two-dimensional NOESY and COSY spectra (Table S1) of the 6-methyl derivative 2. ^1^H NMR spectra also identified the product as 8-substituted quinobenzothiazine 2. The proton signals of the quinoline moiety were observed at low field (above 7.2 ppm) and appeared as a doublet with an ortho coupling constant (proton H-4), a singlet (proton H-12), a triplet with two ortho couplings (proton H-2), and a multiplet consisting of the signals from protons H-1 and H-3. The proton signals of the benzene ring were observed at high field (below 7.23 ppm) and were observed as two signals with a shape and multiplicity depending on the proton to which they were assigned. The proton signals H-10 and H-9 appeared as one multiplet with a multiplicity of 2, while the proton signal H-7 appeared as a broad singlet. To fully confirm the structure of the obtained quinobenzothiazine system, a ^13^C NMR spectrum and two-dimensional HSQC and HMBC spectra were also obtained. These spectra allowed the assignment of the appropriate C atoms to the individual signals. As a result, the products were identified as 8-trifluoromethylquino[3,2-b]benzo[1,4]thiazine (8-trifluoromethylbenzo[b]-1-azaphenothiazine) (Table S2).
Moreover, characteristic splitting signals originating from the CF_3_ group were observed in the ^13^C NMR spectra, as expected. The CF_3_ carbon gave signals at a shift of approximately 122.7–124.8 ppm, depending on the derivative, as quartets. The coupling constants for these signals were in the range ^1^JCF = 214–279 Hz. Similarly, the C-8 carbon with a CF_3_ substituent in the benzene ring was observed as quartets with δ from 129.5 to 130.9, depending on the derivative. The coupling constants for these signals were in the range ^2^JCF = 29.7–34.5 Hz. This effect of the CF_3_ substituent on the ^13^C NMR spectrum is also consistent with previously published data for compounds containing a CF_3_ substituent [63].
The use of 2-amino-4-trifluoromethylbenzenethiol A and 3-bromo-2-chloroquinoline B as starting materials allowed for the selective and efficient synthesis of 6H-8-trifluoromethylquinobenzothiazine, which was subsequently used to obtain derivatives containing N,N-dialkylaminoalkyl substituents at the 6-position, including the quinoline analogs triflupromazine, trifluoperazine, and fluphenazine. N,N-dialkylaminoalkyl derivatives were obtained by N-alkylation reactions of selected acyclic and cyclic dialkylaminoalkyl chloride hydrochlorides. The reactions were carried out in refluxing 1,4-dioxane in the presence of sodium hydroxide. These syntheses allowed for the preparation of eleven different 6-dialkylaminoalkyl derivatives 3–13 with good efficiency (Scheme 2).
Next, the cytotoxic potential of newly synthesized triflupromazine, trifluoperazine and fluphenazine derivatives 3–13 (Scheme 2) and compounds 1 and 2 (Scheme 1) was evaluated against three cancer cell lines: breast adenocarcinoma (MDA-MB-231), pancreatic cancer (Mia-PaCa-2), lung cancer (A-549), as well as on normal HaCaT keratinocytes (Table 1). Among the thirteen new, tested 6-substituted 8-trifluoromethylquinobenzothiazines 1–13, three derivatives—8 (with N-methylpiperidine ethyl substituent), 12 (with hydroxyethylpiperazinylpropyl substituent), and 6 (with N-methylpyrrolidinoethyl substituent)—were distinguished by favorable cytotoxicity and selectivity profiles, justifying further investigation of their anticancer potential. Compound 8 demonstrated uniform and moderate activity against three cancer cell lines: MDA-MB-231 (human breast adenocarcinoma), Mia-PaCa-2 (human pancreatic cancer), and A-549 (human lung carcinoma), achieving IC_50_ values in the range of 6.98–9.96 µM with relatively low toxicity to normal HaCaT keratinocytes (13.06 µM), which translates into a selectivity index (SI) in the range of 1.3–1.9. This combination of moderate potency and advantage over cancer cells makes compound 8 a particularly interesting candidate for further development. From a translational perspective, the relevance of this activity is strengthened by prior evidence demonstrating that ER-negative breast cancer cells can be effectively targeted by phenothiazine-derived agents. In a study conducted by Jeannine S. Strobl et al. [64], thioridazine markedly suppressed the proliferation of the triple-negative breast cancer cell line MDA-MB-231 following 72 h of exposure, demonstrating antiproliferative efficacy comparable to that of pimozide. Importantly, the authors proposed thioridazine as a potential non-cytotoxic alternative to tamoxifen for tamoxifen-resistant breast cancer. Thioridazine elicited also dose-dependent cytotoxicity in A549 sphere cells (lung cancer stem-like cells), inducing caspase-dependent apoptosis and G0/G1 cell cycle arrest, with significant inhibitory effects on xenografts in mice [65]. Guanhua Qian et al. [66] demonstrated that thioridazine induced cell death in A549 cells and enhanced cisplatin sensitivity through mitochondria-dependent apoptosis, with the combination treatment producing the highest percentage of dead cells [66]. Compound 12 showed the highest activity against A-549 cells (IC_50_ = 4.22 µM) and Mia-PaCa-2 (IC_50_ = 5.16 µM), whereas its activity against MDA-MB-231 was weaker (IC_50_ = 10.76 µM), suggesting a relative dependence of activity on tumor type. Its activity toward MDA-MB-231 is consistent with previous observations for fluphenazine, which inhibits proliferation, migration, and invasion of this cell line, although its efficacy varies depending on cancer type. The enhanced activity of compound 12 in lung and pancreatic cancer cells may indicate the involvement of molecular targets particularly relevant in these tumor types [67]. Quinobenzothiazine 6, although it had significantly higher IC_50_ values (approx. 43–48 µM for Mia-PaCa-2 and A-549), was non-toxic to HaCaT (IC_50_ > 100 µM), resulting in SI values exceeding 2. Such a wide therapeutic window, despite its lower potency, makes this compound an interesting scaffold for future structural modifications aimed at enhancing its anticancer activity while maintaining high selectivity. A comparative analysis of selectivity indices further highlights the distinct profiles of compounds 8 and 12 relative to the reference drug doxorubicin. Doxorubicin exhibited submicromolar IC_50_ values against MDA-MB-231 (0.5 µM) and A-549 (0.26 µM), confirming its high intrinsic potency. However, its cytotoxicity toward normal HaCaT keratinocytes (IC_50_ = 0.3 µM) resulted in low selectivity indices (SI = 0.60 for MDA-MB-231 and SI = 1.15 for A-549), indicating a narrow therapeutic margin. Notably, in the Mia-PaCa-2 model, doxorubicin showed markedly reduced activity (IC_50_ = 24.2 µM) combined with extremely poor selectivity (SI = 0.01), reflecting comparable or even greater toxicity toward normal cells than pancreatic cancer cells in this experimental setting. In contrast, compound 8 achieved SI values of 1.31–1.87, while compound 12 reached SI values of 1.40 (Mia-PaCa-2) and 1.71 (A-549), exceeding that of doxorubicin in the lung carcinoma model. Although their absolute potency was lower, both compounds demonstrated a more advantageous therapeutic window. Compound 8, containing a substituted piperidine ring, and compound 12, incorporating a piperazine derivative bearing a hydroxyethyl fragment, demonstrated higher cytotoxic potency than simple aliphatic analogues. Cyclic amines introduce conformational restriction and defined spatial orientation of the nitrogen atom, which may favor binding to protein targets involved in proliferation pathways. In contrast, more flexible side chains may lead to reduced target specificity. An especially interesting case is compound 6, which displayed moderate antiproliferative potency but minimal toxicity toward normal cells (IC_50_ > 100 µM). Consequently, the calculated selectivity indices were SI > 2.30 for Mia-PaCa-2 and SI > 2.09 for A-549, representing the highest selectivity observed within the series. Although compound 6 is less potent than doxorubicin in terms of absolute IC_50_ values, its markedly reduced cytotoxicity toward non-malignant cells suggests a substantially improved safety margin and identifies this scaffold as a promising candidate for further structural optimization. Derivative 11 (with methylpiperazinylpropyl substituent), a quinoline analogue of trifluoperazine, was characterized by an IC_50_ in the range of 11.31–17.88 µM against cancer cells, but its toxicity towards normal cells was comparable (IC_50_ = 11.29 µM), resulting in an SI close to 1 or <1. This indicates no preferential treatment for transformed cells. Derivatives 5 (with a pyrrolidinoethyl substituent) and 7 (with a piperidinoethyl substituent), which do not contain additional substituents in the cyclic amine fragment, showed moderate cytotoxic activity against all three cancer lines (IC_50_~19–50 µM), but their selectivity towards normal cells was low (SI in the range of 0.4–1.3). This means that their activity does not significantly differentiate between transformed and normal cells, which significantly reduces the translational potential of these compounds.
Introduction of morpholino substituents (derivatives 9 and 10) abolished anticancer activity while retaining cytotoxicity toward normal cells. No significant differences were observed with respect to the linker length between the thiazine and morpholine rings.
A direct comparison with the parent scaffold clearly demonstrates that N-alkylation is essential for biological activity. The unsubstituted 6H-8-trifluoromethylquinobenzothiazine 1 and its simple N-methyl derivative 2 exhibited negligible cytotoxic and antibacterial effects, whereas introduction of dialkylaminoalkyl substituents led to a substantial increase in activity. This observation confirms that the quinobenzothiazine core alone does not constitute a fully active pharmacophore and requires appropriate side-chain functionalization to exert biological effects.
In studies of the antibacterial activity of newly synthesized derivatives, molecule 3 containing a dimethylaminopropyl moiety was the only compound tested to exhibit antibacterial activity within the concentration range assessed. Quinobenzothiazine 3 inhibited the growth of S. aureus reference strains and clinical isolates with MIC values of 7.8–15.6 µg/mL. The corresponding MBC values were equal to the MIC or 2× MIC. Compound 3 also showed activity against E. faecalis ATCC 29212. In contrast, its activity against Gram-negative bacteria was limited: for E. coli ATCC 25922 the MIC and MBC were 125 µg/mL (MBC/MIC = 1), whereas no inhibitory effect against P. aeruginosa ATCC 15442 was observed at the highest concentration tested (MIC > 250 µg/mL). Overall, the MBC/MIC ratios for compound 3 ranged from 1 to 2 for all strains where MBC could be determined, consistent with a bactericidal effect under the test conditions. OD-based growth kinetics demonstrated a clear, concentration-dependent inhibition of S. aureus following exposure to compound 3 (0.97–62.5 µg/mL), with optical density measurements recorded every 2 h. For all strains increasing compound 3 concentrations progressively reduced the OD increase compared with the growth control (inoculated medium without the compound). At low quinobenzothiazine 3 concentrations (0.97–3.9 µg/mL), the growth curve of S. aureus ATCC 6538 and T5591 remained comparable to the growth control, whereas the remaining staphylococcal strains exhibited a pronounced shift of the growth curve relative to the control and a reduction in maximum optical density, indicating a higher susceptibility of these strains to the tested compound. This pattern reflects a sub-inhibitory effect characterized by impaired growth kinetics, while the bacterial population retained the capacity for continued proliferation. At the highest 3 concentrations (31.25–62.5 µg/mL), OD trajectories remained close to baseline throughout the monitoring period, indicating strong suppression of bacterial growth. Figure 2 presents OD-based growth inhibition of S. aureus strains exposed to compound 3.
S. aureus is a major cause of severe multiorgan infections, and methicillin-resistant strains (MRSA) rank among the leading bacterial contributors to mortality attributable to antimicrobial resistance worldwide [68]. Quinobenzothiazine 3 demonstrated a clear preference for Gram-positive bacteria, as reflected by low MIC values against both reference and clinical S. aureus strains, including an MRSA isolate expressing an inducible MLSB phenotype (iMLSB), as well as comparable activity against E. faecalis. The corresponding MBC/MIC ratios (1–2) indicate that concentrations required to inhibit growth were bactericidal under in vitro conditions. In contrast, compound 3 showed reduced activity against E. coli and no measurable inhibitory effect against P. aeruginosa within the tested concentration range. Against the backdrop of the global burden of bacterial infections and the limitations of currently available therapies, there remains a clear need for continued discovery of novel antibacterial compounds and chemical optimization of promising lead structures to improve efficacy across a range of clinically relevant pathogens.
4. Materials and Methods
4.1. Chemical Part
Melting points were measured in open capillary tubes using a Boetius melting point apparatus (Stuart Equipment, Stone, UK) and are reported without correction. NMR spectra were acquired on Bruker Avance instruments (Bruker, Billerica, MA, USA), operating at 600 MHz for ^1^H, 150 MHz for ^13^C and 564 MHz for ^19^F nuclei in CDCl_3_ as solvents. For selected compounds, two-dimensional NMR experiments (COSY, NOESY, HSQC, and HMBC) were performed at 600 MHz employing the COSYGPSW, NOESYGPPHSW, HSQCGPPH, and HMBCGP pulse sequences. High-resolution mass spectra (HRMS) were obtained using electron impact (EI) ionization on a Bruker Impact II mass spectrometer (Bruker, Billerica, MA, USA). The ^1^H NMR, ^13^C NMR, and HRMS spectra are provided in the Supplementary Materials. Thin-layer chromatography (TLC) analyses were carried out on neutral aluminum oxide 60 F_254_ plates (type E; Merck 1.05581, Darmstadt, Germany) with dichloromethane (CH_2_Cl_2_) as the eluent.
General procedure for the synthesis of 8-trifluoromethylquinobenzothiazine derivatives:
- Synthesis of 6H-8-trifluoromethylquinobenzothiazine 1.
A solution of 0.24 g (1 mmol) of 3-bromo-2-chloroquinoline B in 10 mL of dry DMF was prepared and heated to reflux, followed by the addition of 0.16 g (1 mmol) of 2-amino-4-trifluoromethylthiophenol A. The mixture was heated at reflux for 1 h. It was then cooled and poured into 30 mL of water. The precipitate was filtered off, washed with water, dried, and purified by crystallization from ethanol to give 6H-8-trifluoromethylquinobenzothiazine 1:
Yield: 88%. M.p.: 220–221 °C. ^1^H NMR (CDCl_3_) δ: 7.00 (s, 1H, H-7), 7.06–7.07 (d, 1H, H-10, J = 6.5 Hz), 7.16–7.18 (d, 1H, H-9, J = 8 Hz), 7.37 (t, 1H, H-2, J = 6.5 Hz), 7.53 (d, 1H, H-1, J = 6.5 Hz), 7.57 (t, 1H, H-3, J = 6.5 Hz), 7.63 (s, 1H, H-12), 7.64 (d, 1H, H-4, J = 8.5 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 112.68, 115.67, 120.29, 121.18, 123.40, 124.42, 125.34, 125.99, 126.33, 126.70, 130.24, 130.64, 132.79, 137.56, 142.80, 149.53. HRMS (ESI) calcd for C_16_H_10_F_3_N_2_S [M+H]^+^: 319.0517, found: 319.0519.
Synthesis of 6-methyl-8-trifluoromethyl-quinobenzothiazine 2.
A suspension of 0.16 g (0.5 mmol) of 6H-8-trifluoromethylquinobenzothiazine 1 and 0.06 g of NaH (2.5 mmol) in 5 mL of dry DMF was prepared and stirred at room temperature for 0.5 h. After this time, 0.05 mL of methyl iodide (0.75 mmol) was added and stirring was continued for 24 h. The reaction mixture was then poured into 30 mL of water. The resulting precipitate was filtered off, washed with water, and purified by column chromatography (alumina, CH_2_Cl_2_) to give 6-methyl-8-trifluoromethylquinobenzothiazine 2:
Yield: 76%. M.p.: 115–116 °C. ^1^H NMR (CDCl_3_) δ: 3.65 (s, 3H, CH_3_), 7.10 (s, 1H, H-7), 7.19–7.23 (m, 2H, H-9, H-10), 7.32 (t, 1H, H-2, J = 6 Hz), 7.54–7.57 (m, 2H, H-1, H-3), 7.69 (s, 1H, H-12), 7.81 (d, 1H, H-4, J = 6 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 33.87, 111.81 (C-7, ^3^J = 4.5 Hz), 117.46 (C-10a), 119.30 (C-9, ^3^J = 4.5 Hz), 123.99 (CF_3_, ^1^J = 271.5 Hz), 124.65 (C-2), 125.35 (C-5a), 125.98 (C-4a), 126.33 (C-3), 126.72 (C-10), 127.48 (C-4), 129.49 (C-1), 130.03 (C-8, ^2^J = 31.5 Hz), 132.40 (C-12), 143.37 (C-6a), 145.74 (C-12a), 152.48 (C-11a). HRMS (ESI) calcd for C_17_H_12_F_3_N_2_S [M+H]^+^: 333.0673, found: 333.0682.
Synthesis of 6-dialkylaminoalkyl-8-trifluoromethylquinobenzothiazines 3–13.
A mixture of 6H-8-trifluoroquinobenzothiazine 1 (0.16 g, 0.5 mmol) and sodium hydroxide (0.30 g, 7.5 mmol) in 5 mL of dioxane was heated at reflux for one hour. Then, dialkylaminoalkyl chloride hydrochloride (1.5 mmol, 3-dimethylaminopropyl—0.24 g,2-diethylaminoethyl—0.26 g, 2-(1-pyrrolidyl)ethyl—0.26 g, 2-(1-methylpyrrolidin-2-yl)ethyl—0.27 g, 2-(1-piperidyl)ethyl—0.26 g, 2-(1-methyl-2-piperidinyl)ethyl—0.30 g, 2-(1-morpholinyl)ethyl—0.28 g, 3-(1-morpholinyl)propyl—0.30 g, 3-(4-methylpiperazin-1-yl)propyl 0.38 g, 3-(4-hydroxyethylpiperazin-1-yl)propyl 0.42 g, 4-(3-chlorophenyl)piperazin-1-yl)propyl—0.47 g) was added to the reaction system and heating at reflux was continued for 3 h. Then the reaction mixture was cooled and poured into water (25 mL). The resulting solution was extracted with chloroform. The combined extracts were washed with water to pH = 7 and dried over Na_2_SO_4_. The chloroform was evaporated in vacuo, and the residue was purified by column chromatography (Al_2_O_3_, CHCl_3_) to afford compounds 3–13:
- N,N-dimethyl-3-(8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazin-6-yl)propan-1-amine (3):
Yield: 65%. M.p.: 75–77 °C. ^1^H NMR (CDCl_3_) δ: 2.07–2.09 (m, 2H, CH_2_), 2.36 (s, 6H, 2CH_3_), 2.54–2.57 (m, 2H, CH_2_), 4.25–4.27 (m, 2H, CH_2_), 7.10–7.11 (m, 2H, H-7, H-9), 7.17 (s, 1H, H-10), 7.26 (t, 1H, H-2, J = 6 Hz), 7.46–7.51 (m, 2H, H-1, H-3), 7.54 (s, 1H, H-12), 7.70 (d, 1H, H-4, J = 6 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 23.89, 43.93, 45.26, 57.20, 112.03 (^3^J = 3.0 Hz), 117.26, 119.07 (^3^J = 3.0 Hz), 124.53 (^1^J = 273.0 Hz), 125.06, 126.07, 126.20, 126.62, 126.85, 127.48, 129.38, 129.91 (^2^J = 31.3 Hz), 132.01, 141.96, 145.73, 151.40. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.83. HRMS (ESI) calcd for: C_21_H_21_F_3_N_3_S [M+H]^+^: 404.1408, found: 404.1401.
N,N-diethyl-2-(8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazin-6-yl)ethan-1-amine (4):
Yield: 71%. M.p.: Yellow oil. ^1^H NMR (CDCl_3_) δ: 1.18–1.25 (m, 6H, 2CH_3_), 2.76 (m, 4H, 2CH_2_), 2.94 (m, 2H, CH_2_), 4.33 (m, 2H, CH_2_), 6.98 (d, 1H, H-7), 7.05–7.08 (m, 2H, H-9, H-10), 7.21 (t, 1H, H-2, J = 6 Hz), 7.43–7.45 (m, 2H, H-1, H-3), 7.53 (s, 1H, H-12), 7.62 (d, 1H, H-4, J = 6 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 11.47, 29.72, 47.05, 47.82, 112.23 (^3^J = 3.0 Hz), 117.06, 120.29 (^3^J = 3.0 Hz), 123.63 (^2^J = 279.0 Hz), 124.36, 126.13, 126.23, 126.68, 126.79, 127.38, 129.45, 129.93 (^1^J = 31.3 Hz), 131.95, 145.66, 149.68, 150.95. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.88. HRMS (ESI) calcd for: C_22_H_23_F_3_N_3_S [M+H]^+^: 418.1565, found: 418.1574.
6-(2-(pyrrolidin-1-yl)ethyl)-8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazine (5):
Yield: 67% M.p.: Yellow oil. ^1^H NMR (CDCl_3_) δ: 2.17–2.20 (m, 4H, 2CH_2_), 3.50–3.53 (m, 4H, 2CH_2_), 3.86–3.88 (m, 2H, CH_2_), 4.76 (t, 2H, CH_2_, J = 7.2 Hz), 7.14 (d, 1H, H-7, J = 3.6 Hz), 7.19–7.21 (m, 2H, H-9, H-10), 7.28 (t, 1H, H-2, J = 7.2 Hz), 7.48–7.50 (m, 2H, H-1, H-3), 7.63–7.69 (m, 2H, H-4, H-12). ^13^C NMR (150 MHz, CDCl_3_) δ: 23.46, 41.45, 50.69, 53.70, 112.13 (^3^J = 3.3 Hz), 117.63, 120.30 (^3^J = 3.5 Hz), 124.25, 124.43 (^1^J = 214.0 Hz), 125.35, 126.21, 126.52, 127.21, 127.43, 129.86, 130.72 (^2^J = 32.7 Hz), 132.78, 141.35, 145.16, 150.78. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.75. HRMS (ESI) calcd for: C_22_H_21_F_3_N_3_S [M+H]^+^: 416.1408, found: 416.1411.
6-(2-(1-methylpyrrolidin-2-yl)ethyl)-8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazine (6):
Yield: 71% M.p.: Yellow oil. ^1^H NMR (CDCl_3_) δ: 1.83–1.79 (m, 2H, CH_2_), 1.83–1.93 (m, 2H, CH_2_), 2.04–2.07 (m, 2H, CH_2_), 2.17–2.22 (m, 2H, CH_2_), 2.34 (s, 3H, CH_3_), 4.19 (t, 2H, CH_2_, J = 5.9 Hz), 7.04 (m, 3H, H-7, H-9, H-10), 7.18 (t, 1H, H-2, J = 3.8 Hz), 7.40–7.42 (m, 2H, H-1, H-3), 7.50 (s, 1H, H-12), 7.60 (d, 1H, H-4, J = 6 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 22.11, 28.95, 30.19, 40.23, 42.86, 56.87, 65.02, 111.93 (^3^J = 3 Hz), 117.22, 119.07 (^3^J = 3.5 Hz), 123.54, 124.32 (^1^J = 233.7 Hz), 124.60, 126.07, 126.20, 126.89, 127.46, 129.39, 129.84 (^2^J = 32.6 Hz), 132.02, 141.83, 145.71, 151.31. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.67. HRMS (ESI) calcd for: C_23_H_23_F_3_N_3_S [M+H]^+^: 430.1565, found: 430.1567.
6-(2-(piperidin-1-yl)ethyl)-8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazine (7):
Yield: 73%. M.p.: Yellow oil. ^1^H NMR (CDCl_3_) δ: 1.82–1.88 (m, 4H, 2CH_2_), 2.24–2.28 (m, 2H, CH_2_), 2.82–2.83 (m, 2H, CH_2_), 3.41 (m, 2H, CH_2_), 3.66–3.67 (m, 2H, CH_2_), 4.87 (t, 2H, CH_2_, J = 6.6 Hz), 7.20 (m, 3H, H-7, H-9, H-10), 7.30 (t, 1H, H-2, J = 7.8 Hz), 7.50–7.53 (m, 2H, H-1, H-3), 7.67 (s, 1H, H-12), 7.76 (d, 1H, H-4, J = 9 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 23.36, 24.56, 29.71, 53.94, 54.47, 112.39, 117.29, 119.51, 124.01 (^1^J = 245.3 Hz), 124.85, 126.13, 126.29, 126.67, 126.95, 127.42, 129.52, 130.23 (^2^J = 32.3 Hz), 132.17, 141.72, 145.52, 150.96. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.47. HRMS (ESI) calcd for: C_23_H_23_F_3_N_3_S [M+H]^+^: 430.1565, found: 430.1579.
6-(2-(1-methylpiperidin-2-yl)ethyl)-8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazine (8):
Yield: 68%. M.p.: 115–116 °C. ^1^H NMR (CDCl_3_) δ: 1.73–1.78 (m, 4H, 2CH_2_), 1.85–1.93 (m, 2H, CH_2_), 2.13–2.21 (m, 2H, CH_2_), 2.28–2.34 (m, 2H, CH_2_), 2.53 (s, 3H, CH_3_), 4.16–4.37 (m, 2H, CH_2_), 7.09 (s, 1H, H-7), 7.13 (s, 2H, H-9, H-10), 7.28 (t, 1H, H-2, J = 4.0 Hz), 7.49–7.52 (m, 2H, H-1, H-3), 7.57 (s, 1H, H-12), 7.68 (d, 1H, H-4, J = 8.6 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 23.83, 25.24, 27.99, 30.43, 42.82, 56.79, 62.64, 111.57 (^3^J = 3.8 Hz), 117.00, 118.98 (^3^J = 3.5 Hz), 123.92 (^2^J = 246.6 Hz), 124.58, 124.84, 126.05, 126.18, 126.81, 127.36, 129.38, 129.30 (^2^J = 32.9 Hz), 131.84, 141.73, 145.74, 150.92. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.75. HRMS (ESI) calcd for: C_24_H_25_F_3_N_3_S [M+H]^+^: 444.1721, found: 444.1727.
4-(3-(8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazin-6-yl)ethyl)morpholine (9):
Yield: 64%. M.p.: 105–106 °C. ^1^H NMR (CDCl_3_) δ: 2.74–2.95 (m, 8H, 4CH_2_), 3.80–3.91 (m, 2H, CH_2_), 4.38–4.51 (m, 2H, CH_2_), 6.89 (s, 1H, H-7), 7.08 (m, 2H, H-9, H-10), 7.22 (t, 1H, H-2, J = 7.2 Hz), 7.43–7.46 (m, 2H, H-1, H-3), 7.55 (s, 1H, H-12), 7.63 (d, 1H, H-4, J = 8.7 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 29.71, 53.06, 53.62, 66.16, 112.48 (^3^J = 3.2 Hz), 117.29, 119.46 (^3^J = 3.3 Hz), 122.74 (^1^J = 247.8 Hz), 124.84, 126.13, 126.29, 126.75, 126.95, 127.42, 129.54, 130.09 (^2^J = 32.3 Hz), 132.21, 141.83, 145.55, 151.12. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.50. HRMS (ESI) calcd for: C_22_H_21_F_3_N_3_OS [M+H]^+^: 432.1357, found: 432.1360.
4-(3-(8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazin-6-yl)propyl)morpholine (10):
Yield: 64%. M.p.: Yellow oil. ^1^H NMR (CDCl_3_) δ: 2.14 (m, 2H, CH_2_), 2.60 (m, 4H, 2CH_2_), 3.79–3.85 (m, 6H, 3CH_2_), 4.35 (m, 2H, CH_2_), 7.12 (s, 1H, H7), 7.15–7.18 (m, 2H, H-9, H-10), 7.30 (t, 1H, H-2, J = 7.1 Hz), 7.52–7.55 (m, 2H, H-1, H-3), 7.63 (s, 1H, H-12), 7.70 (d, 1H, H-4, J = 8.6 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 43.61, 53.61, 56.31, 66.56, 111.98 (^3^J = 4.0 Hz), 117.37, 119.11, 119.13, 124.64, 124.75 (^1^J = 224.8 Hz), 126.09, 126.25, 126.93, 127.37, 129.46, 129.85 (^2^J = 29.7 Hz), 132.1013, 142.00, 145.70, 151.50. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.67. HRMS (ESI) calcd for: C_23_H_23_F_3_N_3_OS [M+H]^+^: 446.1514, found: 446.1523.
6-(3-(4-methylpiperazin-1-yl)propyl)-8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazine (11):
Yield: 70%. M.p.: 108–109 °C. ^1^H NMR (CDCl_3_) δ: 2.06–2.09 (m, 2H, CH_2_), 2.35–2.38 (m, 5H, CH_2_, CH_3_), 2.51–2.55 (m, 2H, CH_2_), 2.60 (s, 4H, 2CH_2_), 3.61 (m, 2H, CH_2_), 4.31 (m, 2H, CH_2_), 7.12 (s, 1H, H7), 7.13–7.16 (m, 2H, H-9, H-10), 7.29 (t, 1H, H-2, J = 5.8 Hz), 7.51–7.54 (m, 2H, H-1, H-3), 7.61 (s, 1H, H-12), 7.69 (d, 1H, H-4, J = 8.7 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 23.39, 43.72, 45.68, 52.79, 54.90, 55.69, 112.03 (^3^J = 3.5 Hz), 117.40, 119.01 (^3^J = 3.5 Hz), 124.00 (^1^J = 270.9 Hz), 125.23, 126.07, 126.21, 126.86, 127.42, 129.39, 129.78 (^2^J = 31.9 Hz), 129.99, 132.00, 142.07, 145.75, 151.54. ^19^F NMR (564 MHz, CDCl_3_) δ: −63.00. HRMS (ESI) calcd for: C_24_H_26_F_3_N_4_S [M+H]^+^: 459.1830, found: 459.1820.
2-(4-(3-(8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]thiazin-6-yl)propyl)piperazin-1-yl)ethan-1-ol (12):
Yield: 67%. M.p.: Yellow oil. ^1^H NMR (CDCl_3_) δ: 1.96–1.98 (m, 2H, CH_2_), 2.06–2.10 (m, 2H, CH_2_), 2.51–2.54 (m, 4H, 2CH_2_), 2.60–2.62 (m, 3H, OH, CH_2_), 3.60–3.62 (m, 2H, 2CH_2_), 3.65–3.68 (m, 4H, 2CH_2_), 4.32 (s, 2H, CH_2_), 7.12 (s, 1H, H-7), 7.15–7.16 (m, 2H, H-9, H-10), 7.30 (t, 1H, H-2, J = 7.8 Hz), 7.52–7.54 (m, 2H, H-1, H-3), 7.63 (s, 1H, H-12), 7.70 (d, 1H, H-4, J = 8.7 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 23.35, 29.77, 43.12, 43.72, 52.95, 57.60, 59.35, 112.03 (^3^J = 3.4 Hz), 117.40, 119.04 (^3^J = 4.0 Hz), 123.63 (^1^J = 267.4 Hz), 124.58, 125.26, 126.07, 126.23, 126.88, 127.39, 129.41, 129.76 (^2^J = 32.0 Hz), 132.04, 142.06, 145.74, 151.55. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.72. HRMS (ESI) calcd for: C_25_H_28_F_3_N_4_OS [M+H]^+^: 489.1936, found: 489.1937.
6-(3-(4-(3-chlorophenyl)piperazin-1-yl)propyl)-8-(trifluoromethyl)-quino[3,2-b]benzo[1,4]-thiazine (13):
Yield: 75%. M.p.: Yellow oil. ^1^H NMR (CDCl_3_) δ: 2.16 (m, 2H, CH_2_), 2.71 (m, 6H, 3CH_2_), 3.261–3.31 (m, 4H, 2CH_2_), 4.37 (m, 2H, CH_2_), 6.80–6.84 (m, 2H, 2H-Ph), 6.89 (s, 1H, H-7), 7.14 (s, 1H, 1H-Ph), 7.17–7.20 (m, 3H, H-9, H-10, 1H-Ph), 7.31 (t, 1H, H-2, J = 7.2 Hz), 7.54–7.55 (m, 2H, H-1, H-3), 7.64 (s, 1H, H-12), 7.72 (d, 1H, H-4, J = 7.8 Hz). ^13^C NMR (150 MHz, CDCl_3_) δ: 29.73, 43.712, 48.48, 53.05, 55.91, 112.00 (^3^J = 3.9 Hz), 113.97, 115.91, 117.39, 119.12 (^3^J = 3.5 Hz), 119.42, 123.07, 123.85, 124.64 (^1^J = 270.4 Hz), 125.28, 126.10, 126.25, 126.94, 127.41, 129.47, 129.99 (^2^J = 34.5 Hz), 130.05, 132.11, 134.95, 142.02, 145.73, 151.54. ^19^F NMR (564 MHz, CDCl_3_) δ: −62.64. HRMS (ESI) calcd for: C_29_H_27_ClF_3_N_4_S [M+H]^+^: 555.1597, found: 555.1595.
4.2. Biological Assays
4.2.1. Cell Line and Culture
The human cell lines MDA-MB-231 (breast cancer), Mia-Paca-2 (pancreatic cancer) A-549 (lung cancer), and HaCaT (immortalized keratinocytes) were sourced from the American Type Culture Collection (ATCC) in Rockville, MD, USA. Culturing conditions varied: A-549, MDA-MB-231, and Mia-Paca-2, HaCaT cells were cultured in DMEM High Glucose (Biowest SAS, France). The growth medium consisted of 10% fetal bovine serum (FBS, Sigma-Aldrich, St. Louis, MO, USA), 20 mM HEPES (Biowest, Nuaillé, France), and antibiotics (100 U/mL of penicillin and 100 μg/mL of streptomycin) from Gibco, Grand Island, NY, USA. All cells were maintained in a humidified incubator at 37 °C with a 5% CO_2_ atmosphere until they reached 80–90% confluence.
4.2.2. MTT Assay
Cytotoxic activity was evaluated using the MTT assay as previously described [69]. The tested compounds were dissolved in DMSO to prepare 10 mM stock solutions and subsequently diluted in culture medium to obtain the required working concentrations. Untreated control cells were incubated in culture medium without test compounds. The final DMSO concentration in the wells was kept constant across all experimental conditions and did not exceed 1% at the highest tested compound concentration. At this DMSO level, no effect on cell viability was observed. Cancer cell lines (MDA-MB-231, Mia-PaCa-2, A-549) and normal HaCaT keratinocytes were seeded in 96-well plates at a density of 1 × 10^4^ cells per well and allowed to adhere overnight. The tested compounds were added at concentrations ranging from 5 to 140 µM and incubated for 72 h. Following incubation, MTT reagent was added and the formation of formazan crystals was quantified spectrophotometrically. Cell viability was expressed as a percentage relative to untreated control cells. All experiments were performed in three independent experiments, each conducted in triplicate (n = 3). IC_50_ values were calculated by nonlinear regression analysis of dose–response curves. Data are presented as mean ± standard deviation (SD). Statistical significance testing was not performed for IC_50_ comparisons, as the analysis was intended to be descriptive and to assess the relative activity of the tested compounds.
4.2.3. In Vitro Antibacterial Studies
Bacterial Strains
The antibacterial activity of newly synthesized compounds 3, 5, and 8 was evaluated against a panel of reference strains obtained from the American Type Culture Collection (ATCC) and the National Collection of Type Cultures (NCTC), as well as two clinical isolates of Staphylococcus aureus. The reference strains included Staphylococcus aureus ATCC 6538 and NCTC 4163, Enterococcus faecalis ATCC 29212, Escherichia coli ATCC 25922, and Pseudomonas aeruginosa ATCC 15442. Clinical isolates comprised S. aureus 2273 (MSSA) and S. aureus T5591 (MRSA, iMLSB phenotype). All strains were stored at −80 °C in appropriate cryoprotective media. Prior to susceptibility testing, bacteria were subcultured twice on Columbia agar (bioMérieux, Marcy-l’Étoile, France) to ensure viability and phenotypic stability.
Inoculum Preparation
Fresh bacterial colonies were suspended in sterile 0.85% NaCl and adjusted to a turbidity equivalent to 0.5 McFarland standard (approximately 1 × 10^8^ CFU/mL) using a densitometer. The suspension was subsequently diluted in Mueller–Hinton broth (MHB; bioMérieux, Marcy-l’Étoile, France) to obtain a final inoculum of 5 × 10^5^ CFU/mL, in accordance with standard susceptibility testing procedures.
Test Compounds
Tested compounds were dissolved in analytical-grade dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) and further diluted in MHB. The final concentration of DMSO in test wells did not exceed 5% (v/v). Two-fold serial dilutions were prepared to achieve final compound concentrations ranging from 1.95 to 250 μg/mL.
MICs were determined by broth microdilution in 96-well plates (Wuxi NEST Biotechnology, Wuxi, China) according to EUCAST recommendations [70] and EN ISO 20776-1:2020 [71]. MIC was defined as the lowest concentration completely inhibiting visible growth (turbidity or a pellet). Each test included a growth control (no compound), sterility control (no inoculum), and a solvent control (DMSO at the corresponding final concentration). DMSO control showed no inhibition vs. growth control.
MBC was defined as the lowest concentration resulting in ≥99.9% (3 log_10_) reduction in viable count relative to the initial inoculum. MBCs were determined by subculturing aliquots from wells without visible growth onto agar plates and recording colony formation after incubation. Experiments were performed in triplicate.
The MBC/MIC ratio was used to classify antimicrobial action. A ratio ≤ 4 was interpreted as bactericidal activity, while >4 indicated a bacteriostatic effect [72].
Bactericidal Kinetics Assay
The time-dependent antibacterial activity of compound 3 was assessed using a turbidity-based growth kinetics assay. This analysis was performed exclusively for staphylococcal strains (both reference and clinical isolates), as compound 3 demonstrated the highest activity against this genus.
Kinetic analysis was not performed for enterococci; therefore, conclusions regarding E. faecalis remain preliminary due to the inclusion of a single strain only.
Wells containing the bacterial inoculum without compound 3 served as positive growth controls, whereas wells containing MHB supplemented with compound 3 but lacking bacterial inoculation were used for blank correction. Optical density at 595 nm (OD_595_) was measured at 37 °C every 2 h for 18 h using a microplate reader (Thermo Scientific, Waltham, MA, USA).
4.2.4. Statistical Analysis
Statistical analysis was performed using GraphPad Prism 8.0.1 software. Data are presented as mean ± SD from at least three independent experiments. Differences between groups were analyzed using one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test. The significance level, i.e., p value < 0.05 was considered statistically significant.
5. Conclusions
In the present study, an efficient method for the synthesis of 6H-8-trifluoromethylquinobenzothiazine was developed and successfully used to obtain a series of eleven new N-substituted derivatives structurally related to trifluoromethyl-substituted phenothiazine drugs. Comprehensive spectroscopic characterization confirmed the proposed structures and substitution patterns of the synthesized compounds. Biological evaluation revealed that selected derivatives exhibit biologically relevant activities in both, anticancer and antibacterial models. Compounds 8 and 12 demonstrated the most balanced anticancer profiles, combining low micromolar cytotoxicity against lung and pancreatic cancer cell lines with moderate selectivity toward malignant cells relative to normal keratinocytes. Although compound 6 displayed lower intrinsic cytotoxic potency, its lack of toxicity toward normal cells resulted in a favorable selectivity index, highlighting this scaffold as a promising starting point for further optimization aimed at improving efficacy while preserving safety. In parallel, antibacterial screening identified compound 3 as a potent agent against Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus. Its low MIC values, bactericidal MBC/MIC ratios, and concentration-dependent inhibition of bacterial growth kinetics underscore the relevance of the quinobenzothiazine framework for antimicrobial drug discovery, particularly in the context of increasing resistance among staphylococcal pathogens. Overall, the presented results demonstrate that structural modification of trifluoromethyl-substituted phenothiazines by incorporation of a quinoline moiety yields compounds with diversified and therapeutically relevant biological activities. Future studies will focus on expanding the library of 8-trifluoromethylquinobenzothiazine derivatives to include analogues bearing piperazine and piperidine ring substituents, elucidating their anticancer mechanism of action, and determining lipophilicity parameters to support further structure–activity relationship optimization.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bray F. Laversanne M. Sung H. Ferlay J. Siegel R.L. Soerjomataram I. Jemal A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries CA. Cancer J. Clin.20247422926310.3322/caac.2183438572751 · doi ↗ · pubmed ↗
- 2Belay W.Y. Getachew M. Tegegne B.A. Teffera Z.H. Dagne A. Zeleke T.K. Wondm S.A. Abebe R.B. Gedif A.A. Fenta A. Antimicrobial Resistance with a Focus on Antibacterial, Antifungal, Antimalarial, and Antiviral Drugs Resistance, Its Threat, Global Priority Pathogens, Prevention, and Control Strategies: A Review Ther. Adv. Infect. Dis.2025122049936125134014410.1177/2049936125134014440416942 PMC 12103682 · doi ↗ · pubmed ↗
- 3Ohlow M.J. Moosmann B. Phenothiazine: The Seven Lives of Pharmacology’s First Lead Structure Drug Discov. Today 20111611913110.1016/j.drudis.2011.01.00121237283 · doi ↗ · pubmed ↗
- 4Mosnaim A.D. Ranade V.V. Wolf M.E. Puente J. Antonieta Valenzuela M. Phenothiazine Molecule Provides the Basic Chemical Structure for Various Classes of Pharmacotherapeutic Agents Am. J. Ther.20061326127310.1097/01.mjt.0000212897.20458.6316772768 · doi ↗ · pubmed ↗
- 5Shen W.W. A History of Antipsychotic Drug Development Compr. Psychiatry 19994040741410.1016/S 0010-440X(99)90082-210579370 · doi ↗ · pubmed ↗
- 6Ban T.A. Fifty Years Chlorpromazine: A Historical Perspective Neuropsychiatr. Dis. Treat.2007349550019300578 PMC 2655089 · pubmed ↗
- 7Posso M.C. Domingues F.C. Ferreira S. Silvestre S. Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review Molecules 20222727610.3390/molecules 2701027635011508 PMC 8746661 · doi ↗ · pubmed ↗
- 8Jana S. Empel C. Pei C. Vinh Nguyen T. Koenigs R.M. Gold-Catalyzed C−H Functionalization of Phenothiazines with Aryldiazoacetates Adv. Synth. Catal.20203625721572710.1002/adsc.202000962 · doi ↗