Cytoprotection as a Unifying Strategy for Hemorrhage and Thrombosis: The Role of BPC 157 and Related Therapeutics
Predrag Sikiric, Ivan Barisic, Mario Udovicic, Martina Lovric Bencic, Diana Balenovic, Dean Strinic, Gordana Zivanovic Posilovic, Sandra Uzun, Hrvoje Vranes, Ivan Krezic, Marin Lozic, Vasilije Stambolija, Ivica Premuzic Mestrovic, Lidija Beketic Oreskovic, Luka Kalogjera

TL;DR
This paper explores how cytoprotection, using BPC 157, can address both bleeding and clotting issues in a unified way, offering a new approach to vascular health.
Contribution
The paper introduces BPC 157 as a full cytoprotective agent that simultaneously counteracts hemorrhage and thrombosis.
Findings
BPC 157 reduces both hemorrhage and thrombosis without affecting the coagulation cascade.
BPC 157 promotes wound healing, arrhythmia control, and normalization of Virchow’s triad.
Conventional drugs provide only partial cytoprotection compared to BPC 157.
Abstract
This review presents an innovative and timely exploration of how cytoprotection can serve as a cohesive therapeutic approach by which to address the hemorrhage–thrombosis paradox. Presenting counteraction of both hemorrhage and thrombosis as phase-dependent outcomes of vascular dysregulation, the manuscript synthesizes conceptual, experimental, and clinical evidence into a unified systems-level model focused on the stable gastric pentadecapeptide BPC 157, which acts as a cytoprotective mediator. In rodents, BPC 157 can simultaneously counteract hemorrhage and thrombosis without directly affecting the coagulation cascade (aggregometry, thromboelastometry). This cytoprotective framework (decreased hemorrhage, decreased thrombosis) stands with presentation of both hemorrhage and thrombosis in the wound, arrhythmias, and Virchow triad, and resolution of these disturbances. As proof of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
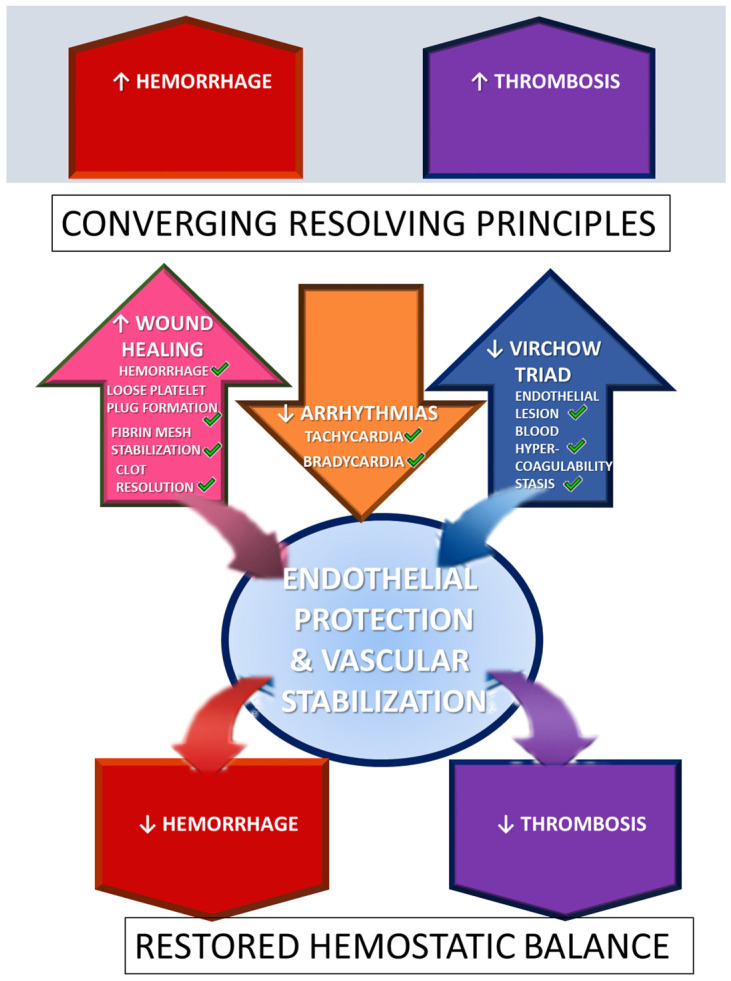 Figure 1
Figure 1- —University of Zagreb
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlatelet Disorders and Treatments · Coagulation, Bradykinin, Polyphosphates, and Angioedema · Neuropeptides and Animal Physiology
1. Introduction
This review attempts to present an innovative and timely exploration of how cytoprotection [1,2,3,4,5,6,7,8,9] can serve as a cohesive therapeutic approach to tackle the hemorrhage–thrombosis paradox [10,11,12,13,14,15]. Presenting counteraction of both hemorrhage and thrombosis as phase-dependent outcomes of vascular dysregulation, the manuscript synthesizes conceptual, experimental, and clinical evidence into a unified systems-level model focused on the stable gastric pentadecapeptide BPC 157, which acts as a cytoprotective mediator [16,17,18,19,20,21,22,23,24,25,26]. In rodents, BPC 157 can simultaneously counteract hemorrhage and thrombosis without directly affecting the coagulation cascade (aggregometry, thromboelastometry) [16,17,18,19,20,21,22,23,24,25,26].
We advocate that the cytoprotection concept, as a process to preserve and reestablish homeostasis [1,2,3,4,5,6,7,8,9], challenges the classical view of hemostasis and thrombosis as opposing pathological states [10,11,12]. Within this conventional framework, anticoagulants prevent thrombosis but induce bleeding, whereas procoagulants control bleeding while promoting thrombosis [13]. In contrast, the cytoprotection concept of vascular integrity frames both hemostasis and thrombosis as processes with an essential dual capacity to preserve tissue integrity and restore homeostasis, irrespective of the nature of the injury. Accordingly, in bleeding/thrombosis disorders, true cytoprotection is the bidirectional normalization of vascular and cellular homeostasis, counteracting both hemorrhage and thrombosis without inducing opposite-pathology toxicity. Importantly, as a universal reversal strategy, this approach would transcend the heterogeneity of anticoagulants and the involvement of distinct coagulation pathways. At present, unlike specific antagonists, no single agent is applicable to counteract both heparin- and warfarin-induced bleeding [14,15], a common antidote is lacking.
Translating this concept into practice through the application of a cytoprotective agent (i.e., the stable gastric pentadecapeptide BPC 157 [16,17,18,19,20,21,22,23,24,25,26]) may be important. This implies that a true cytoprotective therapy should normalize pathological bleeding or thrombosis in either direction without provoking opposing toxicity [16,17,18,19,20,21,22,23,24,25,26]. Such coordinated and balanced control of bleeding and thrombosis raises the longstanding question of an “ideal agent” capable of preserving therapeutic efficacy while avoiding adverse effects.
From a cytoprotective viewpoint—where agents may share partial cytoprotective properties (e.g., antioxidative, gastroprotective, cardioprotective, or organ-protective effects)—partial solutions have been achieved with conventional anticoagulants, antiplatelet agents, and fibrinolytics [10,11,12,13,14,15]. Ultimately, however, given that the cytoprotection concept (cell protection) was born in the stomach [1,2,3,4,5,6,7,8,9], the stable gastric pentadecapeptide BPC 157 has emerged as a particularly relevant cytoprotective therapy [16,17,18,19,20,21,22,23,24,25,26]. This study also includes other agents exhibiting cytoprotective properties, including prostaglandin analogues [1,2,3,4,5,6,7,8,9], antioxidants [27,28,29], angiotensin-converting enzyme inhibitors [30], statins [31,32], beta blockers [33,34], and calcium channel blockers [35,36]. These illustrate that cytoprotection is not a distinct pharmacological class but rather a shared therapeutic property across diverse drug groups. Peptides such as PDGF-BB [37], TGF-β1 [38], FGF-2/bFGF [39], VEGF [40], IGF-1 [41], and BMPs [42] were not incorporated into this framework. Thus, cytoprotection currently represents a conceptual therapeutic effect rather than a standardized clinical drug category.
Stable Gastric Pentadecapeptide BPC 157
Among cytoprotective agents, BPC 157 has accumulated the most extensive preclinical evidence [16,17,18,19,20,21,22,23,24,25,26]. Unlike other compounds, it is native and stable in human gastric juice for more than 24 h and has been proposed as a mediator of cytoprotection capable of transmitting gastrointestinal mucosal integrity to systemic organ protection [16,17,18,19,20,21,22,23,24,25,26]. This distinctive property underlies its pleiotropic beneficial effects. This includes efficacy via multiple routes, including oral administration. These effects are attributed to its modulatory interactions with multiple molecular pathways [43,44,45,46,47,48,49,50,51,52,53,54], particularly the nitric oxide (NO) system [16,55,56,57]. Toxicological studies have demonstrated an exceptionally favorable safety profile, with no lethal dose achieved (LD_1_ not reached) even at doses of 2 g/kg i.v. or i.g. in mice [16,17,18,19,20,21,22,23,24,25,26]. Human data remain limited but encouraging. BPC 157 was found to be effective and well tolerated in phase II clinical trials in ulcerative colitis and subsequently in small clinical studies addressing knee pain and interstitial cystitis, all without reported adverse effects [58,59,60,61]. These findings are supported by toxicological evaluations [62,63] and patient reports [64,65,66], consistent with previous assessments [16].
In hemorrhage- and thrombosis-focused experimental studies, bleeding and thrombosis were treated as specific cytoprotection targets [16,17,18,19,20,21,22,23,24,25,26], incorporating decreased hemorrhage and decreased thrombosis as part of a cytoprotection framework. The first direct evidence of BPC 157’s antithrombotic action was demonstrated in a rat abdominal aorta anastomosis model, where it counteracted thrombosis formation and reversed established clots [67]. Its capacity to normalize prolonged bleeding was subsequently shown in models involving heparin, warfarin, aspirin, and traumatic injury [68,69]. Aggregometry and thromboelastometry studies further indicated that BPC 157 restores platelet function without affecting coagulation pathways [70].
Additional evidence (decreased hemorrhage, decreased thrombosis) derives from severe models characterized by the coexistence of organ hemorrhage and widespread thrombosis, including thrombohemorrhagic disorders, major vessel occlusion, occlusion-like syndromes, and multiorgan failure [71,72,73,74,75,76,77]. These conditions were found to be induced by vascular occlusion, noxious procedures [78,79,80,81], or toxic agents [82,83,84,85,86,87], and were consistently reversed by BPC 157 therapy. A distinctive rescue mechanism involves the rapid activation of collateral circulation, particularly via the azygos vein, leading to restoration of systemic hemodynamics. Importantly, BPC 157 exhibits a modulatory interaction with the NO system, counteracting both NO inhibition and NO overactivity [16], and consistently antagonizes the adverse vascular effects of non-steroidal anti-inflammatory drugs [88].
2. Cytoprotection, Anticoagulants, Antiplatelet Agents, and Fibrinolytics
2.1. Anticoagulants and Cytoprotection: Direct Oral Anticoagulants (DOACs), Unfractionated Heparin, and Low-Molecular-Weight Heparins (LMWHs)
Many anticoagulants exhibit cytoprotective or pleiotropic effects beyond anticoagulation, including endothelial stabilization, anti-inflammatory activity, and modulation of oxidative, fibrotic, and cellular injury pathways [89,90,91,92,93,94]. Although not always described as “cytoprotection,” these actions align with the concept of preserving cellular integrity [1,2,3,4,5,6,7,8,9].
DOACs (dabigatran, rivaroxaban, apixaban, edoxaban) confer endothelial protection, reduce inflammation, prevent oxidative damage, limit fibrosis, and preserve tissue integrity via PAR signaling pathways [89,90,91]. Clinical evidence suggests that oral anticoagulants can lower inflammatory biomarkers, consistent with non-hemostatic cytoprotective effects [90]. DOACs preserve tight junction proteins and reduce endothelial permeability in vitro, further supporting their protective role [91].
LMWHs (e.g., enoxaparin) inhibit complement activation and neutrophil elastase, protecting cells in extracorporeal circulation models [92]. Factor Xa inhibition modulates PAR signaling on endothelial and immune cells, mitigating inflammation [93]. Heparin additionally protects the endothelial glycocalyx and mediates anti-inflammatory interactions independently of antithrombin, contributing to vascular protection [94].
2.2. Warfarin and Cytoprotection
Warfarin, in contrast, lacks cytoprotective effects and may promote vascular dysfunction [95,96,97,98,99,100,101]. By inhibiting vitamin K-dependent matrix Gla protein, warfarin accelerates arterial and valvular calcification, increases susceptibility to oxidative stress, and impairs endothelial survival [95,96,97,98,99]. Consequently, warfarin therapy reduces the vasculature’s intrinsic cytoprotective potential [100,101].
2.3. Antiplatelet Agents and Cytoprotection
Aspirin and P2Y12 inhibitors (clopidogrel, ticagrelor) provide antithrombotic effects along with endothelial protection, anti-inflammatory activity, and reduction of oxidative stress [102,103,104,105,106,107]. Aspirin plays a mechanistic role in foundational gastric cytoprotection studies, demonstrating prostaglandin-mediated adaptive defense via cyclooxygenase inhibition [1,3].
2.4. Fibrinolytics and Indirect Cytoprotection
Fibrinolytic agents (tPA, streptokinase, urokinase) primarily dissolve thrombi but can indirectly protect tissues by restoring perfusion and limiting ischemic injury, apoptosis, oxidative stress, and secondary inflammation [108,109,110,111,112]. In myocardial and cerebral ischemia, fibrinolysis reduces infarct size and tissue damage, although cytoprotection is secondary to thrombus removal [112].
2.5. Hemorrhage and Thrombosis: Anticoagulant Cytoprotection
Despite their preventive intent, anticoagulants can paradoxically induce thrombosis under certain conditions. Examples include heparin-induced thrombocytopenia (HIT), where antibodies against the heparin–PF4 complex cause massive platelet activation [113], and warfarin-induced skin necrosis from transient hypercoagulability due to protein C/S depletion [114,115]. DOACs may trigger thrombosis in cases of underdosing (e.g., renal impairment), and abrupt anticoagulant withdrawal can produce rebound hypercoagulability [116].
Even with low-risk alternatives (LMWHs, fondaparinux, heparinoids), residual HIT risk persists [117,118,119]. Warfarin initiation often requires bridging with UFH or LMWH to mitigate early hypercoagulability [120,121]. Emergency reversal agents (protamine for heparin and idarucizumab for dabigatran), which lack intrinsic prothrombotic properties, rapidly restore hemostasis but may be associated with thrombotic events [122,123,124,125,126]. These events are most plausibly attributable to abrupt neutralization of anticoagulation and re-exposure of the patient’s underlying prothrombotic state rather than to the direct procoagulant effects of the reversal agents.
2.6. Hemorrhage and Thrombosis: Antiplatelet Cytoprotection
Antiplatelet agents, though designed to prevent thrombosis, can also cause rare paradoxical events, including thrombocytopenia-induced thrombosis (e.g., GPIIb/IIIa inhibitors, thienopyridines) [127,128,129] and rebound platelet hyperreactivity after abrupt cessation, especially around percutaneous coronary interventions [130]. Temporary discontinuation of low-dose aspirin for surgery may increase cardiovascular morbidity and mortality [131].
2.7. Hemorrhage and Thrombosis: Fibrinolytic Cytoprotection
Fibrinolytics can paradoxically induce thrombotic events despite their clot-dissolving activity. Rapid clot lysis exposes procoagulant surfaces, activates platelets, thrombin, and coagulation factors, and may precipitate reocclusion or recurrent thrombosis, termed the “thrombolytic paradox” [132,133,134,135,136]. Plasmin generated during fibrinolysis can additionally activate platelets or endothelial cells under inflammatory conditions, temporarily increasing thrombotic risk [136].
These points are summarized in Table 1.
2.8. Thrombohemorrhagic Disorders: A Hemostatic Paradox of Simultaneous Thrombosis and Hemorrhage
Thrombohemorrhagic disorders, in which thrombosis and bleeding occur concurrently, reflect profound dysregulation of vascular and cellular homeostasis rather than simple deficiencies or excesses of clotting factors. Conceptually, this represents a failure of cytoprotective mechanisms that normally preserve endothelial integrity and maintain hemostatic balance. Conventional therapies have not systematically leveraged cytoprotection to address these syndromes.
Disseminated intravascular coagulation (DIC) exemplifies this paradox: uncontrolled thrombin generation causes microvascular thrombosis, while consumption of platelets and clotting factors produces bleeding. Triggers include sepsis, trauma, malignancy, and obstetric complications, involving excessive procoagulant activation, impaired anticoagulant pathways, endothelial injury, and variable fibrinolysis. Therapy focuses on treating the underlying cause, which is most effective in restoring hemostasis [137]. Supportive interventions—platelets, fresh frozen plasma, cryoprecipitate—address critical deficiencies but do not prevent ongoing microthrombosis. Low-dose heparin may be considered when thrombosis predominates, but bleeding risk must be carefully weighed [138].
Thrombotic microangiopathies (TMAs), such as thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic uremic syndrome (aHUS), similarly demonstrate this dual risk. Widespread microvascular platelet-rich thrombi cause thrombocytopenia, hemolytic anemia, and organ ischemia while predisposing to bleeding. Plasma exchange in TTP removes ultra-large von Willebrand factor multimers and pathogenic autoantibodies, reducing thrombosis and mortality [139]. Complement inhibition with eculizumab in aHUS prevents microthrombus formation and preserves hematologic and renal function [140,141]. Platelet transfusions are generally reserved for life-threatening hemorrhage.
Effective management requires early recognition and mechanism-directed therapy, as supportive measures alone are insufficient. Interventions—anticoagulation, plasma exchange, complement inhibition—must be tailored to dominant pathophysiology, continuously balancing thrombosis and hemorrhage, guided by close laboratory monitoring.
These syndromes illustrate a fundamental limitation of conventional anticoagulants, antiplatelets, and fibrinolytics: by acting on discrete hemostatic components, they inevitably exchange thrombotic risk for bleeding risk or vice versa. This paradox highlights the absence of a true cytoprotective mechanism capable of preserving endothelial integrity, maintaining cellular homeostasis, and restoring physiologic hemostasis.
Agents such as BPC 157, with demonstrated pleiotropic cytoprotective effects, may offer a therapeutic approach that directly addresses this hemostatic paradox by simultaneously supporting vascular and cellular homeostasis while counteracting both thrombosis and hemorrhage.
2.9. BPC 157 Cytoprotection
Conceptual theory implementation (decreased hemorrhage, decreased thrombosis) holds the cytoprotection concept [1,2,3,4,5,6,7,8,9], though it is not organ specific (although formed in the stomach). A systemic protection (cytoprotection → organoprotection, i.e., gastrointestinal tract maintenance transmitted to other organs therapy) is achieved by administering cytoprotective agents and via the innate pleiotropic beneficial effects attributed to those agents [1,2,3,4,5,6,7,8,9]. This principle is much more attributable to BPC 157 than to other cytoprotection agents mentioned before [16,17,18,19,20,21,22,23,24,25,26].
There, the stable gastric pentadecapeptide BPC 157, holds particular cardioprotection, anti-thrombotic, anti-hemorrhagic, anti-arrhythmic, and vascular recovery potential, particularly during ischemia/reperfusion injury recovery [16,17,18,19,20,21,22,23,24,25,26]. Specifically, principle verification (decreased hemorrhage, decreased thrombosis) includes a complex experimental model range [16,17,18,19,20,21,22,23,24,25,26] that encompasses vessel anastomosis [67], tail or leg amputation, anticoagulants, antiplatelets [68,69], organ perforation [81,142], vessel occlusion [71,72,73,74,75,76], severe injury induction [78,79,80,81], agent application [82,83,84,85,86,87], and occlusion/occlusion-like syndrome. Also included are counteractions of those aggravated by NO/NO synthase (NOS) blockade (L-NAME) or NO overactivity (L-arginine) [69,85]. Notably, as pointed out, this range also includes the simultaneous presentation of organ hemorrhage and thrombosis and counteraction. Additional evidence that BPC 157 may rescue thrombocyte function without affecting coagulation pathways [68,69] includes aggregometry and thromboelastometry studies [70]. Notably, BPC 157 may recover prostaglandin function, given the counteraction of NSAID-induced adverse effects [88].
2.10. Verification of the Cytoprotective Effects (Decreased Hemorrhage, Decreased Thrombosis) Through Three Confirmatory Principles: (i) Wound, (ii) Arrhythmias, and (iii) Virchow Triad
These cytoprotective effects, decreased hemorrhage and decreased thrombosis, conceptually anchor three confirmatory principles that provide presentation of both hemorrhage and thrombosis—(i) wound, (ii) arrhythmias, and (iii) Virchow triad. These (decreased hemorrhage, decreased thrombosis) have to be reversed in order to achieve (i) wound healing, (ii) arrhythmia mitigation, and (iii) normalization of Virchow’s triad (see Figure 1). BPC 157 therapy full accomplishes all of these points [16,17,18,19,20,21,22,23,24,25,26], which is significant given that both hemorrhage and thrombosis regularly need to be counteracted simultaneously.
Wound healing, in particular, encompasses all four major hemostatic events—vascular constriction, loose platelet plug formation, fibrin mesh stabilization, and clot resolution—which occur in a coordinated sequence after vascular injury [143,144,145]. Accordingly, in hemorrhage/thrombosis terms, the principle ↑ wound healing = ↓ hemorrhage and ↓ thrombosis applies, as agents that promote wound healing, such as BPC 157, effectively reduce both bleeding and thrombotic risk. Taken as a conceptual model, these confirmatory endpoints also provide a framework for evaluating other cytoprotective agents, although none matches the comprehensive systemic efficacy of BPC 157.
Converging resolving principles. Considering that hemorrhage and thrombosis are both involved in the wound, arrhythmia, and Virchow triad, the effects on the circumstances of wound healing, arrhythmia, and Virchow triad were considered as key proof of the agent’s counteracting activity (green tick marks) on either hemorrhage or thrombosis, or counteraction of both hemorrhage and thrombosis [16,17,18,19,20,21,22,23,24,25,26,145].
3. Wound Healing
3.1. BPC 157: Wound Healing as a Resolving Principle for Hemorrhage/Thrombosis Therapy
Wound healing has long been recognized as an inherent component of the cytoprotection concept and a central mediator of cytoprotective agent activity [1,2,3,4,5,6,7,8,9,16,17,18,19,20,21,22,23,24,25,26]. In the context of BPC 157 therapy, wound healing provides a unifying framework for resolving the apparent paradox of simultaneously counteracting hemorrhage and thrombosis [145]. This can be particularly seen given the comparative studies reviewed [145]. As a conceptual principle, we have proposed that any agent truly promoting wound healing must, by necessity, normalize both bleeding and thrombosis, depending on the phase of vascular repair [68].
Early BPC 157 studies demonstrated endothelium maintenance. Initially, this was in the context of alcohol-induced gastric lesions [146], and later in models of thrombosis and prolonged bleeding, including rat abdominal aorta anastomosis [67], anticoagulant (heparin, warfarin) and antiplatelet exposure [68,69], limb amputation, organ perforation [81,142], vessel occlusion [71,72,73,74,75,76,77], severe injury induction [78,79,80,81], and agents’ application [82,83,84,85,86,87] in occlusion/occlusion-like syndromes [71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87]. These models also included NO/NOS dysregulation, with L-NAME or L-arginine, and conditions where hemorrhage and thrombosis occurred simultaneously [69,85]. BPC 157 restored thrombocyte function without affecting coagulation pathways [70] and counteracted NSAID-induced prostaglandin dysfunction [88].
The dual therapeutic effect of BPC 157, context-dependent, aligns with the four sequential phases of physiological wound healing: (i) vascular constriction, (ii) loose platelet plug formation, (iii) fibrin mesh stabilization, and (iv) clot resolution with restoration of vascular patency [143,144,145,147]. Effective wound healing, therefore, inherently supports early hemostasis, thrombus formation, and timely clot dissolution. Failure in any phase can result in persistent hemorrhage or pathological thrombosis, highlighting that bleeding and thrombosis are not opposing events but temporally and functionally linked stages of the repair process [145]. This principle establishes the therapeutic design requirement for phase-sensitive, cytoprotective interventions with robust wound healing capacity as the primary endpoint.
Experimental evidence (decreased hemorrhage, decreased thrombosis) confirms BPC 157’s potent wound healing activity across multiple tissues and injury types. These include skin wounds, deep burns [53,54,63,148,149,150,151,152], tendons [50,51,153,154,155,156,157,158], ligaments [159], muscle injuries [47,158,160,161,162,163], osteotendinous and myotendinous junctions [153,154,157,164], and muscle-to-bone attachments [165]. Corneal ulcer healing, including restoration of transparency and counteraction of neovascularization, further exemplifies its multi-tissue efficacy [21,22,23,26,166]. Internal wound repair is also robust, as demonstrated by recovery of anastomoses [167] and fistulas [168], both external [169,170,171,172,173] and internal [174,175,176,177]. These outcomes confirm early hemostasis, subsequent clot removal, and microvascular restoration.
Overall, BPC 157’s systemic wound healing capacity substantiates its dual role in decreasing hemorrhage and thrombosis and provides a conceptual and experimental framework for evaluating other cytoprotective agents, though none match its comprehensive efficacy.
Notably, as part of the increasing interest for soft tissue injuries healing and BPC 157 efficacy, several reviews have recently appeared [178,179,180,181,182,183,184,185,186,187,188,189,190].
3.2. Cytoprotective Effects of Prostaglandins, Statins, ACE Inhibitors, Beta Blockers, Ca Channel Blockers, and NO Modulators as Possibilities for Wound Healing
When examined from the perspective of other pharmacological agents that exhibit partial cytoprotective or pleiotropic effects, supportive evidence for wound healing is present but notably inconsistent. Unlike BPC 157, these agents were not developed as cytoprotective therapies and do not uniformly engage the coordinated, multistage repair program required for effective wound healing.
Prostaglandins, which historically represent the first mediators of the cytoprotection concept, demonstrate a biphasic influence on wound repair [191]. Under physiological conditions, prostaglandins generally promote healing through vasodilation, angiogenesis, and epithelial restitution [192,193,194]. However, excessive prostaglandin signaling—particularly elevated PGE_2_—may exacerbate inflammation, fibrosis, and scarring, resulting in impaired or dysregulated healing [195].
Statins also exhibit dose- and context-dependent effects. At low-to-moderate doses, statins may enhance wound healing via anti-inflammatory actions, endothelial stabilization, and improved nitric oxide bioavailability. In contrast, higher doses have been associated with delayed tissue repair, likely due to excessive inhibition of cellular proliferation and prenylation pathways essential for regeneration [196].
ACE inhibitors generally support wound healing, presumably through improved microcirculation, bradykinin-mediated vasodilation, and modulation of inflammatory responses [197,198,199].
Beta blockers exhibit complex effects: while some studies report accelerated wound closure—possibly through reduced stress signaling and catecholamine effects—others report delayed healing, reflecting interference with perfusion, angiogenesis, or keratinocyte migration [200,201,202,203,204].
Calcium channel blockers are more consistently associated with improved wound healing, likely through enhanced microvascular perfusion, reduced vasospasm, and favorable effects on fibroblast and endothelial function [205,206,207,208,209,210,211].
Nitric oxide (NO) plays a central physiological role in wound repair, regulating angiogenesis, inflammation, fibroblast activity, collagen deposition, and re-epithelialization [212,213]. Accordingly, NO donors and restoration of nitric oxide synthase (NOS) activity accelerate wound closure [214,215,216], whereas NOS inhibition or NO deficiency delays healing, establishing NO modulation as a pro-healing strategy [217,218,219].
Overall, although these agents can influence selected components of wound repair, their effects are heterogeneous, dose-dependent, and mechanistically restricted, lacking the coordinated control of all of the phases of healing that are characteristic of true cytoprotection.
3.3. Prostaglandins, Statins, ACE Inhibitors, Beta Blockers, Ca Channel Blockers, and NO Modulators: Wound Healing as an Inconsistent Resolving Principle for Hemorrhage/Thrombosis Therapy
When the wound-healing capacity of these agents is examined in relation to the proposed cytoprotection principle linking enhanced healing with reduced hemorrhage and thrombosis (↑ wound healing = ↓ hemorrhage and ↓ thrombosis), the relationship proves inconsistent and context dependent. Unlike BPC 157, these agents do not reliably translate partial wound-healing effects into balanced hemostatic normalization.
Prostaglandins, despite their pro-healing actions, may increase bleeding risk through platelet inhibition and vasodilation [220,221,222]. Prostacyclin analogs such as iloprost are clinically used to prevent thrombosis in peripheral vascular disease and dialysis grafts, illustrating antithrombotic efficacy but at the expense of increased bleeding tendency [220].
ACE inhibitors may increase bleeding risk, particularly gastrointestinal bleeding, more frequently than angiotensin receptor blockers, consistent with bradykinin-mediated increases in vascular permeability [223,224]. Nevertheless, large cardiovascular trials demonstrate that ACE inhibitors reduce myocardial infarction and stroke incidence, indicating overall neutral or modestly antithrombotic effects [225,226,227]. Rare reports of thrombosis exist but are considered context dependent and not intrinsic pharmacological properties of this drug class [228,229].
Beta blockers generally reduce bleeding risk [229,230], yet earlier generations may reduce coronary flow at lower heart rates, potentially increasing thrombotic susceptibility [231,232]. Moreover, excessive beta blockade, as reported with sotalol overdose, may precipitate severe occlusion or occlusion-like syndromes accompanied by widespread thrombosis [84].
For calcium channel blockers, epidemiological data suggest a possible association with gastrointestinal bleeding [233], while, in patients with nonvalvular atrial fibrillation receiving oral anticoagulants and dihydropyridine, calcium channel blocker use has been associated with a higher incidence of ischemic stroke compared with non-use [234].
Statins, in contrast, are associated with reduced bleeding risk [235] and a lower incidence of venous thromboembolism in humans [236], yet these effects do not consistently align with wound-healing outcomes.
Nitric oxide represents a physiological antiplatelet and anticoagulant mediator; thus, enhancement of NO signaling, particularly at systemic or high doses, may increase bleeding risk. Conversely, NOS inhibition with L-NAME is prothrombotic, underscoring the narrow therapeutic window of NO-based modulation [237,238].
Collectively, these observations indicate that, while many pleiotropic cardiovascular agents can influence wound healing, bleeding, or thrombosis individually, they do so by shifting isolated pathways rather than restoring vascular homeostasis as an integrated process. Their inability to coordinate all four phases of wound healing—hemostasis, platelet function, fibrin stabilization, and timely clot resolution—explains why they fail to consistently resolve the hemorrhage–thrombosis paradox. This limitation sharply contrasts with the cytoprotective profile of BPC 157, which uniquely fulfills the wound-healing–hemostasis continuum as a unified therapeutic principle.
These comparative effects on wound healing, and consequently, on hemorrhage and thrombosis of prostaglandins, beta blockers, Ca channel blockers, ACE inhibitors, and NO modulators were summarized in Table 2.
Thereby, as shown in Table 2, it seems that, with all mentioned agents—prostaglandins, beta blockers, Ca channel blockers, ACE inhibitors, and NO modulators—that are supposed to have some cytoprotective capabilities, and which are thereby elaborated for wound healing/hemorrhage/thrombosis relation, the ↑wound healing = ↓ hemorrhage ↓ thrombosis remains unachieved and is not combined in a consequent chain of events.
3.4. Cytoprotective Effects of Anticoagulants, Antiplatelets, and Fibrinolytics as Possibilities for Wound Healing
In contrast to BPC 157, anticoagulants, antiplatelet agents, and fibrinolytics do not consistently satisfy the principle that enhanced wound healing translates into reduced hemorrhage and reduced thrombosis. Rather than coordinating the sequential phases of vascular repair, these agents act predominantly through unidirectional interference with individual hemostatic components, thereby altering wound healing in a context- and phase-dependent, but non-integrative, manner.
Heparin illustrates this limitation. While some studies demonstrate enhanced wound healing, attributed to anti-inflammatory effects, growth factor interactions, or endothelial modulation [239,240,241], others report impaired healing, delayed epithelialization, or increased wound complications [242,243,244]. These divergent outcomes reflect the absence of phase-sensitive regulation: heparin may facilitate early events but interfere with later stabilization or remodeling phases, preventing consistent restoration of vascular integrity.
Warfarin more uniformly delays wound healing [245,246,247], consistent with its inhibition of vitamin K-dependent proteins involved in tissue repair, extracellular matrix regulation, and vascular cell survival. This effect underscores a fundamental limitation of anticoagulation strategies that suppress physiological repair mechanisms rather than modulating them.
Antiplatelet agents similarly delay wound healing [248,249,250], reflecting the impaired platelet-derived signaling required for early hemostasis, growth factor release, and subsequent tissue regeneration. Fibrinolytic agents likewise delay wound healing [251,252,253], as premature or excessive clot dissolution disrupts the scaffold formation necessary for stable repair.
Collectively, these findings indicate that conventional antithrombotic agents do not support wound healing as a unified physiological process. Instead, they selectively inhibit or exaggerate specific phases, thereby failing to integrate bleeding control, clot formation, stabilization, and resolution into a coherent reparative sequence.
3.5. Concluding Remarks on Wound Healing–Hemorrhage–Thrombosis Relations
Wound healing represents an inherent physiological organizing principle of cytoprotection, encompassing all four essential hemostatic events—vascular constriction, loose platelet plug formation, fibrin clot stabilization, and clot dissolution—which occur in a tightly regulated temporal sequence following vascular injury. Successful healing requires that each phase is initiated, supported, and terminated appropriately; failure at any stage results in persistent hemorrhage, pathological thrombosis, or defective tissue repair.
Within this framework, hemorrhage and thrombosis are not opposing pathological states but phase-dependent manifestations of the same repair process. Accordingly, the relationship ↑ wound healing = ↓ hemorrhage and ↓ thrombosis reflects restoration of physiological homeostasis rather than pharmacological suppression of coagulation or platelet activity.
The consistent observation that BPC 157 simultaneously reduces bleeding and thrombosis therefore indicates a unique capacity to integrate wound healing with hemostatic regulation. In cytoprotection terms, this implies early hemostasis when bleeding predominates, timely clot resolution when thrombosis emerges, microvascular restoration, and normalization of platelet function—without direct interference with coagulation pathways. The outcome is a balanced, phase-sensitive modulation that follows physiological repair logic.
Other agents may influence individual components of wound healing or hemostasis, but none consistently achieves both reduced hemorrhage and reduced thrombosis in a coordinated, phase-dependent manner. Thus, BPC 157 exemplifies a cytoprotective strategy that aligns therapeutic intervention with endogenous repair mechanisms, redefining hemorrhage and thrombosis as resolvable phases of vascular healing rather than mutually exclusive targets.
3.6. Concluding Remarks on Wound Healing/Hemorrhage/Thrombosis Relations and BPC 157/NO System Relation
It can also be argued that such a balance would require an applicable dual effect that can be seen in both heparin and warfarin, given that they are essential as hemorrhage models [254,255,256,257,258].
Notably, unlike BPC 157 therapy studies with both heparin and warfarin [68,69], none of the mentioned agents, sharing some cytoprotection capabilities, prostaglandins, beta blockers, Ca channel blockers, and ACE inhibitors, were tested with heparin or warfarin bleeding. Likewise, they were not used in combination with targeted modulation of the NO system.
The only direct study combining NO agents with heparin and warfarin is that with regard to the effects of BPC 157 [69]. Thereby, the formula by which BPC 157 ↑ wound healing = ↓ hemorrhage and ↓ thrombosis stands with the consistent dual context-dependent effect, on both heparin and warfarin, in which BPC 157 counteracts both L-NAME pro-thrombotic effects, as well as an L-arginine anti-coagulant effect [69]. These effects occur along with the counteraction of other effects of NO agents, i.e., L-NAME-induced hypertension, and L-arginine-induced hypotension [16,54,55,56]. Notably, such counteracting effects have occurred in a large number of targets (i.e., more than 80), suggesting a general BPC 157/NO system relation [16,56]. Additionally, BPC 157 induces the NO release, resistant to L-NAME, that counteracts the NO release induced by L-arginine [16,54,55,56]. Indicating restoration of redox balance [16], BPC 157 ↑ wound healing = ↓ hemorrhage and ↓ thrombosis correlates with the findings that BPC 157 therapy normalizes NO tissue levels, either increased or decreased, along with a counteraction of the increased MDA values [16].
In conclusion, indicative therapy occurred for hemorrhage, thrombosis, hypertension, hypotension, and thrombocytes’ function (without affecting the coagulation cascade) [16,54,55,56,67,68,69,70]. In addition, BPC 157 therapy affects signaling pathways controlling vasomotor tone [45,46,47] (VEGFR2-Akt-eNOS and Src-Caveolin-1-eNOS). At the general level, this dual (modulatory) action (i.e., either of pro-coagulant effects, anti-coagulant effects, hypertension, and hypotension reversed toward normal) also applies to the NO system’s effects as a whole [16,54,55,56,67,68,69,70]. Such a role for BPC 157 therapy could be essential amid the dual role of NO, as both an inhibition and an uncontrolled excess of NO could lead to significant damage [259,260]. Consequently, in providing the essential role of NO to wound healing [261], BPC 157’s ability to restore NO system homeostasis may represent a central mechanism underlying its wide-ranging therapeutic potential, represented as the maintained equation of ↑ wound healing = ↓ hemorrhage and ↓ thrombosis [16,45,46,47,54,55,56,67,68,69,70,145].
4. Arrhythmias
As a conceptual principle, the ↑ wound healing = ↓ hemorrhage and ↓ thrombosis principle reflects a cytoprotective agent’s ability to normalize both extremes of bleeding and thrombosis, restoring vascular homeostasis and orderly healing [145]. Arrhythmias represent another critical facet of this systemic cytoprotection, as endothelial maintenance and vascular integrity extend pleiotropic effects from cytoprotection to organoprotection [1,2,3,4,5,6,7,8,9]. Thus, as a further conceptual point, antiarrhythmic normalization is therefore claimed as an essential component of true cytoprotection [262], and BPC 157’s anti-arrhythmic potential and its counteraction of arrhythmias has been recently reviewed [262].
Arrhythmias, particularly those impairing effective cardiac contraction, promote stasis, contributing to Virchow’s triad (stasis + endothelial dysfunction + hypercoagulability) and increasing thrombotic risk [263,264,265,266,267]. Atrial fibrillation can form left atrial appendage thrombi, while ventricular arrhythmias reduce cardiac output and favor regional or systemic hypoperfusion. Endothelial injury and arrhythmia-mediated hypercoagulability may occur even without ischemia [265,266,267].
Preclinical studies show that BPC 157 counteracts a wide array of arrhythmogenic triggers, including digitalis [268], potassium overdose [269], furosemide [270], lidocaine [271], bupivacaine [272], succinylcholine [273], neuroleptics [83,274], amphetamine [83], domperidone [83], metoclopramide [274], sotalol [84], isoprenaline [85], sodium laureate [86], alcohol [87], monocrotaline [275], hyperkalemia [269,273], hypokalemia [270], hyperlithiemia [82], and various occlusion/occlusion-like syndromes [71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87]. These effects also include mitigation of arrhythmias aggravated by NO/NOS modulation [85,268,269,270,271,272], supported by HEK293 studies showing direct membrane stabilization [269,270,271,272,276]. Analogous to its ability to normalize both tachycardia and bradycardia [262], BPC 157 restores hemostatic and microvascular balance without direct pro- or anticoagulant activity, integrating arrhythmia control with reduced hemorrhage and thrombosis. Notably, this could also be mobilized against a thrombus via the use of antiarrhythmics, and against hemorrhage in particular, because anticoagulants are utilized as part of an antiarrhythmic application [277,278,279,280]. Notably, as mentioned, BPC 157 is effective against sodium laurate intravenous administration [86]. This can be, as suggested, a cytoprotective therapy that preserves endothelial integrity and microvascular homeostasis [262].
Comparative pharmacology highlights BPC 157’s uniqueness. Intrinsic sympathomimetic β blockers can normalize heart rate bidirectionally [281,282,283], whereas Ca channel blockers modulate heart rate directionally—non-dihydropyridines may induce bradycardia and dihydropyridines may provoke reflex tachycardia—but lack self-normalizing capability [284,285]. ACE inhibitors and prostaglandins may modestly alter heart rate via reflex sympathetic activation [286,287,288]. NO donors or L-arginine enhance vagal tone and limit tachycardia, while NOS inhibition (L-NAME) promotes tachycardia and hypertension [289,290,291]. Statins can reduce elevated heart rates and improve autonomic balance without inducing bradycardia [292,293,294,295,296,297].
In summary, BPC 157 uniquely integrates cytoprotection across wound healing, hemostasis, thrombosis, and arrhythmias by restoring endothelial function, stabilizing cardiac electrical activity, and maintaining microvascular homeostasis. Its effects are context-dependent, bidirectional, and phase-sensitive, exemplifying the systemic principle that functional normalization—not mere inhibition or activation—underlies the resolution of hemorrhage and thrombosis.
5. Virchow Triad
Thrombohemorrhagic disorders, as emphasized in Section 2.7, can present with both thrombosis and hemorrhage simultaneously. Early cytoprotection studies have demonstrated that endothelial lesions, thrombus formation, and stasis precede gastric epithelial hemorrhagic lesions [4,5,6,7], implying that Virchow’s triad conditions co-occur with hemorrhage during lesion development. Consequently, cytoprotection and Virchow triad circumstances are closely linked [16,17,18,19,20,21,22,23,24,25,26], even if not explicitly framed as such initially [4,5,6,7]. As a final conceptual point, the logical extension is that cytoprotective agents, by resolving these conditions, can achieve a ↓ Virchow triad = ↓ hemorrhage + ↓ thrombosis equation, complementing the other cytoprotective principles, ↑ wound healing = ↓ hemorrhage + ↓ thrombosis and ↓ arrhythmias = ↓ hemorrhage + ↓ thrombosis [16,17,18,19,20,21,22,23,24,25,26].
BPC 157 therapy has been systematically evaluated in a broad spectrum of occlusion/occlusion-like models [71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87], where it consistently reversed severe vascular and multiorgan failure. Models included major vessel occlusion [74,75,76,77], bile duct occlusion with acute pancreatitis [80], intra-abdominal hypertension grade III–IV [78,79], ischemia-reperfusion injury [78,79], organ perforation [81], and exposure to cytotoxic agents such as lithium [82], intragastric alcohol [87], intravenous sodium laurate [86], isoprenaline [85], sotalol [84], neuroleptics [83], and amphetamine [83]. With BPC 157 therapy, the described ↓ Virchow triad circumstances = ↓ hemorrhage and ↓ thrombosis equation also occurs with the reversal of Pringle maneuver ischemia–reperfusion [71], Budd–Chiari syndrome models [72], and inferior caval vein ligation [73].
Restoring vascular patency, microcirculation, and endothelial integrity means that BPC 157 promptly counteracts hemorrhage and thrombosis, peripherally and centrally. This means reversing secondary lesions in the brain, heart, lung, liver, kidney, and gastrointestinal tract. Likewise, this means attenuation/elimination of various arrhythmias (bradycardias and tachycardias); intracranial, portal, and caval hypertension; and aortal hypotension. The particular reversal includes the major vessels that had failed, the inferior caval vein and superior mesenteric vein, which were congested, and the aorta and azygos vein, which were collapsed [74,75,76,77,78,79,80,81,82,83,84,85,86,87], all of which fully recovered (i.e., azygos vein activated, direct blood flow delivery). The circumstances of Virchow’s triad are regularly reversed.
A key mechanistic feature is collateral vessel recruitment, particularly the azygos vein, which rapidly redistributes venous blood when primary pathways are blocked, alleviating venous hypertension and arterial hypotension. BPC 157 also stabilizes endothelial and cardiac membranes, acts as a free radical scavenger, and maintains cellular junctions, counteracting “leaky gut” phenomena [44,145]. These vascular protective effects are closely linked to the NO system, with BPC 157 normalizing NO levels, mitigating both L-NAME-induced prothrombotic effects and L-arginine-induced hypotension/anti-coagulation [16,54,55,56,69]. Rapid vascular wall remodeling, evidenced by FTIR studies, occurs in parallel with restored perfusion, and may be responsible for the noted chain of events, commonly counteracted (i.e., resolved bradycardia or tachycardia, and reversal of peripheral and central hemorrhage and thrombosis) [298,299].
As emphasized before, these therapy effects occur even while the underlying insult persists (i.e., occlusion of major blood vessels [71,72,73,74,75,76,77], increased intra-abdominal pressure [78], grade III, grade IV) or the injury is already highly advanced and fully established [79]. This indicates a functional action at the final common pathway, whatever the molecular construct [43,44,45,46,47,48,49,50,51,52,53,54]. This explains BPC 157’s therapeutic effects, fully specified, i.e., in inferior caval vein syndrome (induced by infrarenal ligation of inferior caval vein), where there is a counteraction of the full syndrome [73]. This includes cases where there is a direct vein injury, thrombosis, thrombocytopenia, and prolonged bleeding. Rapid presentation of collaterals means instant redistribution of the trapped blood volume amid counteracted venous hypertension and arterial hypotension and tachycardia. This occurs with counteraction of oxidative stress (as a result of the lysis of endothelial cells [73]) and low NO level in the inferior caval vein. Particular gene expression (i.e., EGR, NOS, SRF, VEGR, AKT1, PLCɣ, KRAS) appears as a likely special point to explain how the dysfunction and its counteraction are causal to or a result of each other [73].
Importantly, the BPC 157 effect is conditional and context-dependent, appearing even with established and severe insults, such as complete vascular occlusion or advanced highly increased intra-abdominal pressure (grade III, grade IV), highlighting action at a final common pathway irrespective of the initial molecular insult [43,44,45,46,47,48,49,50,51,52,53,54]. Molecular studies implicate the modulation of critical signaling pathways, including VEGFR2-Akt-eNOS and Src-Caveolin-1-eNOS, supporting vasomotor tone regulation and endothelial recovery [16,45,46,47,54,55,56,67,68,69,70]. These effects occur without the need for exogenous ligands or shear stress, suggesting a direct interaction of BPC 157 with cellular targets and gasotransmitter pathways [300].
In contrast, comparative cytoprotective agents—prostaglandins, beta blockers, Ca channel blockers, ACE inhibitors, and statins—have not been tested in these occlusion/occlusion-like models. Thus, BPC 157 uniquely demonstrates a consistent ability to reverse Virchow’s triad, while simultaneously normalizing hemodynamic parameters and microvascular perfusion.
The convergence of the three cytoprotective principles—↑ wound healing, ↓ arrhythmias, and ↓ Virchow triad—suggests that BPC 157 functions as a system-level modulator, rather than a single-pathway pharmacologic agent. By integrating endothelial stabilization, collateral recruitment, NO system homeostasis, oxidative stress reduction, and vasomotor regulation, BPC 157 achieves bidirectional, phase-sensitive restoration of vascular homeostasis without pushing physiology toward either extreme [16].
This BPC 157 hemorrhage/thrombosis relation is summarized in Table 3.
Likewise, being native and stable in human gastric juice (more than 24 h) while acting as a cytoprotective mediator likely allows for the translation of the maintenance of the gastrointestinal mucosa to other organ therapies, broadening the practical applicability of this peptide (i.e., pleiotropic beneficial effects, which are always applied alone without carrier), including per-oral application [16,17,18,19,20,21,22,23,24,25,26,145,262,301]. Given the consistent beneficial effects so far presented in the mentioned studies, BPC 157 therapy could specifically counteract the cytotoxic and damaging actions of NO, being organ specific. This might be seen as a network of evidence for the physiological significance of the revealed BPC 157/NO system interplay. Notably, BPC 157, native and stable in human gastric juice, was found in in situ hybridization and immunostaining studies in humans to be largely distributed in tissues [16,145,262,301] and may have additional physiological regulatory roles [16,17,18,19,20,21,22,23,24,25,26,145,301] when released from the stomach and sent to other organs. These points, and further application (i.e., efficacy and the lack of adverse effects in limited clinical trials [58,59,60,61,64,65,66]), are supported by the lack of adverse effects in toxicology studies. Notably, for BPC 157, an effective range within 10 µg–10 ng/kg means full confirmation [62,63] that a lethal dose (LD1) was not reached even at 2 g/kg i.v. or i.g. (a harmless limit test, without adverse effects in rodents) (Figure 2).
Finally, Table 4 expresses a summary of the converging resolving principles when cytoprotection was found to decrease hemorrhage and thrombosis as phase-dependent outcomes of the counteracted vascular dysregulation carried out by BPC 157 therapy.
6. Limitations and Future Directions
Some final notations must emphasize the complexity of the problem of the hemorrhage–thrombosis paradox as an inherent limitation for any review.
In general, there is still a dominance of one group within the presented papers [16,17,18,19,20,21,22,23,24,25,26,145,262,301]. Additionally, and in general, the cytoprotection concept [1,2,3,4,5,6,7,8,9], as a concept, is still not implemented in clinics. Taken as a hypothesis-based interpretive model (i.e., “ideal” agent capable of bidirectional regulation), it is elaborated that BPC 157 and other agents have been found to share some cytoprotective properties. Finally, decreased hemorrhage, decrease thrombosis determination goes through three converging resolving principles—wound, arrhythmias, and Virchow triad—meaning ↑ wound healing = ↓ hemorrhage + ↓ thrombosis; ↓ arrhythmia = ↓ hemorrhage + ↓ thrombosis; and ↓ Virchow triad = ↓ hemorrhage + ↓ thrombosis. All evidence combined can provide a unified systems-level model focused on BPC 157.
However, together, these could still oversimplify the multifaceted nature of the hemorrhage/thrombosis issue. Nevertheless, reliance on preclinical models necessitates further clinical validation.
Likewise, and also in general, thrombus formation and the severity, timing, and nature of blood loss—conditions that are almost impossible to standardize in clinical (human) settings—have been examined using rat models. However, it may be that a rat model simplifies these variables, which is scientifically useful but limits direct clinical predictability [302,303,304,305].
Finally, it remains to be seen whether the evidence derived from the majority of experimental work (i.e., initial discovery, mechanistic exploration, and model validation) [16,17,18,19,20,21,22,23,24,25,26,145,262,301] is found within a single research group, providing that it aligns with the confirmatory reports of other groups (i.e., [45,46,47,48,49,50,51,52,53,54,306,307,308,309,310,311,312,313]). In many studies, translational values have emphasized the similar therapeutic effect obtained by different manners of administration in the same model [16,17,18,19,20,21,22,23,24,25,26,145,262,301]. In addition to the clinical data (although still limited), studies have included favorable safety reports [58,59,60,61,62,63,64,65,66] that are comparable with previous notations [16].
There is also a need to search for a resolving analogy (preclinical models → further clinical validation). Although likely hypothetical, and not a substitute for the rigorous clinical validation that should be undertaken, such a pattern agrees with what that has been historically observed with a favorable outcome. Notably, this has already occurred, during the early preclinical phases of several now-established therapies, including the erythropoietin (EPO) [314,315] and glucagon-like peptide-1 (GLP-1) analogs before industrial expansion [316,317,318], and neuropeptides such as pituitary adenylate cyclase-activating polypeptide (PACAP) [319] and vasoactive intestinal peptide (VIP) [320,321].
7. Conclusions
This review addresses the hemorrhage/thrombosis ideal agent problem from a cytoprotective perspective (decreased hemorrhage and thrombosis to preserve and reestablish homeostasis). It proposes the stable gastric pentadecapeptide BPC 157, which acts as a cytoprotective mediator. In rodents, BPC 157 can simultaneously counteract hemorrhage and thrombosis without directly affecting the coagulation cascade (aggregometry, thromboelastometry). Within this framework, presenting both hemorrhage and thrombosis that should be both counteracted, the manuscript synthesizes conceptual, experimental, and clinical evidence. BPC 157’s effective wound healing, arrhythmia control, and normalization of Virchow’s triad represent converging physiological processes for the realization of the principle of decreased hemorrhage and decreased thrombosis to preserve and reestablish homeostasis.
As a comparison from a cytoprotective (partial vs. full) standpoint, conventional agents—including anticoagulants, antiplatelet drugs, fibrinolytics, beta blockers, calcium channel blockers, prostaglandins, NO modulators, ACE inhibitors, and statins—provide only partial, context-dependent protection, typically unidirectional, dose-limited, or achieved at the expense of opposing pathological risks. In contrast, preclinical evidence indicates that BPC 157 functions as a vasoprotective cytoprotective mediator capable of bidirectional regulation. Its effects likely involve the preservation of endothelial integrity, the normalization of microcirculation, the modulation of the NO system, the stabilization of hemostatic balance, and the recruitment of adaptive collateral pathways. Accordingly, with the potential limitations of conceptual points and those regarding preclinical predominance, the relationships among wound healing, arrhythmia control, and Virchow triad normalization may be viewed as interconnected cytoprotective principles contributing to the reduction of both hemorrhage and thrombosis.
Importantly, BPC 157 can counteract the bleeding induced by anticoagulants or antiplatelet agents, while simultaneously mitigating organ hemorrhage and thrombosis under conditions of vascular occlusion or arrhythmia. These properties suggest that wound healing is not merely a local reparative process, but a central organizing principle of systemic vascular homeostasis. Combined with its oral bioavailability, gastric stability, high safety profile, and efficacy in both prophylactic and therapeutic settings, BPC 157 holds substantial translational potential as a novel cytoprotective strategy for complex vascular and hemostatic disorders.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Robert A. Cytoprotection by prostaglandins Gastroenterology 19797776176710.1016/0016-5085(79)90235-X 38173 · doi ↗ · pubmed ↗
- 2Robert A. Current history of cytoprotection Prostaglandins 198121899610.1016/0090-6980(81)90123-47029653 · doi ↗ · pubmed ↗
- 3Robert A. Nezamis J.E. Lancaster C. Davis J.P. Field S.O. Hanchar A.J. Mild irritants prevent gastric necrosis through “adaptive cytoprotection” mediated by prostaglandins Am. J. Physiol.1983245 G 113G 12110.1152/ajpgi.1983.245.1.G 1136869543 · doi ↗ · pubmed ↗
- 4Szabo S. Role of sulfhydryls and early vascular lesions in gastric mucosal injury Acta Physiol. Hung.1984642032146532115 · pubmed ↗
- 5Trier J.S. Szabo S. Allan C.H. Ethanol-induced damage to mucosal capillaries of rat stomach. Ultrastructural features and effects of prostaglandin F 2β and cysteamine Gastroenterology 198792132210.1016/0016-5085(87)90834-13781180 · doi ↗ · pubmed ↗
- 6Pihan G. Majzoubi D. Haudenschild C. Trier J.S. Szabo S. Early microcirculatory stasis in acute gastric mucosal injury in the rat and prevention by 16,16-dimethyl prostaglandin E 2 or sodium thiosulfate Gastroenterology 1986911415142610.1016/0016-5085(86)90195-22945748 · doi ↗ · pubmed ↗
- 7Szabo S. Trier J.S. Brown A. Schnoor J. Early vascular injury and increased vascular permeability in gastric mucosal injury caused by ethanol in the rat Gastroenterology 19858822823610.1016/S 0016-5085(85)80176-13871087 · doi ↗ · pubmed ↗
- 8Szabo S. Experimental basis for a role for sulfhydryls and dopamine in ulcerogenesis: A primer for cytoprotection-organoprotection Klin. Wochenschr.1986641161223560772 · pubmed ↗