Stability and Reactivity of Cyclopentane Nucleoside Analogs in 98% w/w Sulfuric Acid
Sara Seager, Maxwell D. Seager, Ton Visser, Nittert Marinus, Mael Poizat, Jim van Wiltenburg, Martin Poelert, Janusz J. Petkowski

TL;DR
Scientists tested how stable certain DNA-like molecules are in very strong sulfuric acid and found some can stay intact, which might be useful for understanding life in extreme environments like Venus.
Contribution
Identified cyclopentane as a stable linker in nucleoside analogs under concentrated sulfuric acid, with implications for genetic-like polymers in extreme environments.
Findings
Adenine, guanine, and thymine cyclopentane nucleosides remain stable in 98% sulfuric acid for at least two weeks.
Cytosine analogs degrade rapidly, releasing soluble cytosine.
3,3-dimethylcyclopentyl adenine analog (A4) is the most stable, avoiding carbocation-mediated cleavage.
Abstract
We synthesized seven carbocyclic nucleoside analogs featuring a cyclopentane ring in place of the (deoxy)ribose sugar, which serves as a linker in DNA/RNA nucleosides. We assessed the stability of cyclopentane nucleosides in 98% w/w sulfuric acid at room temperature via 1H and 13C NMR spectroscopy. We observe that adenine (A1, A4), guanine (G1) and thymine (T1) cyclopentane nucleoside analogs remain stable for at least two weeks at room temperature, with only minor (~4%) degradation in A1. In contrast, the cytosine analog (C1) rapidly degrades to release a soluble cytosine. Methyl-substituted adenine analogs mimicking polymer backbone attachments at positions prone to tertiary carbocation formation (A2, A3) prove unstable and release soluble adenine. Only the 3,3-dimethylcyclopentyl adenine analog (A4) exhibits sufficient stability. Our findings reveal that cyclopentane serves as a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
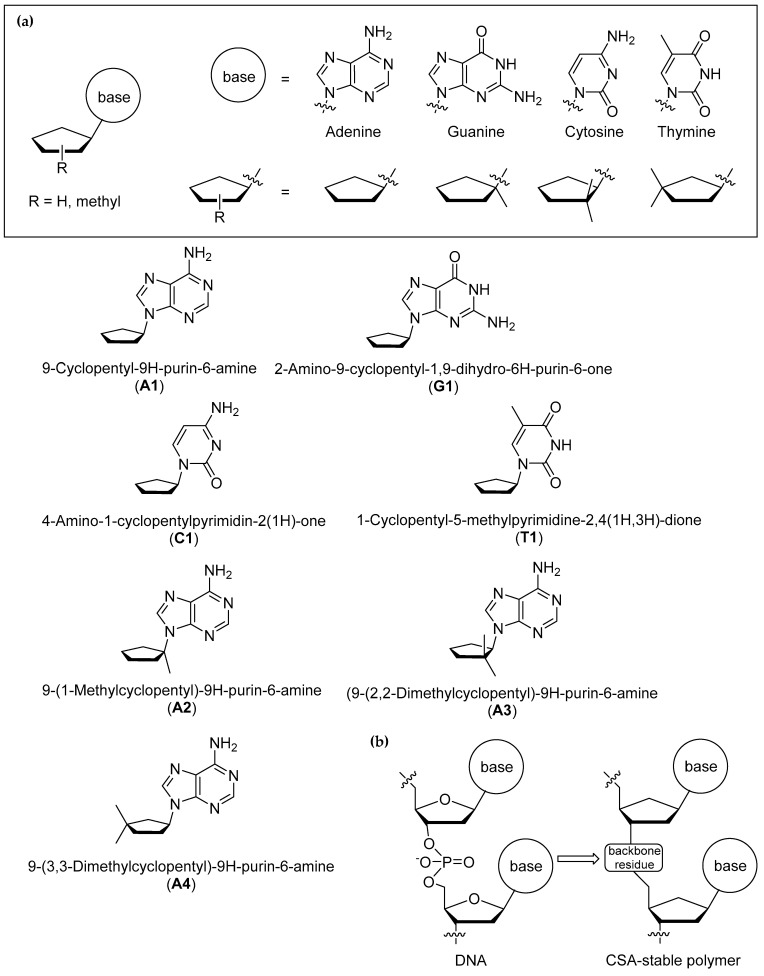 Figure 1
Figure 1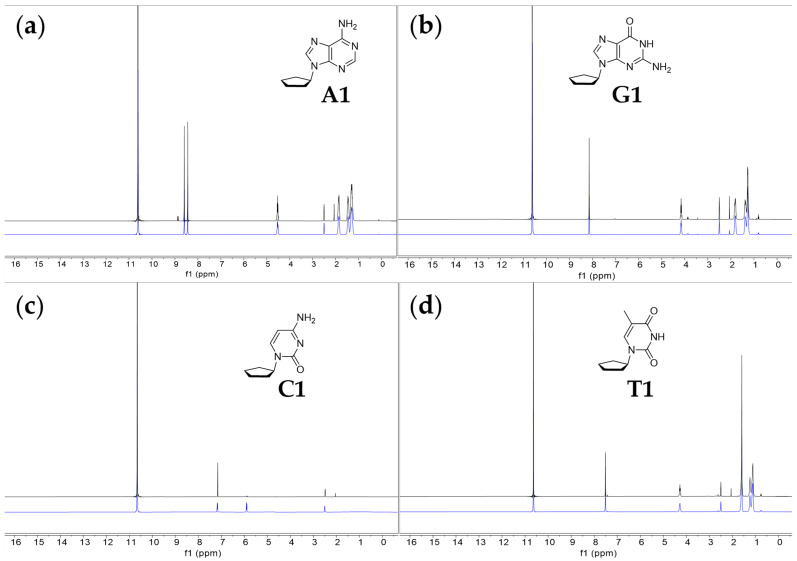 Figure 2
Figure 2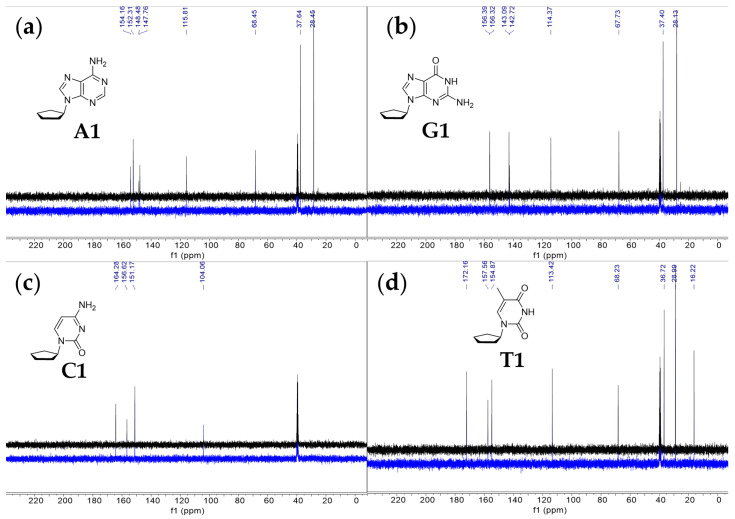 Figure 3
Figure 3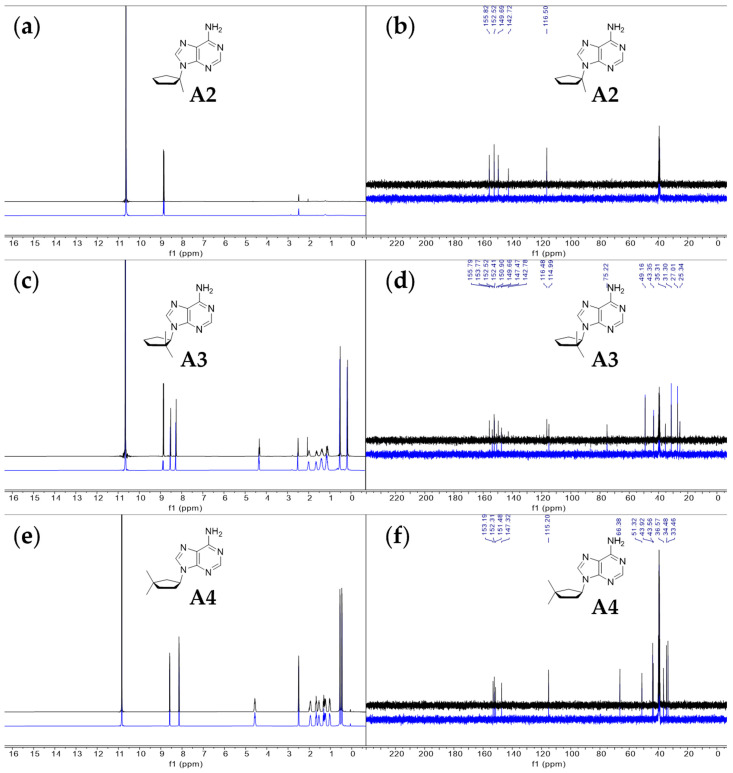 Figure 4
Figure 4 Figure 5
Figure 5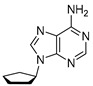 Figure 6
Figure 6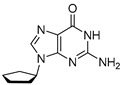 Figure 7
Figure 7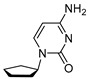 Figure 8
Figure 8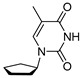 Figure 9
Figure 9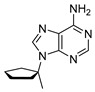 Figure 10
Figure 10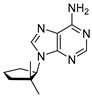 Figure 11
Figure 11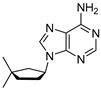 Figure 12
Figure 12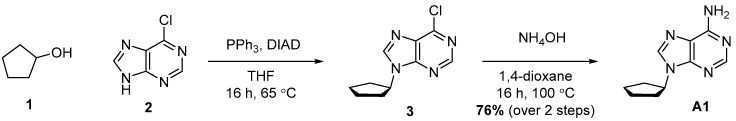 Figure 13
Figure 13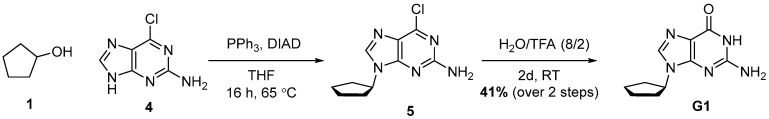 Figure 14
Figure 14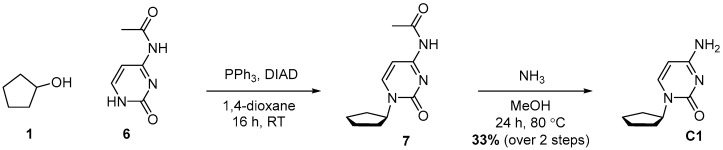 Figure 15
Figure 15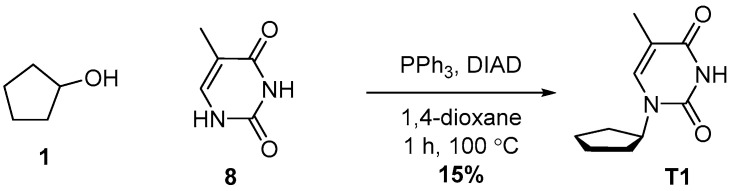 Figure 16
Figure 16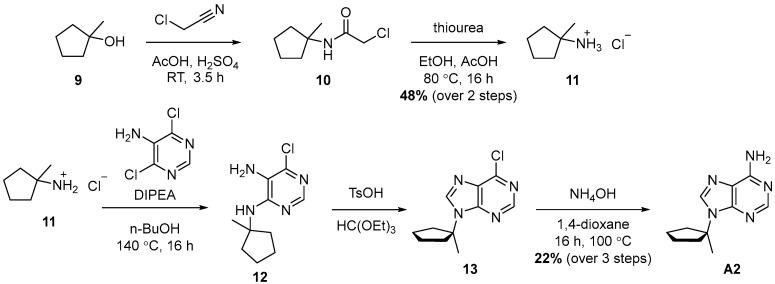 Figure 17
Figure 17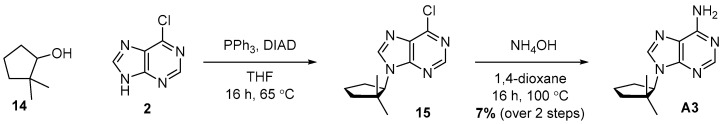 Figure 18
Figure 18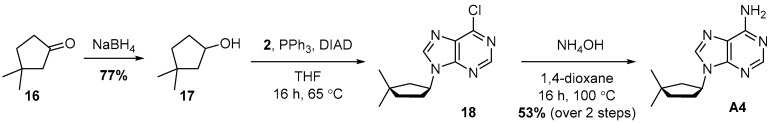 Figure 19
Figure 19- —The Alfred P. Sloan Foundation
- —Nanoplanet Consulting
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA and Nucleic Acid Chemistry · HIV/AIDS drug development and treatment · RNA Interference and Gene Delivery
1. Introduction
Organic chemistry in concentrated sulfuric acid is rarely studied despite its surprising richness. Recent studies, building on isolated reports from decades ago (e.g., [1,2,3,4,5,6,7,8]), have renewed the interest in this area of organic chemistry, showing that complex organic molecules can stably persist in this aggressive solvent.
The resurgence of interest in organic chemistry of concentrated sulfuric acid stems from speculations about possible life in Venus’s atmosphere—not on the scorching 700 K surface but within the sulfuric acid clouds at 48–60 km altitude where temperatures resemble those on Earth’s surface (e.g., [9,10,11,12,13,14,15,16,17,18,19]). Although complex organic chemistry does not equate to life, its presence in a planetary environment provides an essential prerequisite for the possibility of life [20].
Recently, Spacek and Benner have demonstrated that diverse organic reactions spontaneously emerge in concentrated sulfuric acid from simple starting materials like formaldehyde or carbon monoxide [21,22,23]. Our team showed the stability of simple organic chemical building blocks, including nucleic acid bases [24,25], amino acids [26] and dipeptides [27,28]. We have also shown that simple lipids are not only stable but spontaneously form lipid vesicles, micelles and other complex membranous structures in concentrated sulfuric acid at room temperature [29].
This recent work shows promise, yet life requires far more sophisticated molecular structures—particularly complex polymers—to perform biological functions. In particular, the need for genetic polymers with functional and structural properties similar to RNA and DNA seems to be the universal requirement for life, regardless of life’s underlying chemistry [20,30]. Therefore, identifying genetic polymers that resist degradation in concentrated sulfuric acid becomes a critical step to study the possibility of life in environments where sulfuric acid is a dominant liquid.
Here, we explore the cyclopentane motif as a candidate structural analog of the (deoxy)ribose linker in DNA and RNA (Figure 1). Cyclopentane nucleosides (and, more generally, carbocyclic nucleosides) have been studied as potential antiviral drugs for decades [31,32]. The cyclopentene nucleosides carbovir and its prodrug abacavir, as potent and selective anti-HIV agents, are prime examples of the antiviral activity of this family of compounds (reviewed in [33]).
To evaluate cyclopentane as a potential linker residue in genetic-like polymers that are stable in concentrated sulfuric acid, we synthesized seven cyclopentane nucleosides and tested their stability in 98% w/w sulfuric acid at room temperature (RT). We employed ^13^C and ^1^H Nuclear Magnetic Resonance Spectroscopy (NMR) to monitor the integrity of these molecules in 98% w/w sulfuric acid at RT over periods ranging from hours to weeks. We also describe detailed synthetic routes for all seven investigated compounds.
2. Results
We tested the stability of seven cyclopentyl nucleosides dissolved in D_2_SO_4_ (98 wt%, 2 wt% D_2_O). The ^1^H and ^13^C NMR spectra were measured after 1–2 h, 2 days, 7 days and 14 days (Table 1). The stability and reactivity of cyclopentyl nucleosides have not been previously tested in concentrated sulfuric acid. Here, we consider the compounds to be stable if we see no detectable reactivity and degradation of the tested compounds after incubation in 98% w/w sulfuric acid for up to 14 days at room temperature (RT). We first analyzed the compounds after 1 h of incubation in 98% w/w sulfuric acid and next repeated the NMR measurements after two and seven days, and finally after two weeks for an additional final stability test. By stable, we also mean the long-term (~2 weeks) stability of the heavy atom bonding topology in the tested compounds, i.e., the bonding of nonhydrogen atoms to each other, and not a rapid, reversible exchange of protons between the bases and the solvent. Such rapid exchange of hydrogen atoms is expected in sulfuric acid and is not a sign of the instability of the tested compounds [1,6,24,34,35].
We chose 98% w/w concentrated sulfuric acid for the stability experiments for two reasons. First, the concentration of sulfuric acid in the clouds of Venus varies with altitude, from 81% w/w acid in the top clouds, at 56.5–70 km above the surface, to 98% w/w acid in the lower cloud region (47.5–50.5 km) [36,37,38,39]. At 98% w/w, the amount of water is very low and sulfuric acid dominates the chemistry of the solvent, potentially entirely changing the expected chemical behavior of the investigated compounds, as compared with more diluted acid [27,28]. Second, the early experiments on the stability and reactivity of organic molecules, including nucleic acid bases, often focused on similarly high concentrations of acid [1,6,8,34,35].
We first tested the stability of four cyclopentane nucleosides (A1, G1, C1, T1), where the cyclopentane linker bonds directly to four different nucleic acid bases: adenine (A1), guanine (G1), thymine (T1) and cytosine (C1) (see Section 2.1). In Section 2.2, we explore how the position of the methyl group substituent in the cyclopentane ring influences the reactivity of the tested compound in 98% w/w sulfuric acid. In compound A2, we tested the reactivity of the cyclopentyl linker with two substitutions at Position 1 in the cyclopentane ring, i.e., the base and the methyl group bonded to the carbon C1 in the ring. In compound A3, we examined the reactivity of the cyclopentyl linker with two methyl groups bonded to the second (C2) position in the cyclopentane ring. Finally, in compound A4, we tested the reactivity of the cyclopentyl linker with two methyl groups bonded to the third (C3) position in the cyclopentane ring. The addition of the methyl groups at these specific positions in the cyclopentane ring aims to mimic the connection to other nucleosides in the backbone of the polymer.
2.1. Stability and Reactivity of Adenine, Guanine, Cytosine and Thymine Cyclopentane Nucleosides (A1, G1, C1, T1) in 98% w/w Sulfuric Acid
Out of the four tested cyclopentane nucleosides, three are soluble and stable in 98% w/w sulfuric acid (Figure 2 and Figure 3). The ^1^H and ^13^C NMR spectra of the compounds (A1, G1, T1) do not change significantly over the span of the two-week incubation in 98% w/w sulfuric acid. The cytosine cyclopentane nucleoside (C1) is highly reactive in 98% w/w sulfuric acid. The C–N bond connecting the base to the cyclopentane ring readily breaks in 98% w/w sulfuric acid, leaving behind acid-soluble cytosine. The cyclopentane itself is immiscible and insoluble in sulfuric acid. We note that over time, the ^1^H NMR signal corresponding to the H5 hydrogen (~5.8 ppm) in cytosine splits and broadens (Figure 2). The splitting and broadening of the H5 peak indicate an exchange of the H5 proton of the cytosine ring with the D_2_SO_4_ deuterium (i.e., H/D exchange) and is not a sign of an instability of the pyrimidine ring. Such an H/D exchange readily happens in acidic solutions [40]. Splitting and broadening, consistent with efficient H/D exchange and possible carbocation formation, is also visible in the ^13^C NMR spectrum of C1, where the C5 carbon signal of the cytosine nucleobase splits and broadens over time. We expect such splitting to happen, as aromatic systems often undergo efficient hydrogen exchange with the solvent, which could potentially lead to the formation of the stable carbocation [41,42,43]. Formation of the carbocation could also be consistent with the ^1^H NMR spectrum, where the H5 hydrogen peak also splits heavily and broadens. We have observed similar behavior of cytosine nucleic acid bases before, when they were incubated over long period of time in concentrated sulfuric acid [24,25].
We now turn to the detailed NMR assignment of the cyclopentane nucleosides A1, G1 and T1 that are stable in 98% w/w sulfuric acid, focusing on the spectra collected after 2 weeks of incubation in 98% w/w sulfuric acid at RT.
For compound A1, in the ^13^C NMR spectrum (Figure 3), we find five peaks that correspond to the five carbons in the adenine ring and three peaks corresponding to five atoms of the cyclopentane ring. Each of the five peaks corresponding to the adenine ring reside in the region of the NMR spectrum associated with aromatic compounds (peaks δ 154.2, 152.3, 148.4, 147.8 and 115.8). The ^13^C NMR spectrum matches the chemical shifts of adenine from our previous NMR studies in 98% w/w sulfuric acid [24,25]. We assign the three remaining carbon peaks to the aliphatic cyclopentane carbons. The peak at 68.5 ppm is significantly more deshielded than the other two peaks and corresponds to the cyclopentane carbon C1 attached to adenine’s imidazole ring nitrogen. The most upfield peak at 28.5 ppm corresponds to the two symmetric carbon atoms of the cyclopentane ring that are the furthest from the adenine ring. The peak at 37.6 ppm corresponds to the two symmetric carbon atoms of the cyclopentane ring, which experience a moderate deshielding effect and are bonded to the carbon atom that is directly connected to the adenine ring. The ^1^H NMR spectra are also consistent with compound A1 being largely stable at RT for at least 2 weeks. We observe only very slight degradation (~4%) of the compound A1, as estimated from the ^1^H NMR spectrum. We also note that the chemical shift trends for the ^1^H and ^13^C NMR spectra agree with the values of ^13^C and ^1^H NMR collected in CDCl_3_ solvent (see Section 4.1 in Materials and Methods).
For compound G1, in the ^13^C NMR spectrum (Figure 3), we find five peaks that correspond to the five carbons in the guanine ring and three peaks corresponding to five atoms of cyclopentane ring. Each of the five peaks corresponding to the guanine ring reside in the region of the NMR spectrum associated with aromatic compounds (peaks δ 156.4, 156.3, 143.1, 142.7 and 114.4). The ^13^C NMR spectrum matches the chemical shifts of guanine from our previous NMR studies in 98% w/w sulfuric acid [24,25]. We assign the three remaining carbon peaks to the aliphatic cyclopentane carbons. The peak at 67.7 ppm is significantly more deshielded than the other two peaks and corresponds to the cyclopentane carbon attached to guanine’s imidazole ring nitrogen. The most upfield peak at 28.1 ppm corresponds to the two carbon atoms of the cyclopentane ring that are the furthest from the guanine ring. The peak at 37.4 ppm corresponds to the two symmetric carbon atoms of the cyclopentane ring, which experience a moderate deshielding effect and are bonded to the carbon atom that is directly connected to the guanine ring. The ^1^H NMR spectra are also consistent with compound G1 being stable at RT for at least 2 weeks. We observe no degradation of the compound G1, as estimated from the ^1^H NMR spectrum. No new peaks emerge over the span of 2 weeks of incubation, confirming the overall stability of the compound G1 in 98% w/w sulfuric acid. We also note that the chemical shift trends for the ^1^H and ^13^C NMR spectra agree with the values of ^13^C and ^1^H NMR collected in CDCl_3_ solvent (see Section 4.2 in Materials and Methods).
Finally, for compound T1, in the ^13^C NMR spectrum (Figure 3), we find four peaks that correspond to the four carbons in the thymine ring and three peaks corresponding to five carbon atoms of the cyclopentane ring. Each of the four peaks corresponding to the thymine ring reside in the region of the NMR spectrum associated with aromatic compounds (peaks δ 172.2, 157.6, 154.9 and 113.4). The ^13^C NMR spectrum generally matches the chemical shifts of thymine from our previous NMR studies in 98% w/w sulfuric acid [24,25]. We assign the three remaining carbon peaks to the aliphatic cyclopentane carbons. The peak at 68.2 ppm is significantly more deshielded than the other two peaks and corresponds to the cyclopentane carbon attached to thymine’s ring nitrogen. The most upfield peak of the three, at 29.0 ppm, corresponds to the two carbon atoms of the cyclopentane ring that are the furthest from the thymine ring. The peak at 36.7 ppm corresponds to the two symmetric carbon atoms of the cyclopentane ring, which experience a moderate deshielding effect and are bonded to the carbon atom directly connected to the thymine ring. Finally, we assign the peak at 16.2 ppm to the methyl group carbon of the thymine nucleobase. The ^1^H NMR spectra are also consistent with compound T1 being stable at RT for at least 2 weeks. We observe no degradation of the compound T1, as estimated from the ^1^H NMR spectrum. No new peaks emerge over the span of 2 weeks of incubation, confirming the overall stability of the compound T1 in 98% w/w sulfuric acid. We also note that the chemical shift trends for ^1^H and ^13^C NMR spectra agree with the values of ^13^C and ^1^H NMR collected in CDCl_3_ solvent (see Section 4.4 in Materials and Methods).
2.2. Stability and Reactivity of Modified Adenine Cyclopentane Nucleosides (A2, A3, A4) in 98% w/w Sulfuric Acid
Having established that, overall, compounds A1, G1 and T1 remain stable in 98% w/w sulfuric acid, we investigated how the cyclopentane ring substituents affect the stability of the nucleoside structure. We modified compound A1 to include additional methyl groups at various positions in the cyclopentane ring (compounds A2, A3 and A4 in Figure 1). We show that compounds A2 and A3 are unstable in concentrated sulfuric acid, leading to the release of the soluble adenine nucleobase (Figure 4). Cleavage of the branched-carbocycle adenine connection in compound A2 is expected, a tertiary carbocation forms more readily, is more stable, and acts as a better leaving group than a secondary carbocation that forms in the cyclopentane variant A1. Carbocations form as a result of the exchange of H between tertiary carbon and the sulfuric acid solvent. Such exchange leads to the formation of the reactive meta-stable species with positively charged carbon atoms (carbocations). The carbocation formation is only efficient in aromatic compounds (see compound C1 above) or alkyl groups with tertiary carbon groups (in A2) and not in CH_2_ (in A1) or in methyl (CH_3_) groups themselves [42]. Compound A3 degrades slowly over time, and after 14 days, 48% of A3, as estimated by ^1^H NMR, converts to a soluble nucleobase adenine (Figure 4).
Out of the three tested analogs of compound A1, only the cyclopentane nucleoside A4 is stable in 98% w/w sulfuric acid. For compound A4, in the ^13^C NMR spectrum (Figure 4), we find five peaks that correspond to the five carbons in the adenine ring and seven peaks corresponding to five atoms of cyclopentane ring and two methyl group substituents. Each of the five peaks corresponding to the adenine ring reside in the region of the NMR spectrum associated with aromatic compounds (peaks δ 153.2, 152.3, 151.5, 147.3 and 115.2). The ^13^C NMR spectrum matches the chemical shifts of adenine from our previous NMR studies in 98% w/w sulfuric acid [24,25]. The remaining seven carbon peaks (peaks δ 66.4, 51.3, 43.9, 43.6, 36.6, 34.5 and 33.5) correspond to the 3,3-dimethylcyclopentyl ring, with the two most upfield peaks (34.5 ppm and 33.5 ppm) corresponding to the two methyl group substituents. The ^1^H NMR spectra are also consistent with compound A4 being stable at RT for at least 2 weeks. We observe no degradation of the compound A4, as estimated from the ^1^H NMR spectrum. No new peaks emerge over the span of 2 weeks of incubation, confirming the overall stability of the compound A4 in 98% w/w sulfuric acid. We also note that the chemical shift trends for ^1^H and ^13^C NMR spectra agree with the values of ^13^C and ^1^H NMR collected in CDCl_3_ solvent (see Section 4.7 in Materials and Methods).
3. Discussion
Our work is a part of a larger effort towards the design and synthesis of a structural analog of DNA capable of forming a double-helix-like structure in concentrated sulfuric acid. DNA itself rapidly degrades in this aggressive solvent due to the solvolysis of deoxyribose residues and the phosphodiester backbone of the polymer. Therefore, the stability of a new linker (substituting the reactive deoxyribose) and backbone (instead of the phosphate ester) residues need to be designed and tested.
Reactivity tests of the cyclopentane linker, as a structural analog of deoxyribose, are a first step towards this goal. We note that replacing the ribose sugar with a cyclopentane scaffold may reduce the intrinsic chemical and stereochemical functionality available to the backbone itself, even though additional functional groups could, in principle, be introduced elsewhere in the polymer.
Our results show that the reactivity of cyclopentane nucleosides in 98% w/w sulfuric acid varies significantly with respect to the nucleobase attached to the cyclopentane linker (Table 1). The differences in reactivity amongst nucleosides A1, G1, C1 and T1 in 98% w/w sulfuric acid at RT pose a potential problem for the design of the polymer with a cyclopentane linker. If the stability of all four nucleosides is not the same, then such linker–base connections could lead to different sequence-dependent reactivity of the whole polymer. The use of cyclopentane in the design of such a polymer might therefore require substituting reactive canonical bases, like cytosine, with other less reactive counterparts (see the discussion below).
We also explored the differences in the reactivity of the cyclopentane with respect to the position of the attachment of the linker ring to the backbone of the polymer. In compounds A2, A3 and A4, the position of the additional methyl groups corresponds to the potential attachment points to the rest of the polymer (Figure 1). Our reactivity experiments show that the arrangement of the methyl groups in the cyclopentane ring highly influences the stability of the entire nucleoside in 98% w/w sulfuric acid. In other words, the stability of the cyclopentane nucleosides A2, A3 and A4 differs with respect to the position of the additional methyl groups attached to the ring. This observation is important, as the place of attachment of the backbone residue to the cyclopentane linker could influence the stability of the entire polymer. Our reactivity experiments show that the attachment of the backbone residues at the C1 or C2 positions in the cyclopentane ring could result in the instability of the entire polymer, as exemplified by the overall instability of compounds A2 and A3 (Table 1). On the other hand, the fact that compound A4 is stable suggests that the backbone residue connected at position C3 in the cyclopentane linker might lead to a stable polymer structure. In follow-up studies, we plan to explore the reactivity of other substituents at other positions (positions C3 and C4 in compounds A5, A6 and A7 in Figure 5) to fully cover the structural chemical space of possible connections to the stable backbone of the polymer.
More work is needed for a full assessment of the viability of cyclopentanes as potential linker residues that are stable in concentrated sulfuric acid. Specifically, stability assessments of selected modified cyclopentanes at higher temperatures, spanning the 80–100 °C temperature range of the Venusian cloud deck, are needed. For example, we previously demonstrated that Peptide Nucleic Acid (PNA) could be a promising candidate for a genetic polymer that is stable in concentrated sulfuric acid [44]. PNA, however, while stable at room temperature in 98% w/w sulfuric acid, decomposes above 50 °C. PNA’s solvolysis at high temperatures proceeds by cleavage of a single tertiary amide bond in an acetyl group linker that connects the nucleic acid base to the backbone of the polymer. The solvolysis yields two distinct products that appear to be stable to further degradation in 98% w/w sulfuric acid [44]. The thermal instability of PNA’s acetyl group linker prompts us to seek other structural analogs to DNA in parallel, in an effort to identify at least several examples of chemical motifs that could serve as stable backbone and linker residues of a genetic-like polymer in concentrated sulfuric acid. The thermal stability of modified cyclopentane nucleosides at temperatures above 50 °C should be part of these future efforts.
The next stages in the design of the genetic-like polymer also include finding an alternative to the reactive DNA phosphate ester backbone. We plan to explore phosphinic acid and amine backbone polymers. A phosphinic acid-based backbone residue exchanges the –O–P–O– phosphate ester bonds for an acid stable –C–P–C– phosphinic backbone. Such a phosphinic backbone, however, would likely lose its permanent repeating negative charge due to efficient protonation in concentrated sulfuric acid. An amine might therefore be an especially promising candidate for an alternative backbone. In concentrated sulfuric acid, amines are fully and stably protonated. Recall that a permanent repeating charge, no matter if it is negative (as phosphates in DNA in water) or positive (as in amines in concentrated sulfuric acid), is likely a universal requirement for any genetic polymer of life, regardless of its biochemical makeup [45,46,47].
We are aware, however, that even with a stable alternative linker and backbone, such as that of PNA [44], or modified cyclopentane, it is likely that the protonation and tautomeric forms of the adenine, thymine, guanine and cytosine bases will be different in concentrated sulfuric acid than in water. As a result, classical Watson–Crick base pairing may not be possible between complementary strands containing canonical A, T, C, G bases, and alternative bases, that do not rely on hydrogen bonding to pair, may be necessary for life to survive in this solvent [48]. Such alternative nucleobases that pair exclusively via hydrophobic and van der Waals interactions have been routinely explored in synthetic biology (e.g., [49,50,51,52,53,54]).
In summary, we find that cyclopentane is a promising structural motif for the linker residue of a genetic-like polymer that is stable in 98% w/w sulfuric acid at room temperature. Future stability experiments should test the thermal stability of the cyclopentane linker, extend the cyclopentane scaffold to other modified variants to test additional substitution patterns (A5–A7) and culminate in an effort to pair stable cyclopentane linkers with alternative backbone chemistries. Together, our experiments aim to identify at least one robust candidate for a genetic-like polymer that could potentially persist in the sulfuric acid clouds of Venus in order to motivate space missions to search in situ in the Venusian atmosphere for signs of life or life itself.
4. Materials and Methods
We confirmed the structural integrity of the synthesized compounds with Liquid Chromatography–Mass Spectrometry (LCMS) and NMR spectroscopy. We used LCMS as a supporting technique to NMR to identify the intermediate as well as the final products of cyclopentane nucleoside synthesis. We used MNova software, version: 14.3.0-30573 (Mestrelab Research, Barcelona, Spain) to process and analyze the NMR data [55]. The original data for all LCMS and NMR spectra are available on Zenodo at https://zenodo.org/records/18441685.
4.1. Synthesis of 9-Cyclopentyl-9H-purin-6-amine (A1)
Compound A1 was synthesized in two steps starting with a Mitsunobu reaction between Alcohol 1 and Compound 2, followed by amination with ammonia. Compound A1 was obtained with 76% yield over two steps.
Cyclopentanol (500 mg, 527 μL, 1 equiv., 5.81 mmol), 6-chloro-9H-purine (1.08 g, 1.2 equiv., 6.97 mmol) and triphenylphosphine (1.83 g, 1.2 equiv., 6.97 mmol) were dissolved in THF (58 mL). DIAD (1.35 mL, 1.2 equiv., 6.97 mmol) was added and the mixture was refluxed for 16 h. The mixture was concentrated in vacuo and then purified by column chromatography (40 g silica, 0–100% EtOAc/heptane gradient) to obtain the slightly impure Intermediate 3 (1.27 g). Intermediate 3 was dissolved in 1,4-dioxane (6.0 mL) and transferred to a pressure tube. Ammonia (6.0 mL, 25 wt%, 12 equiv., 69 mmol) was added and the reaction mixture was heated to 100 °C for 16 h. The mixture was partitioned between brine and EtOAc. We extracted the aqueous layer once with EtOAc again. Combined organic layers were dried over Na_2_SO_4_, filtered and concentrated in vacuo. The crude extract was purified by column chromatography (25 g silica, 0–4% MeOH/DCM gradient), and Product A1 (900 mg, 4.43 mmol, 76% yield over 2 steps) was obtained as a white solid (Scheme 1). ^1^H NMR (400 MHz, CDCl_3_) had peaks at δ 8.36 (s, 1H), 7.85 (s, 1H), 5.85 (s, 2H), 4.92 (p, J = 7.3 Hz, 1H), 2.36–2.20 (m, 2H) and 2.07–1.71 (m, 6H). ^13^C NMR (101 MHz, CDCl_3_) had peaks at δ 155.5, 152.7, 150.3, 138.8, 120.1, 56.1, 32.9 and 24.0. Compound A1 was dissolved in 98 wt% D_2_SO_4_ and DMSO-d_6_ (9/1 v/v), and NMR spectra were measured: ^1^H NMR (400 MHz, D_2_SO_4_), δ 8.59 (s, 1H), 8.44 (d, J = 1.2 Hz, 1H), 4.58–4.46 (m, 1H), 1.92–1.79 (m, 2H), 1.52–1.39 (m, 2H) and 1.38–1.21 (m, 4H); ^13^C NMR (101 MHz, D_2_SO_4_), δ 154.2, 152.3, 148.4, 147.8, 115.8, 68.5, 37.6 and 28.5.
4.2. Synthesis of 2-Amino-9-cyclopentyl-1,9-dihydro-6H-purin-6-one (G1)
Compound G1 was synthesized in two steps, starting with a Mitsunobu reaction between Alcohol 1 and Compound 4, followed by hydrolysis with aqueous TFA. Compound G1 was obtained with 41% yield over two steps.
To a mixture of cyclopentanol (1.33 g, 1.40 mL, 1 equiv., 15.4 mmol), 6-chloro-9H-purin-2-amine (3.93 g, 1.5 equiv., 23.2 mmol), triphenylphosphine (6.08 g, 1.5 equiv., 23.2 mmol) and THF (58 mL), DIAD (4.68 g, 4.50 mL, 1.5 equiv., 23.2 mmol) was added at RT. The mixture was then heated to reflux for 16 h and concentrated in vacuo. The crude extract was purified by column chromatography (120 g silica, 0–4% MeOH/DCM gradient) to obtain Intermediate 5 (4.1 g) with triphenylphosphine oxide as an impurity. Impure Intermediate 5 was dissolved in TFA/H_2_O (8/2 v/v, 90 mL) and stirred at RT for 2 days. The mixture was concentrated in vacuo and co-evaporated with toluene. The residue was suspended in Et_2_O by sonication, filtered over a glass filter, washed with Et_2_O and then dried in vacuo. Product G1 (1,41 g, 41% yield over 2 steps) was obtained as a white solid (Scheme 2). ^1^H NMR (400 MHz, DMSO) had peaks at δ 10.78 (s, 1H), 7.74 (s, 1H), 6.48 (s, 2H), 4.59 (p, J = 7.4 Hz, 1H), 2.12–1.96 (m, 2H), 1.94–1.74 (m, 4H) and 1.74–1.56 (m, 2H). ^13^C NMR (101 MHz, DMSO) had peaks at δ 157.5, 153.7, 151.1, 135.3, 116.9, 54.7, 32.0 and 23.4. Compound G1 was dissolved in 98 wt% D_2_SO_4_ and DMSO-d_6_ (9/1 v/v), and the NMR spectra were measured: ^1^H NMR (400 MHz, D_2_SO_4_), δ 8.14 (s, 1H), 4.26–4.07 (m, 1H), 1.91–1.75 (m, 2H) and 1.33 (d, J = 44.5 Hz, 6H); ^13^C NMR (101 MHz, D_2_SO_4_), δ 156.4, 156.3, 143.1, 142.7, 114.4, 67.7, 37.4 and 28.1.
4.3. Synthesis of 4-Amino-1-cyclopentylpyrimidin-2(1H)-one (C1)
Compound C1 was synthesized in two steps, starting with a Mitsunobu reaction between Alcohol 1 and Compound 6, followed by deacylation with ammonia. Compound C1 was obtained with 33% yield over two steps.
To a mixture of cyclopentanol (609 mg, 642 μL, 1 equiv., 7.07 mmol), N-(2-oxo-1,2-dihydropyrimidin-4-yl)acetamide (1.19 g, 1.1 equiv., 7.78 mmol), triphenylphosphine (2.23 g, 1.2 equiv., 8.48 mmol) and 1,4-dioxane (70 mL), DIAD (1.65 mL, 1.2 equiv., 8.48 mmol) was added dropwise over 5 min. The mixture was then sonicated for 1 min and stirred at RT for 16 h. The mixture was concentrated in vacuo, and the crude extract was purified by column chromatography (120 g silica, 0–40% EtOAc/heptane gradient) to obtain Intermediate 7. Compound 7 was dissolved in MeOH (6.0 mL) and transferred to a pressure tube, and NH_3_ in MeOH (4.7 g, 6.0 mL, 7.0 molar, 5.9 equiv., 42 mmol) was added. The mixture was heated to 80 °C for 24 h and then concentrated in vacuo. The crude mixture was purified by column chromatography (40 g silica, 20–40% EtOAc/heptane gradient) to obtain Product C1 (417 mg, 33% yield over 2 steps) as a white solid (Scheme 3). ^1^H NMR (400 MHz, CDCl_3_) had peaks at δ 8.00 (d, J = 5.7 Hz, 1H), 6.06 (d, J = 5.7 Hz, 1H), 5.34 (tt, J = 6.1, 3.0 Hz, 1H), 4.86 (s, 2H), 2.00–1.71 (m, 6H) and 1.69–1.51 (m, 2H). ^13^C NMR (101 MHz, CDCl_3_) had peaks at δ 165.2, 164.7, 157.5, 99.1, 79.2, 33.0 and 23.9. Compound C1 was dissolved in 98 wt% D_2_SO_4_ and DMSO-d_6_ (9/1 v/v), and the NMR spectra were measured. Only cytosine was visible.
4.4. Synthesis of 1-Cyclopentyl-5-methylpyrimidine-2,4(1H,3H)-dione (T1)
Compound T1 was synthesized in one step via a Mitsunobu reaction between Alcohol 1 and Compound 8. Compound T1 was obtained with 15% yield after multiple purifications.
Cyclopentanol (1.4 g, 2 mL, 1.0 equiv., 16 mmol), 5-methylpyrimidine-2,4(1H,3H)-dione (2.0 g, 1 equiv., 16 mmol) and triphenylphosphine (6.2 g, 1.5 equiv., 24 mmol) were dissolved in 1,4-dioxane (40 mL). The mixture was then heated to reflux, after which DIAD (4.8 g, 4.6 mL, 1.5 equiv., 24 mmol) was added in dropwise fashion. After addition, refluxing was continued for another hour, after which LCMS showed complete conversion to the desired product. The mixture was concentrated in vacuo, and the crude extract was subjected to purification by silica column chromatography to produce 3.5 g of the desired material with low purity. Following this, reverse phase chromatography was used to obtain Product T1 (465 mg, 2.39 mmol, 15%) as a white solid (Scheme 4). ^1^H NMR (400 MHz, CDCl_3_) had peaks at δ 8.66 (s, 1H), 7.02 (d, J = 1.1 Hz, 1H), 4.91 (p, J = 8.2 Hz, 1H), 2.19–2.01 (m, 2H), 1.94 (d, J = 1.2 Hz, 3H) and 1.88–1.53 (m, 6H). ^13^C NMR (101 MHz, CDCl_3_) had peaks at δ 163.7, 151.2, 136.9, 111.0, 56.5, 31.4, 24.3 and 12.8. Compound T1 was dissolved in 98 wt% D_2_SO_4_ and DMSO-d_6_ (9/1 v/v), and the NMR spectra were measured: ^1^H NMR (400 MHz, D_2_SO_4_), δ 7.51 (s, 1H), 4.35–4.19 (m, 1H), 1.59 (s, 5H) and 1.18 (d, J = 47.8 Hz, 6H); ^13^C NMR (101 MHz, D_2_SO_4_), δ 172.2, 157.6, 154.9, 113.4, 68.2, 36.7, 29.0 and 16.2.
4.5. Synthesis of 9-(1-Methylcyclopentyl)-9H-purin-6-amine (A2)
The synthesis of 1-methyl substituted A1 started with the synthesis of tertiary amine 11, because the substitution on tertiary alcohol 9 is not expected to work. Amine 11 was prepared from Alcohol 9 in two steps via a Ritter reaction with 48% yield. Next, Amine 11 was converted to A2 in three steps with 22% yield.
To a mixture of 1-methylcyclopentan-1-ol (2.0 g, 1 equiv., 20 mmol), chloroacetonitrile (2.3 g, 1.9 mL, 1.5 equiv., 30 mmol) and AcOH (2.9 g, 2.8 mL, 2.4 equiv., 49 mmol) at 0 °C, H_2_SO_4_ was added (5.2 g, 2.8 mL, 2.6 equiv., 53 mmol) dropwise over 2–3 min. The mixture was stirred for 3.5 h at RT, whereupon ice-cold water (50 mL) and tBME (50 mL) were added. The organic layer was washed twice with sat. NaHCO_3_, with brine, dried over Na_2_SO_4_, filtered and concentrated in vacuo. The resulting crude mixture of compound 10 (3.1 g), thiourea (1.5 g, 1 equiv., 20 mmol), EtOH (30 mL) and AcOH (6.0 mL) were heated to 80 °C for 16 h. The mixture was allowed to cool to RT, and water (60 mL) was added. The mixture was filtered, washed with water and then basified to pH 10 with NaOH (30 wt%). The aqueous layer was extracted with tBME (3x), and the combined organic layers were washed with brine, dried over Na_2_SO_4_ and filtered. The filtrate was acidified by adding HCl (10 mL, 4 M in dioxane) and then concentrated in vacuo. The green solids were triturated with Et_2_O, dissolved in a minimal amount of MeOH and precipitated by adding it to excess Et_2_O. The resulting precipitate was filtered, washed with Et_2_O and with pentane, and dried in vacuo. Product 11 (1.29 g, 48% yield over 2 steps) was obtained as a white solid (Scheme 5). ^1^H NMR (400 MHz, MeOD) had peaks at δ 1.88–1.73 (m, 8H) and 1.41 (s, 3H).
A mixture of 1-methylcyclopentan-1-amine hydrochloride (11) (1.29 g, 1 equiv., 9.51 mmol), 4,6-dichloropyrimidin-5-amine (2.34 g, 1.5 equiv., 14.3 mmol), DIPEA (4.92 g, 6.63 mL, 4 equiv., 38.0 mmol) and n-butanol (25 mL) was heated to 140 °C (external) in a pressure tube for 16 h. The mixture was concentrated in vacuo, dissolved in EtOAc and washed with HCl (50 mL, 0.2 M), with sat. NaHCO_3_ and then with brine. The organic layer was dried over Na_2_SO_4_, filtered and concentrated in vacuo. The residue was purified by column chromatography (80 g silica, 0–30% EtOAc/heptane gradient) to obtain Intermediate 12 (650 mg, 2.87 mmol, 30%). To Compound 12 (650 mg) triethyl orthoformate (8.3 mL, 50 mmol) and p-toluenesulfonic acid monohydrate (38 mg, 0.20 mmol) were added, and the mixture was stirred at RT for 24 h. The mixture was concentrated in vacuo and to the resulting orange solid (13) ammonia (3.0 mL, 25 wt%, 39 mmol) and 1,4-dioxane (3 mL) were added. This mixture was heated in a pressure tube at 100 °C for 16 h. The reaction mixture was partitioned between EtOAc (25 mL) and brine (25 mL), and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na_2_SO_4_, filtered and concentrated in vacuo. The residue was purified by column chromatography (25 g silica, 0–5% MeOH/DCM gradient) to obtain Product A2 (458 mg, 2.11 mmol, 22% yield over 3 steps) as a white solid (Scheme 5). ^1^H NMR (400 MHz, CDCl_3_) had peaks at δ 8.35 (s, 1H), 7.86 (s, 1H), 5.81 (s, 2H), 2.49 (dt, J = 11.6, 5.5 Hz, 2H), 2.24–2.08 (m, 2H), 1.82 (dq, J = 6.5, 3.2 Hz, 4H) and 1.69 (s, 3H). ^13^C NMR (101 MHz, CDCl_3_) had peaks at δ 155.6, 152.1, 150.4, 139.0, 121.1, 66.8, 38.7, 26.4 and 22.8. Compound A2 was dissolved in 98 wt% D_2_SO_4_ and DMSO-d_6_ (9/1 v/v), and the NMR spectra were measured. Only adenine was visible.
4.6. Synthesis of (9-(2,2-Dimethylcyclopentyl)-9H-purin-6-amine (A3)
Compound A3 was synthesized in two steps, starting with a Mitsunobu reaction between Alcohol 14 and Compound 2, followed by amination with ammonia. Compound A3 was obtained with 7% yield over two steps. The synthesis of A3 was low-yielding because the first Mitsunobu step towards 15 is troublesome due to the steric hindrance of Alcohol 14.
To a mixture of 2,2-dimethylcyclopentan-1-ol (451 mg, 1 equiv., 3.95 mmol), 6-chloro-9H-purine (1.22 g, 2 equiv., 7.90 mmol), triphenylphosphine (2.07 g, 2 equiv., 7.90 mmol) and THF (45 mL), DIAD (1.54 mL, 2 equiv., 7.90 mmol) was added at RT. The mixture was heated to 65 °C for 16 h and then concentrated in vacuo. The crude mixture was purified by column chromatography (80 g silica, 0–70% EtOAc/heptane gradient) to obtain impure Intermediate 15 (755 mg). This material was dissolved in 1,4-dioxane (4.0 mL) and transferred to a pressure tube, then ammonia (4.0 mL, 25 wt%, 12 equiv., 46 mmol) was added and the resulting mixture was heated to 100 °C for 16 h. The reaction mixture was partitioned between EtOAc and brine, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na_2_SO_4_, filtered and concentrated in vacuo. The residue was purified by column chromatography (25 g silica, 0–4% MeOH/DCM gradient) to obtain slightly impure Product A3 (64 mg, 0.28 mmol, 7% yield over 2 steps) as a white solid (Scheme 6). ^1^H NMR (400 MHz, CDCl_3_) had peaks at δ 8.36 (s, 1H), 7.84 (s, 1H), 5.87 (s, 2H), 4.71 (t, J = 8.1 Hz, 1H), 2.44–2.18 (m, 2H), 2.04–1.81 (m, 2H), 1.74–1.65 (m, 2H), 1.11 (s, 3H) and 0.75 (s, 3H). ^13^C NMR (101 MHz, CDCl_3_) had peaks at δ 155.6, 152.8, 151.0, 139.6, 119.6, 63.8, 43.2, 39.2, 30.1, 27.7, 23.0 and 20.9. Compound A3 was dissolved in 98 wt% D_2_SO_4_ and DMSO-d_6_ (9/1 v/v) and the NMR spectra were measured: ^1^H NMR (400 MHz, D_2_SO_4_), δ 8.52 (s, 1H), 8.26 (s, 1H), 4.33 (t, J = 7.3 Hz, 1H), 2.05–1.91 (m, 1H), 1.70–1.56 (m, 1H), 1.47–1.29 (m, 2H), 1.21–1.03 (m, 2H), 0.51 (s, 3H) and 0.16 (s, 3H); ^13^C NMR (101 MHz, D_2_SO_4_) δ 153.8, 152.4, 150.7, 147.5, 115.0, 75.3, 49.2, 43.3, 35.3, 31.3, 27.0 and 25.3. Note that A3 degraded slowly over time to adenine in 98% w/w D_2_SO_4_.
4.7. Synthesis of 9-(3,3-Dimethylcyclopentyl)-9H-purin-6-amine (A4)
Synthesis of A4 started with sodium borohydride reduction of Ketone 16 to Alcohol 17. Alcohol 17 was obtained in 77% yield and then substituted with 2 under Mitsunobu conditions towards 18. Treatment of 18 with ammonia gave A4 with 53% yield over two steps.
To a mixture of 3,3-dimethylcyclopentan-1-one (1.95 g, 1 equiv., 17.4 mmol) in methanol (60 mL) at 0 °C, NaBH_4_ was added (789 mg, 1.2 equiv., 20.9 mmol) in small batches over 1 min. The mixture was stirred at RT for 3 h and then partitioned between water (75 mL) and Et_2_O (75 mL). The organic layer was washed with brine, dried over Na_2_SO_4_, filtered and concentrated in vacuo. The crude mixture was purified by column chromatography (40 g silica, 0–4% MeOH/DCM gradient) to obtain Product 17 (1.53 g, 77%) as a transparent oil. ^1^H NMR (400 MHz, CDCl_3_) had peaks at δ 4.38 (tdd, J = 9.1, 4.5, 3.2 Hz, 1H), 2.06–1.94 (m, 1H), 1.79 (dd, J = 13.4, 6.8 Hz, 1H), 1.69–1.59 (m, 2H), 1.50 (s, 1H), 1.44–1.34 (m, 2H), 1.11 (s, 3H) and 0.96 (s, 3H).
To a mixture of 3,3-dimethylcyclopentan-1-ol (17) (654 mg, 1 equiv., 5.73 mmol), 6-chloro-9H-purine (1.77 g, 2 equiv., 11.5 mmol), triphenylphosphine (3.00 g, 2 equiv., 11.5 mmol) and THF (40 mL), DIAD was added (2.23 mL, 2 equiv., 11.5 mmol) at RT. The mixture was heated to 65 °C for 16 h and then concentrated in vacuo. The crude extract was purified by column chromatography (120 g silica, 0–70% EtOAc/heptane gradient) to obtain impure Intermediate 18 (2.3 g). This material was dissolved in 1,4-dioxane (11.5 mL) and transferred to a pressure tube, then ammonia was added (11.5 mL, 25 wt%, 133 mmol) and then the resulting mixture was heated to 100 °C for 16 h. The reaction mixture was partitioned between EtOAc and brine, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na_2_SO_4_, filtered and concentrated in vacuo. The residue was purified by column chromatography (40 g silica, 0–4% MeOH/DCM gradient) to obtain Product A4 (740 mg, 3.20 mmol, 56% yield over 2 steps) as a white solid (Scheme 7). ^1^H NMR (400 MHz, CDCl_3_) had peaks at δ 8.36 (s, 1H), 7.89 (s, 1H), 5.83 (s, 2H), 5.02 (dq, J = 9.7, 8.1 Hz, 1H), 2.43 (dtd, J = 13.9, 7.9, 6.3 Hz, 1H), 2.20–2.12 (m, 1H), 2.11–2.02 (m, 1H), 1.92–1.75 (m, 2H), 1.63 (dtd, J = 13.0, 7.7, 0.8 Hz, 1H), 1.20 (s, 3H) and 1.12 (s, 3H). ^13^C NMR (101 MHz, CDCl_3_) had peaks at δ 155.4, 152.5, 150.2, 138.7, 120.0, 55.2, 47.4, 39.2, 38.3, 32.4, 30.2 and 29.2. Compound A4 was dissolved in 98 wt% D_2_SO_4_ and DMSO-d_6_ (9/1 v/v), and the NMR spectra were measured: ^1^H NMR (400 MHz, D_2_SO_4_), δ 8.58 (d, J = 4.0 Hz, 1H), 8.14 (d, J = 4.1 Hz, 1H), 4.57 (td, J = 9.1, 3.5 Hz, 1H), 2.04–1.86 (m, 1H), 1.73–1.62 (m, 1H), 1.62–1.49 (m, 1H), 1.37–1.18 (m, 2H), 1.10–0.97 (m, 1H), 0.55 (d, J = 3.9 Hz, 3H) and 0.46 (d, J = 3.8 Hz, 3H); ^13^C NMR (101 MHz, D_2_SO_4_), δ 153.2, 152.3, 151.5, 147.3, 115.2, 66.4, 51.3, 43.9, 43.6, 36.6, 34.5, and 33.5.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Benoit R.L. Frechette M. 1H and 13C nuclear magnetic resonance and ultraviolet studies of the protonation of cytosine, uracil, thymine, and related compounds Can. J. Chem.1986642348235210.1139/v 86-387 · doi ↗
- 2Albright L.F. Houle L. Sumutka A.M. Eckert R.E. Alkylation of isobutane with butenes: Effect of sulfuric acid compositions Ind. Eng. Chem. Process Des. Dev.19721144645010.1021/i 260043 a 020 · doi ↗
- 3Habeeb A. The reaction of sulphuric acid with lysozyme and horse globin Can. J. Biochem. Physiol.196139314310.1139/o 61-00513710223 · doi ↗ · pubmed ↗
- 4Miron S. Lee R.J. Molecular Structure of Conjunct Polymers J. Chem. Eng. Data 1963815016010.1021/je 60016 a 043 · doi ↗
- 5Reitz H.C. Ferrel R.E. Fraenkel-Conrat H. Olcott H.S. Action of sulfating agents on proteins and model substances. I. Concentrated sulfuric acid J. Am. Chem. Soc.1946681024103110.1021/ja 01210 a 03620985614 · doi ↗ · pubmed ↗
- 6Schumacher M. Günther H. Beiträge zur 15N-NMR-Spektroskopie Protonierung und Tautomerie in Purinen: Purin und 7-und 9-Methylpurin Chem. Ber.19831162001201410.1002/cber.19831160526 · doi ↗
- 7Steigman J. Shane N. Micelle Formation in Concentrated Sulfuric Acid as Solvent J. Phys. Chem.19656996897310.1021/j 100887 a 047 · doi ↗
- 8Städeli W. Bigler P. von Philipsborn W. 15N, 1H coupling constants in pyridines and pyrimidines An application of the INEPT pulse sequence Org. Magn. Reson.19811617017210.1002/mrc.1270160223 · doi ↗