Pharmacogenetic Associations with Statin Regimen Modification, Intolerance, and Adverse Outcomes in Coronary Artery Disease Patients
Rania Abdel-latif, Shaban Mohammed, Mohamad Saad, Khalid Kunji, Wadha Al-Muftah, Ayman El-Menyar, Jassim Al Suwaidi

TL;DR
This study examines how genetic factors influence statin use and side effects in heart disease patients, finding that clinical factors are more important than genetic ones in adjusting statin regimens.
Contribution
The study evaluates the real-world impact of cumulative pharmacogenetic burden on statin regimen changes and adverse outcomes.
Findings
PGx burden was not statistically significantly associated with statin intolerance or adverse outcomes.
Higher PGx burden showed a directional tendency toward dose de-escalation and lower likelihood of escalation.
Clinical factors, like statin intensity and myopathy, were the main drivers of regimen modification.
Abstract
Background: Statins are central to primary and secondary prevention of atherosclerotic cardiovascular disease but are often underutilized due to myopathy and intolerance. While individual pharmacogenetic (PGx) variants, particularly in SLCO1B1, are linked to statin-associated muscle symptoms, the real-world impact of both clinical and cumulative PGx burden on regimen modification and adverse outcomes remains unclear. We aimed to evaluate the existing uncertainty regarding whether combined PGx scores can effectively guide statin dose titration and regimen modification, thereby filling a key clinical gap. Methods: A retrospective cohort study of 911 statin-treated patients with coronary artery disease was conducted from the Qatar Cardiovascular Biorepository with available whole-genome sequencing data. Variants in SLCO1B1, ABCG2, and CYP2C9 were combined into a functional PGx burden…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
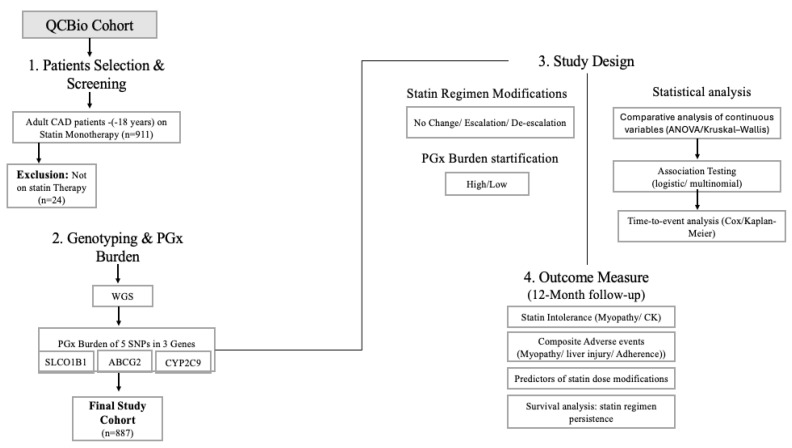 Figure 1
Figure 1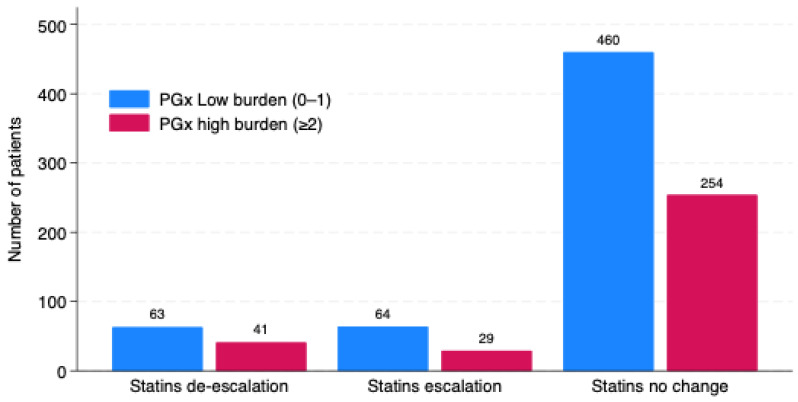 Figure 2
Figure 2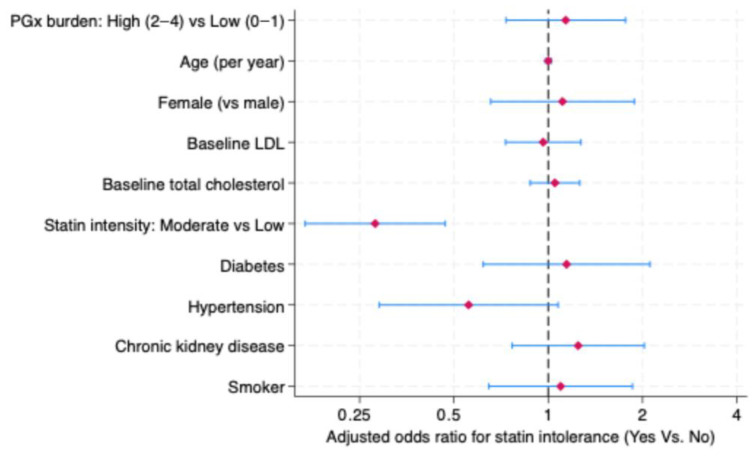 Figure 3
Figure 3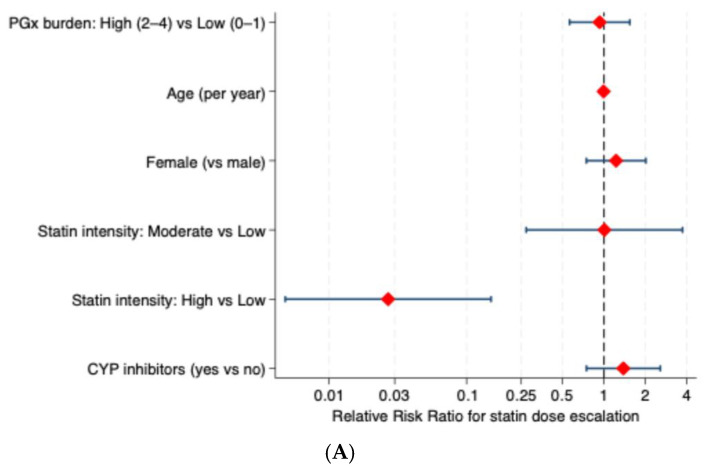 Figure 4
Figure 4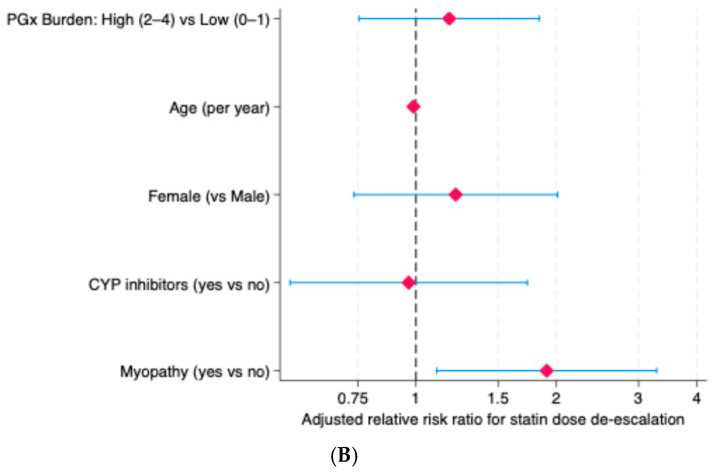 Figure 5
Figure 5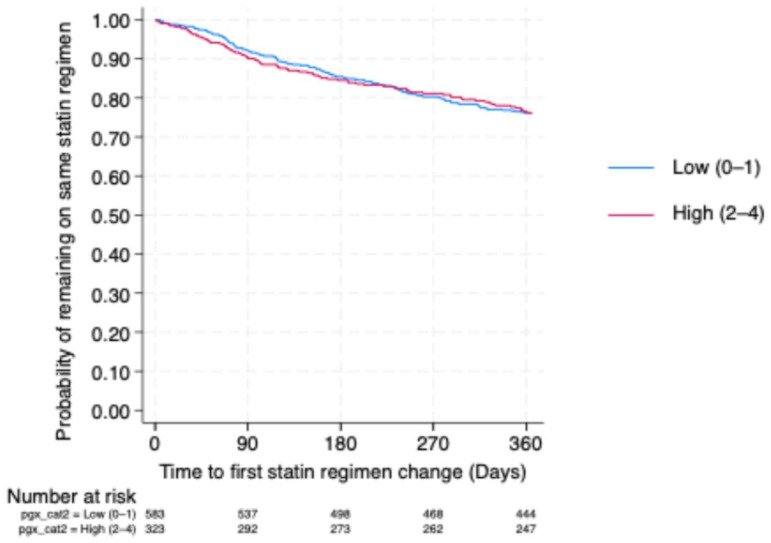 Figure 6
Figure 6- —Qatar Precision Health Institute
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLipoproteins and Cardiovascular Health · Pharmacogenetics and Drug Metabolism · Genetic Associations and Epidemiology
1. Introduction
Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, are the cornerstone of lipid-lowering therapy and are widely prescribed for the primary and secondary prevention of atherosclerotic cardiovascular disease [1,2]. By inhibiting the rate-limiting step of hepatic cholesterol biosynthesis, statins effectively lower plasma low-density lipoprotein cholesterol (LDL-C) levels, resulting in substantial reductions in cardiovascular morbidity and mortality, as consistently demonstrated in large randomized clinical trials [3,4]. Despite their well-established efficacy and generally favorable safety profile, statin intolerance remains a significant clinical challenge. In routine clinical practice, up to 30–50% of patients discontinue or modify statin therapy within 1–2 years, most commonly due to statin-associated muscle symptoms (SAMS) [5,6,7]. SAMS is influenced by multiple factors, including drug–drug interactions, comorbidities, and genetic variability, leading to poor adherence and increased residual cardiovascular risk [8,9]. In 2020, the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline highlighted the role of multiple pharmacokinetic genes, including SLCO1B1, ABCG2, and CYP2C9, in guiding statin therapy, underscoring the extensive investigations into the contribution of genetic variability to interindividual susceptibility to SAMS [10]. The strongest and most consistent evidence relates to SLCO1B1, where the reduced-function c.521T>C (*5, p.V174A, rs4149056) variant impairs hepatic uptake via OATP1B1, leading to higher circulating statin concentrations and a well-established increase in myopathy risk, especially with simvastatin and, to a lesser extent, atorvastatin [11,12]. In addition, the c.388A>G variant (rs2306283) has been reported to modify SLCO1B1 transport activity and, in combination with rs4149056, further influence the plasma concentration of the statins [13,14]. Consistent with this evidence, CPIC guidelines classify carriers of SLCO1B1 decreased or poor function phenotypes, including the *5 and *15 haplotypes, as being at increased risk for SAMS. These guidelines recommend avoiding simvastatin and favoring lower doses or alternative statins, with consideration of creatine kinase monitoring when clinically indicated [10]. Variants in ABCG2, most notably c.421C>A (rs2231142), reduce efflux activity of the breast cancer resistance protein (BCRP), leading to increased systemic exposure to several statins, specifically atorvastatin and rosuvastatin [15,16]. CYP2C9 reduced-function alleles (*2, *3) influence the metabolism of statins such as fluvastatin and contribute modestly to variability in statin exposure [17]. Evidence linking these variants to SAMS is limited, but CPIC incorporates CYP2C9 genotype into dosing recommendations to minimize the risk of toxicity, particularly in the presence of interacting drugs [10]. The functional impact and clinical relevance of SLCO1B1, ABCG2, and CYP2C9 variants in statin therapy are summarized in Table 1.
Given the multifactorial nature of SAMS, genetic predisposition alone, particularly when limited to a single SLCO1B1 variant, is insufficient to explain clinical intolerance [18]. Beyond genetics, SAMS risk is influenced by concomitant medications, comorbid conditions, adherence behavior, and other real-world treatment factors [19]. Clinicians often manage statin-associated complications by transitioning patients from lipophilic agents to hydrophilic alternatives or to agents with a distinct metabolic profile. These swaps can minimize certain drug–drug interactions, but do not resolve the substantial interindividual variability in systemic exposure due to pharmacogenetic factors, along-side physiological and environmental determinants [20,21,22]. Although the pharmacokinetic effects of key statin pharmacogenetics are well established, most prior studies have focused on single-gene associations or statin-specific toxicity rather than cumulative genetic susceptibility within real-world prescribing contexts. Moreover, the clinical relevance of integrating multiple actionable variants with drug–drug interactions and patient-level characteristics remains incompletely defined [18]. Therefore, we investigated the combined impact of SLCO1B1, ABCG2, and CYP2C9 genotypes, along with clinical and treatment factors, on statin intolerance and statin regimen modification in a large Middle Eastern cohort of patients with coronary artery disease (CAD) receiving statins for secondary prevention. This study aims to capture gene–drug–environment interactions and advance the translation of pharmacogenetics (PGx) into personalized statin therapy. As there is limited evidence, we sought to explore whether integrating multiple actionable PGx variants can meaningfully inform statin dose titration and regimen modification in routine clinical practice.
2. Results
2.1. Baseline Clinical and Treatment Characteristics
A total of 911 patients were included, of whom 10.21% underwent dose escalation, 11.42% underwent dose de-escalation, and 78.38% remained on the same statin drug and dose throughout a 12-month follow-up period. Figure 1 shows the study workflow.
Baseline demographic characteristics were largely comparable across groups, with no significant differences in age or smoking status. The proportion of males differed significantly, with the escalation group showing the lowest representation (60.22%, p = 0.025). Among comorbidities, asthma was most frequent in the escalation group (29.55%, p = 0.023), whereas diabetes, hypertension, chronic kidney disease, and heart failure showed no significant between-group differences.
For laboratory parameters, triglyceride levels demonstrated a statistically significant difference (p = 0.003), with higher levels observed particularly in the de-escalation group. In contrast, levels of cholesterol, high-density lipoprotein (HDL), low-density lipoprotein (LDL), creatinine, Alanine transaminase (ALT), and Aspartate transaminase (AST) were similar across groups.
Treatment-related characteristics showed the most apparent separation. Statin type distribution differed significantly (p = 0.001), with atorvastatin being most prescribed, but rosuvastatin was more frequent in the de-escalation group. Statin intensity also varied markedly (p = 0.001): the escalation group mostly initiated therapy at moderate intensity (79.7%), whereas the de-escalation group predominantly received high-intensity statins (75.8%). Concomitant use of enzyme inhibitors or inducers did not differ significantly between groups (Table 2).
2.2. Pharmacogenomic Profiles and Their Distribution Across Statin Regimen Change Groups
Genotype frequencies for SLCO1B1 and ABCG2 were broadly similar across the three-statin regimen change groups, with no statistically significant differences observed. In contrast, CYP2C9 reduced-function alleles were significantly more prevalent in the escalation group, occurring 1.3 times more frequently than in the other groups. Specifically, the escalation group had a notable 43.01% proportion of reduced-function carriers (p = 0.006) (Table 3).
Phenotype-based classification of SLCO1B1 diplotypes showed no significant association with statin regimen change (p = 0.49). Normal-function phenotypes accounted for approximately two-thirds of each group, whereas decreased- and poor-function phenotypes were distributed similarly, with no meaningful deviation (Table 4).
The distribution of the PGx burden score (0–4), representing the cumulative number of risk alleles across key statin-related genes, as shown in Figure 2, showed a predominance of low burden (scores 0–1) in all groups. A similar pattern across the escalation, de-escalation, and no-change groups was observed, without evidence of enrichment for higher PGx burden in any specific regimen-change category.
2.3. Clinical Outcomes and Muscle/Liver Biomarkers Across Statin Regimen Change Groups and Their Association with Pharmacogenomic Burden
Across statin regimen groups, rates of myopathy changed significantly (p = 0.027), with the highest rate observed in the de-escalation change group (23.08%). PGx burden categories showed no meaningful association with myopathy risk (all p > 0.44). Statin escalation was significantly associated (p = 0.002) with a higher frequency of liver injury, particularly among patients with low PGx burden. Poor adherence was most prevalent among patients whose statin regimen was not modified, particularly in those with low PGx burden. Table 5 shows that muscle and liver biomarkers did not differ significantly between statin regimen groups. Across all biomarkers, PGx burden categories showed no statistically significant associations.
2.4. PGx Burden and Composite Outcomes
As shown in Table 6, the composite adverse outcome occurred similarly in patients with low PGx burden (0–1) and those with high PGx burden (2–4). Composite events were observed in 40.7% of the low-burden group and 42.6% of the high-burden group. In the unadjusted analysis, patients with a high PGx burden showed a trend toward a higher risk of composite adverse outcomes; however, this did not reach statistical significance (OR = 1.08; 95% CI: 0.82–1.42; p = 0.582).
2.5. Multivariable Predictors of Statin Intolerance
Patients with a high PGx burden showed a numerically higher risk of statin intolerance compared with those with a low burden. However, this association did not reach statistical significance (adjusted OR = 1.14; 95% CI: 0.73–1.76). Initiating a moderate-intensity statin was independently associated with lower odds of developing statin intolerance (OR = 0.28; 95% CI: 0.17–0.47). High-intensity statin therapy was omitted from the model due to collinearity. No significant associations were observed for age, gender, lipid parameters, cardiometabolic comorbidities, or smoking status after multivariate adjustment (Figure 3).
2.6. Predictors of Statin Dose Modification
PGx burden was not significantly associated with either statin dose escalation or de-escalation relative to no change. However, the direction of effect differed by outcome, with high PGx burden showing a tendency toward dose de-escalation (adjusted RRR = 1.18, 95% CI: 0.76–1.84) and a lower relative likelihood of dose escalation (adjusted RRR = 0.93, 95% CI: 0.56–1.54), although neither association reached statistical significance (Figure 4).
For escalation, Baseline statin intensity showed a strong differential effect. High-intensity statin therapy was associated with a substantially lower relative risk of subsequent dose escalation compared with low-intensity therapy (adjusted RRR = 0.03, 95% CI: 0.005–0.15), whereas moderate-intensity statin therapy was not significantly associated with escalation risk (adjusted RRR = 1.01, 95% CI: 0.27–3.73). Other demographic or clinical covariates did not independently influence escalation decisions in this cohort. Furthermore, Statin-associated myopathy was independently associated with an increased likelihood of dose de-escalation (adjusted RRR = 1.91, 95% CI: 1.11–3.28), indicating that clinical intolerance, rather than pharmacogenetic burden, primarily drove dose reduction decisions.
2.7. Association of Pharmacogenetic Burden with Time to Regimen Change
At 12 months, the estimated probability of remaining on the initial statin regimen was approximately 75–78% in both PGx burden categories. The survival curves for both groups largely overlapped throughout the follow-up period, with no evident early separation or divergence over time (Figure 5). Consistent with this, no significant association was observed between PGx burden and regimen persistence (HR 1.02, 95% CI: 0.77–1.35; p = 0.884).
3. Discussion
This study evaluated the influence of genetic and clinical factors on statin regimen modification and statin-associated adverse outcomes in a large real-world Middle Eastern CAD cohort. By focusing on well-characterized statin pharmacogenetics and functional alleles, alongside clinically relevant treatment variables, we sought to clarify how genetic susceptibility and routine prescribing practices interact to shape statin use, tolerability, and downstream outcomes in everyday clinical care. While CPIC recommendations are primarily statin- and variant-specific [10], a cumulative multi-gene approach may better reflect the multifactorial nature of statin disposition, particularly in real-world settings where multiple pathways influence exposure and tolerability.
To capture cumulative genetic susceptibility, we constructed a weighted functional PGx burden score that reflects the relative contributions of key statin disposition pathways. SLCO1B1 was assigned a greater weight because impaired OATP1B1-mediated hepatic uptake represents the most consistently validated and clinically impactful mechanism underlying statin-associated muscle toxicity [10,12,23]. Although numerous SLCO1B1 variants have been described, functional effects are largely driven by two common non-synonymous SNPs (c.388A>G and c.521T>C), with the latter defining the *5 and *15 haplotypes associated with reduced transporter function due to altered hepatic trafficking [23].
Notably, population-level analyses from the Qatar Genome Program have demonstrated that the minor allele frequency of SLCO1B1 c.521T>C in the Qatari population is relatively high compared with several global subpopulations (up to 0.2322), further underscoring its clinical relevance in this cohort. In contrast, ABCG2 c.421C>A exhibits lower frequency across most populations except East Asians. Additionally, CYP2C92* and *3 alleles also demonstrate relatively low and stable frequencies across ancestries [24]. Accordingly, ABCG2 and CYP2C9 were weighed more modestly, reflecting their statin-specific or pathway-limited effects on efflux transport and metabolic clearance. This pragmatic approach balances biological plausibility, clinical relevance, and statistical stability, yielding a composite score applicable across statin types in real-world practice while acknowledging heterogeneity in gene–statin interactions.
In this cohort, most patients remained on their initial statin regimen during follow-up, with dose escalation or de-escalation occurring in a minority, consistent with the overall stability of statin prescribing in routine care [25]. Baseline demographic and comorbidity profiles were largely comparable across modification groups, suggesting that regimen changes were not primarily driven by global cardiovascular risk or systemic disease burden. Instead, statin-related characteristics emerged as the dominant determinants of dose modification. Escalation was strongly associated with initiation of moderate-intensity therapy, consistent with guideline-directed stepwise titration toward lipid targets [26]. Conversely, de-escalation was predominantly linked to high-intensity statin use, supporting the interpretation that dose reduction frequently reflects tolerability concerns or biochemical changes rather than a lack of efficacy [27]. Our multinomial regression analyses further clarified these patterns. De-escalation was associated with myopathy, reinforcing the central role of muscle-related symptoms in prompting dose reduction, whereas escalation was driven primarily by statin intensity.
In the current cohort, none of the group studies showed high PGx burden mirroring the lack of genotype-driven dosing in practice. This finding further supports the argument that PGx burden does not distinctly influence regimen changes, underscoring the potential gap in applying genetic insights to clinical decision-making
Although high PGx burden was not statistically associated with dose escalation or de-escalation, patients with higher PGx burden showed a greater likelihood of de-escalation and a lower likelihood of escalation. Importantly, this pattern is consistent with real-world prescribing behavior, in which dose intensification may be avoided or reduced in response to early clinical or laboratory signals, rather than being guided by pre-emptive PGx information [28,29]. The lack of statistical significance likely reflects a combination of limited statistical power, heterogeneity in statin exposure, and the absence of preemptive PGx-guided prescribing, suggesting that genetic risk may influence clinical practice indirectly or in an attenuated manner in real-world settings. Previous studies showed that genotype may influence some statins prescribing changes but not uniformly [29]. In support, Ferri et al. emphasize that genetic predisposition alone is insufficient, and clinical factors must also be considered while considering the multifactorial nature of SAMS [18].
This interpretation is further supported by the time-to-event analysis, which demonstrated comparable 12-month persistence on the initial statin regimen across PGx burden categories, with no evidence of differential regimen switching. Together, these findings reinforce the notion that cumulative PGx risk alone does not strongly influence decisions to modify statin therapy once treatment has been initiated in routine clinical practice. PGx burden reflects probabilistic variation in statin disposition that may increase exposure without consistently reaching clinical thresholds that prompt regimen changes, particularly when moderate-intensity statins or conservative dose titration strategies are employed [30,31].
Evaluation of adverse outcomes, including myopathy, liver injury, and non-adherence, provided additional insight into the clinical impact of PGx burden. Although de-escalation of doses was more frequently associated with myopathy, the overall influence of PGx burden on the composite adverse outcome, statin intolerance and biomarker profile was modest, suggesting that genetic susceptibility alone is insufficient to drive clinical events in the absence of overt manifestations.
These findings align with prior large-scale observational studies. Analyses from the UK Biobank and other real-world cohorts have shown that SLCO1B1 variants and myopathy exert largely independent effects, with no strong interaction detected, and that SLCO1B1 variation does not uniformly predict statin-associated muscle symptoms or tolerability [32,33]. On the other hand, some statin-specific analyses have demonstrated clearer associations between reduced-function SLCO1B1 alleles and intolerance, particularly for simvastatin; these effects appear to be exposure- and drug-dependent rather than class-wide [16,34,35]. In our stratified analyses of simvastatin and atorvastatin users, PGx burden was likewise not significantly associated with statin intolerance; however, consistently higher event rates suggest a directional tendency toward increased risk. Our findings are consistent with prior studies, underscoring that PGx is not a standalone predictor of adverse drug events but a complementary tool that, when integrated with clinical factors, helps identify at-risk patients and enables more precise, individualized therapy [36].
4. Materials and Methods
4.1. Study Population and Data Sources
Participants in this retrospective cohort study were recruited from the Qatar Cardiovascular Biorepository (QCBio) at Hamad Medical Corporation (HMC) [37], Doha, Qatar. This study involved patients with coronary artery disease from multiple sites, including the Cardiac Catheterization Laboratory, Coronary Care Unit, and Heart Hospital outpatient clinics. Participants underwent standardized phenotyping, clinical data collection, and biospecimen sampling were performed according to predefined biobank protocols. Genomic DNA was extracted using validated automated protocols and quantified prior to sequencing. Whole-genome sequencing was performed using high-throughput sequencing platforms. Variant calling, alignment to the reference genome, and quality-control filtering followed established pipelines as previously described [38].
The study protocol received approval from the Qatar Biobank Institutional Review Board (IRB Protocol #: E -2018-QF-QBB-RES-ACC-0077-0057). Written informed consent was obtained from all participants before enrollment in the QCBio cohort.
4.2. Statin User Identification
From the QCBio cohort, 911 statin users were identified. Statin use was defined as treatment with simvastatin, atorvastatin, pravastatin, rosuvastatin, or fluvastatin as monotherapy among QCBio participants.
4.3. Clinical and Medication Data Collection
Demographic characteristics, clinical diagnoses, laboratory measurements, and medication prescriptions were extracted from the electronic health records (EHR) of HMC. Detailed information on statin type, dose, treatment duration, and concomitant medications, with particular attention to drugs known to interact with statins, was obtained.
4.4. Assessment of Statin Exposure and Adherence
Pharmacy dispensing records were used to assess statin switching, discontinuation, and medication adherence. Adherence was estimated using the medication possession ratio (MPR), calculated as (MPR = cumulative medications dispensed within follow-up period/total days of follow-up). Patients with an MPR ≥ 80% were classified as adherent, whereas those with an MPR < 80% were classified as non-adherent, consistent with established definitions [39].
4.5. Definition of Statin Regimen Modification
Statin regimen changes were classified according to the American College of Cardiology (ACC) and the American Heart Association (AHA) statin intensity definitions [2]. Dose escalation was defined as a change from a lower- to a higher-intensity statin regimen, whereas dose de-escalation was defined as a change from a higher- to a lower-intensity regimen. No change was defined as continuation of the same statin type at the same dose, corresponding to unchanged statin intensity, throughout the follow-up period. Statin intensity categories and corresponding doses used for classification are summarized in Table 7.
4.6. Definition of Statin Intolerance Phenotypes
Biochemical evidence of statin intolerance was characterized by elevated creatine kinase (CK) levels, with sex-specific upper limits of normal of 120 IU/L for women and 180 IU/L for men [4,5]. Statin intolerance was further characterized by a 3-fold elevation of CK, accompanied by myopathy findings or statin dose deescalation or change in statin more than 3 times [40,41].
4.7. Definition of Myopathy and Composite Adverse Events
Recorded myopathy in EHR refers to disorders in which the primary pathology involves skeletal muscle, resulting in muscle weakness or dysfunction. Clinical phenotypes of myopathy were classified according to definitions adapted from ACC/AHA and the National Heart, Lung, and Blood Institute [42]. The spectrum of muscle-related adverse events included myalgia, myositis, rhabdomyolysis, asymptomatic myopathy, and statin-induced myopathy [7]. The composite adverse events were defined as the occurrence of any statin-related adverse event, including statin-associated myopathy, liver injury, or poor medication adherence, during the follow-up period. Patients were classified as having experienced the composite outcome if at least one of these events occurred.
4.8. PGx Burden Score
We constructed a PGx burden score from five loci SLCO1B1 rs4149056 [12:21331549:T:C], SLCO1B1 rs2306283 [12:21329738:A:G], ABCG2 rs2231142 [4:89052323:G:T], CYP2C92* rs1799853 [10:96702047:C:T] and CYP2C93* rs4986893 [10:96540410:A:C] based on functional characterization and pharmacokinetic studies [10,20,43]. A PGx burden score was calculated based on the established functional patterns of the incorporated alleles. SLCO1B1 was modeled using diplotype-derived transporter function phenotype, with normal function assigned 0 points, decreased function 1 point, and poor function 2 points. rs2231142 homozygosity was rare; therefore, ABCG2 was modeled under a dominant framework where 0 was assigned for non-carriers, and 1 was assigned for any variant allele carrier to ensure stable estimation and avoid sparse-data bias. Similarly, CYP2C9 reduced-function alleles (*2 or *3) were coded under a dominant model (0 = *1/*1; 1 = any reduced-function allele) to capture any reduction in enzymatic activity relevant to statin metabolism. The overall PGx burden score was calculated as the sum of these components. The weighted functional score was explored, assigning greater weight to SLCO1B1 due to its more substantial and more consistent evidence for clinically relevant statin exposure and myopathy risk [10] (Table 8).
4.9. Statistical Analysis
Continuous variables were summarized as mean ± standard deviation (SD) or median (interquartile range; IQR), as appropriate, and compared across statin regimen-change groups using one-way ANOVA or the Kruskal–Wallis test. Categorical variables were summarized as counts and percentages and compared using the χ^2^ test. Associations between PGx variables and clinical outcomes were assessed using χ^2^ tests. Multivariable logistic regression models were constructed to assess the independent association between PGx burden and statin intolerance. Multinomial logistic regression was used to evaluate predictors of statin regimen modification. Multinomial and logistic regression analyses were adjusted for demographic and clinical covariates, including age, sex, baseline LDL-C levels, diabetes status, baseline statin intensity, and concomitant medications, to account for potential confounding effects. Time to first statin regimen change within 12 months was analyzed using Cox proportional hazards models, with Kaplan–Meier curves for visualization and the log-rank test for comparison. Data were expressed as odds ratio (OR), relative risk ratio (RRR), Hazard ratio (HR), and 95% confidence interval (CI); whenever applicable. Analyses were performed using Stata version 18.5 (StataCorp, College Station, TX, USA), with p < 0.05 considered statistically significant.
Study limitation: The retrospective nature of this study and single-center data are limitations that cannot rule out the selection bias and unmeasured confounders. This study was conducted in a real-world CAD population with substantial comorbidity and polypharmacy, which may have attenuated the independent effects of PGx burden despite multivariable adjustment. Although the performed analyses were based on prespecified hypotheses, we acknowledge that multiple statistical comparisons were performed, and findings should be interpreted cautiously within the context of the overall study design and the specific population examined. The multiple comparisons could generate a type I error, and the findings should be interpreted cautiously. Furthermore, statin intolerance and myopathy were identified from EHR data and laboratory thresholds, which may not fully capture patient-reported muscle symptoms. It is worth noting that the cohort was predominantly treated with atorvastatin and rosuvastatin, limiting statin-specific analyses, particularly for simvastatin, where PGx effects are most pronounced. In addition, PGx testing was not implemented preemptively, and prescribing decisions were made without access to genetic information, likely reducing the observable clinical impact of PGx burden.
5. Conclusions
The study findings suggest that PGx burden may shape the direction of treatment modification, favoring de-escalation over escalation rather than the overall frequency of regimen changes. The modest effect observed in this retrospective, non-genotype-guided cohort likely reflects routine clinical practice where dosing decisions were not informed by PGx results. The clinical impact of PGx information is therefore likely to be maximized when integrated into structured PGx-guided workflows, combined with standardized intolerance phenotyping, adherence assessment, and exposure-matched dosing strategies. Prospective studies incorporating pre-emptive PGx testing and clinical decision support are needed to clarify the utility of PGx burden in optimizing statin therapy and reducing adverse outcomes in routine practice.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tahaineh L. Alsou’b N. Adherence to the 2018 American College of Cardiology and American Heart Association Guideline on the Management of Blood Cholesterol in ambulatory care settings: A cross-sectional study Pharmacia 2024711710.3897/pharmacia.71.e 125449 · doi ↗
- 2Grundy S.M. Stone N.J. Bailey A.L. Beam C. Birtcher K.K. Blumenthal R.S. Braun L.T. De Ferranti S. Faiella-Tommasino J. Forman D.E. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/A Ph A/ASPC/NLA/PCNA guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines J. Am. Coll. Cardiol.201973 e 285e 35010.1016/j.jacc.2018.11.00330423393 · doi ↗ · pubmed ↗
- 3Stone N.J. Robinson J.G. Lichtenstein A.H. Bairey Merz C.N. Blum C.B. Eckel R.H. Goldberg A.C. Gordon D. Levy D. Lloyd-Jones D.M. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines J. Am. Coll. Cardiol.2014632889293410.1016/j.jacc.2013.11.00224239923 · doi ↗ · pubmed ↗
- 4Cholesterol Treatment Trialists Collaboration Baigent C. Blackwell L. Emberson J. Holland L.E. Reith C. Bhala N. Peto R. Barnes E.H. Keech A. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials Lancet 20103761670168110.1016/S 0140-6736(10)61350-521067804 PMC 2988224 · doi ↗ · pubmed ↗
- 5Rodriguez F. Maron D.J. Knowles J.W. Virani S.S. Lin S. Heidenreich P.A. Association of statin adherence with mortality in patients with atherosclerotic cardiovascular disease JAMA Cardiol.2019420621310.1001/jamacardio.2018.493630758506 PMC 6439552 · doi ↗ · pubmed ↗
- 6Toth P.P. Granowitz C. Hull M. Anderson A. Philip S. Long-term statin persistence is poor among high-risk patients with dyslipidemia: A real-world administrative claims analysis Lipids Health Dis.20191817510.1186/s 12944-019-1099-z 31526399 PMC 6747753 · doi ↗ · pubmed ↗
- 7Abed W. Abujbara M. Batieha A. Ajlouni K. Statin Induced Myopathy Among Patients Attending the National Center for Diabetes, endocrinology, & genetics Ann. Med. Surg.20227410330410.1016/j.amsu.2022.10330435145672 PMC 8818528 · doi ↗ · pubmed ↗
- 8Neuvonen P.J. Niemi M. Backman J.T. Drug interactions with lipid-lowering drugs: Mechanisms and clinical relevance Clin. Pharmacol. Ther.20068056558110.1016/j.clpt.2006.09.00317178259 · doi ↗ · pubmed ↗