Pediatric Sjögren Disease: Clinical Features, Diagnostic Challenges, and Outcomes in a Single-Centre Romanian Case Series
Mihaela Sparchez, Ioana Filimon, Mirela Crisan, Lidia Man, Simona Corina Senila, Ionut Iarca, Laura Banias, Andreea Liana Bot (Rachisan)

TL;DR
This study examines the clinical features and diagnostic challenges of childhood-onset Sjögren disease in a Romanian patient group, highlighting the need for better pediatric criteria.
Contribution
The study reports novel clinical presentations and emphasizes the diagnostic challenges of cSjD, particularly its overlap with lupus.
Findings
Extraglandular symptoms were more common than glandular symptoms at diagnosis in children with Sjögren disease.
Lupus-like features led to initial misdiagnosis as systemic lupus erythematosus in several patients.
Salivary gland ultrasound abnormalities and anti-SSA antibodies were highly prevalent in the cohort.
Abstract
Background/Objectives: Childhood-onset Sjögren disease (cSjD) is a rare autoimmune disorder with heterogeneous manifestations and ongoing diagnostic challenges, as there are no validated paediatric criteria. Our study aims to characterise the clinical, laboratory, and imaging features of children diagnosed with cSjD at a single Romanian paediatric rheumatology centre between 2015 and 2025 and contextualise these findings within the most recent literature. Methods: A retrospective review of 15 consecutive cSjD patients was conducted, including clinical features, autoantibodies, imaging, biopsy findings, treatment, and outcomes. Results: Our cohort showed a significant female predominance (80%) and a broad age range at disease onset (3–15 years). Extraglandular manifestations were more common at presentation than glandular phenotypes (53.3% vs. 40%). Lupus-like extraglandular…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
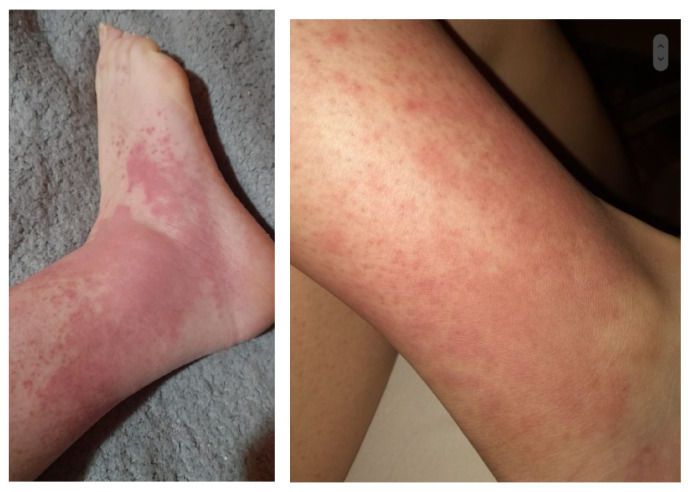 Figure 1
Figure 1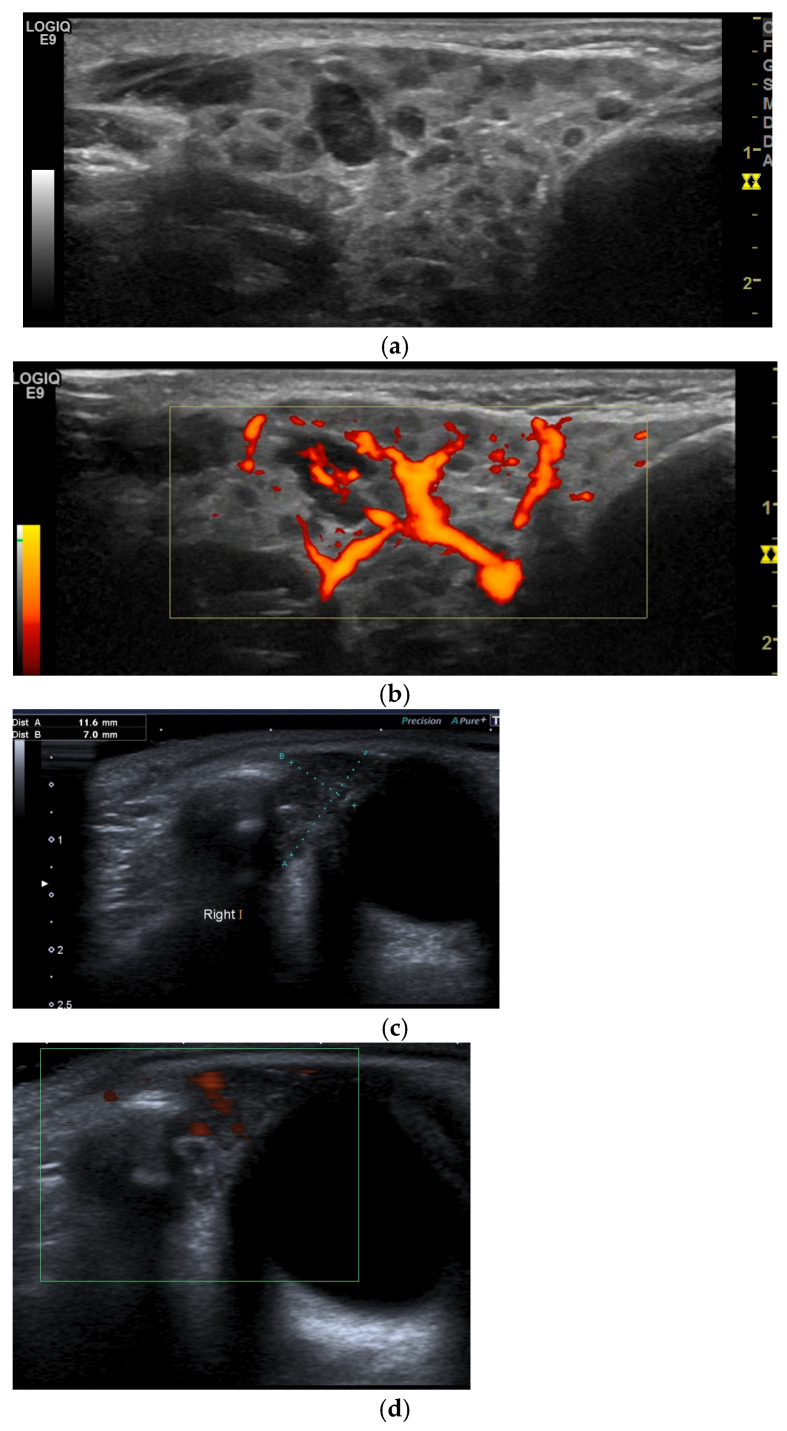 Figure 2
Figure 2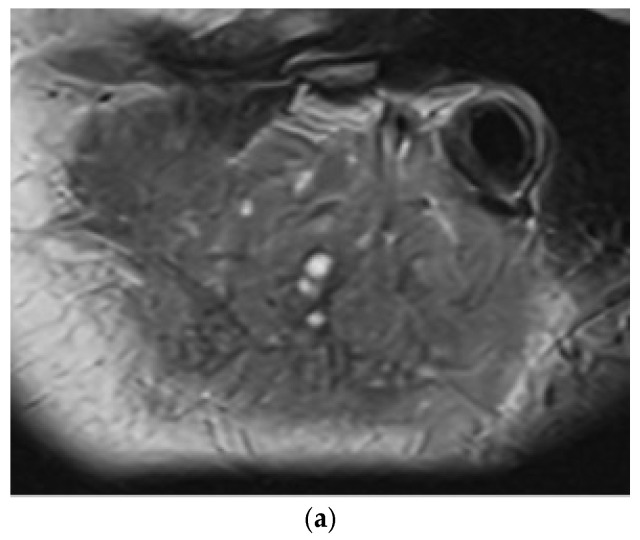 Figure 3
Figure 3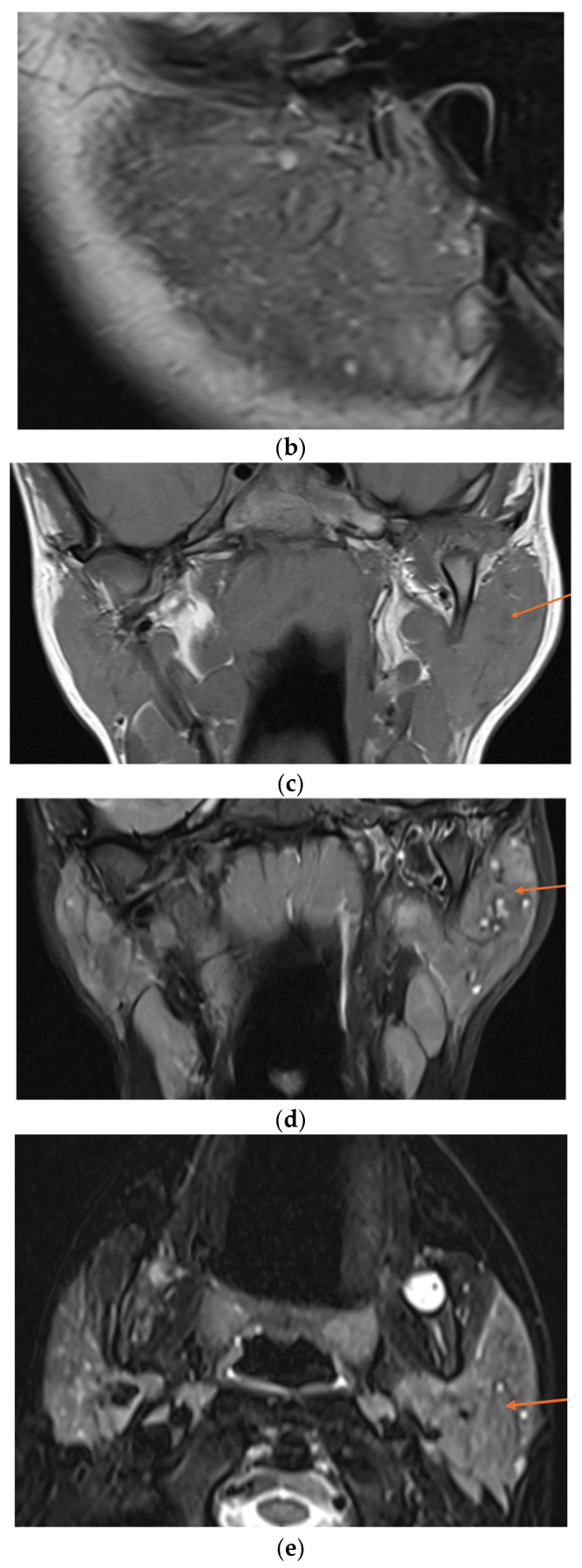 Figure 4
Figure 4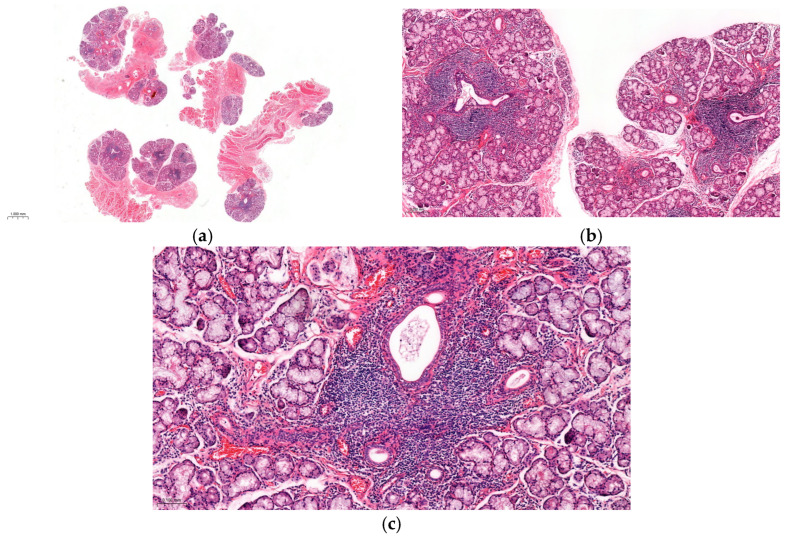 Figure 5
Figure 5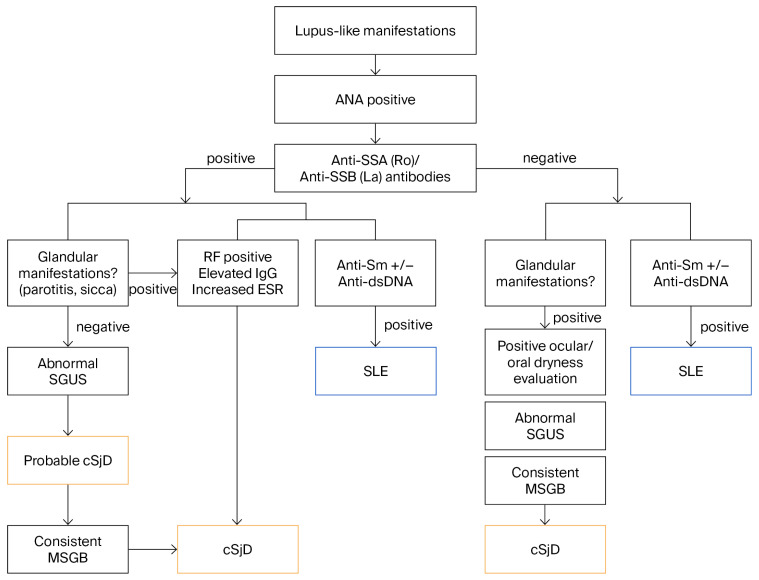 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSalivary Gland Disorders and Functions · Systemic Lupus Erythematosus Research · Diabetes and associated disorders
1. Introduction
Childhood-onset Sjögren’s disease (cSjD) is a rare autoimmune exocrinopathy, comprising about 1% of all Sjögren’s disease cases [1]. Compared to adults, children present with a distinct clinical profile, characterised by more frequent recurrent parotid gland enlargement and systemic symptoms, and fewer sicca symptoms at disease onset [2,3,4,5,6,7,8]. Disease can start as early as 4 years old, although most patients are diagnosed between 5 and 13 years [9,10]. Although recent multicentre studies have significantly broadened the current understanding, cSjD remains underrecognised, as its clinical presentation is highly variable [2,3,4,5,6,7,8].
Adult classification criteria, including the 2016 ACR/EULAR criteria [11], are inadequate for diagnosing SjD in children [3,8,12]. As a result, paediatric-specific diagnostic approaches have been proposed, culminating in a recently published clinical algorithm tailored to cSjD [13,14,15]. Objective investigations—such as serology, minor salivary gland biopsy (MSGB), and ocular or salivary functional tests—are used when feasible but lack validated reference standards for this age group, diagnosis often relies on expert clinical judgment [8,15]. The proposed algorithm emphasises the early use of non-invasive salivary gland ultrasonography (SGUS), which has emerged as a valuable diagnostic and monitoring tool in paediatric disease [15]. Given its invasive nature, MSGB is not routinely indicated; however, it should be considered in cases with atypical or concerning features [15].
The British Society of Rheumatology recently published the first guideline for the management of SjD, including children; however, evidence in children and adolescents remains limited, and most conventional immunosuppressive drugs are used off-label in children [16]. Long-term care focuses on monitoring systemic complications, preventing irreversible damage, and preserving quality of life through multidisciplinary follow-up [3,8,15].
This study aims to describe a single-centre paediatric cohort with cSjD, highlighting the heterogeneity of clinical presentation, diagnostic challenges, and therapeutic strategies, and to contextualise our findings within the existing paediatric literature.
2. Materials and Methods
2.1. Study Design
A retrospective observational study was conducted at the Paediatric Rheumatology Division of the Emergency Clinical Hospital for Children Cluj-Napoca, Romania, a tertiary referral centre. Eligible participants were all consecutive patients diagnosed with cSjD between 2015 and 2025, based on a combined expert opinion that considered clinical evaluation supported by autoantibodies, glandular imaging, or MSGB. Patients with other rheumatic diseases accompanied by potential cSjD overlap were excluded.
We also assessed all newly referred patients during the studied period with suspected cSjD. For those in whom the diagnosis was not confirmed, we reviewed and classified their characteristics and final diagnoses.
Upon admission to our clinic for routine clinical appointments, written informed consent was obtained from parents to use anonymised clinical data.
2.2. Data Collection
For the cSjD patients included in the study, the following data were collected from electronic medical records: demographics; presenting symptoms, signs, and additional disease manifestations developed since presentation to the last assessment, defined as per ESSDAI glossary [17]; laboratory results (ANA, SSA, SSB, RF, IgG, C3, C4, FBC, urinalysis); SGUS and/or magnetic resonance imaging (MRI); ophthalmological evaluation; MSGB analysis; additional autoimmune conditions; and treatment strategies ever used for cSjD and follow-up outcomes. We evaluated patient-reported outcomes, disease activity, and damage at the last study assessment using validated adult instruments: the EULAR Sjögren’s Syndrome Patient-Reported Index (ESSPRI) [18], the EULAR Sjögren’s Syndrome Disease Activity Index (ESSDAI) [19], and the Sjögren’s Syndrome Disease Damage Index (SSDDI) [20].
2.3. Evaluation of Sicca Syndrome
For ocular dryness, objective signs were documented by an experienced ophthalmologist and deemed sufficient. When possible, a Schirmer test was performed to corroborate the clinical findings.
Oral dryness was evaluated based on subjective symptoms, objective evidence of tongue or gingival mucosal changes consistent with hyposalivation as assessed by an experienced oral medicine specialist, or the presence of recurrent dental caries suggestive of salivary dysfunction.
2.4. Ultrasound Evaluation
An experienced ultrasonographer examined the major salivary glands (parotid, submandibular, and, when indicated, lacrimal) using a high-resolution linear transducer (9–15 MHz) to identify abnormal gland architecture at the initial clinic visit and during follow-up assessments as needed. An abnormal SGUS consistent with SjD, such as an inhomogeneous echotexture ranging from a finely granular pattern to multiple hypoechoic areas, with irregular glandular contours, increased parenchymal Doppler flow, and possible hyperechoic fibrotic bands, was considered positive, as SGUS interpretation in children has not been established [15].
For this study, we applied the semiquantitative OMERACT scoring system [21] and reanalysed archived ultrasound images or video clips from the first presentation when available. Accordingly, the scores were defined as follows: score 0, normal parenchyma; score 1, mild inhomogeneity without anechoic or hypoechoic areas and hyperechogenic bands; score 2, moderate inhomogeneity with focal anechoic or hypoechoic areas; and score 3, severe inhomogeneity with diffuse anechoic or hypoechoic areas occupying the entire gland or a fibrous gland [21].
2.5. Histological Evaluation
The MSGB was considered confirmatory for cSjD in the presence of focal lymphocytic sialadenitis (focus score ≥ 1 per 4 mm^2^) [11].
3. Results
3.1. Patient Characteristics
A total of 15 children were identified, 12 (80%) were female, with a median age at symptom onset of 9 years (range 3–15) and a median follow-up of 24 months (range 2 to 56 months) (Table 1). The entire cohort consisted of individuals of White Caucasian ancestry. One patient had a maternal history of SjD, and three additional patients reported a family history of other autoimmune disorders. Thirteen of the 15 individuals ([86.7%]) were diagnosed between 2022 and 2025. Two of them had positive serology for coeliac disease (patients 8 and 13).
3.2. Clinical Presentation at Disease Onset
The most common initial manifestations were extraglandular features, observed in eight patients (53.3%). These included persistent or recurrent facial rash in 5 of 15 patients (33%), arthralgia or arthritis in 3 of 15 (20%), recurrent self-limited episodes of cutaneous vasculitis affecting the lower legs with a typical duration of 3–4 days in 1 of 15 (6.7%) (Figure 1), and recurrent pericarditis in 1 of 15 (6.7%) (Table 1).
Glandular manifestations were present before or at initial presentation in six patients (40%), comprising recurrent parotitis in five patients (33%) and lacrimal gland swelling in one patient (6.7%). Sicca symptoms were observed at diagnosis in 3 of 15 patients (20%), with 1 patient experiencing a prolonged disease course of 7 years before diagnosis. Additionally, oral and/or ocular dryness developed during follow-up in four patients (patients 1, 4, 9, and 10).
Fatigue was reported as an accompanying symptom at presentation in 3 of 15 patients (20%), while the Raynaud phenomenon was documented in 1 patient (6.7%).
3.3. Laboratory Findings
Antinuclear antibodies (ANA ≥ 1:320 by indirect immunofluorescence) were detected in all our patients with cSjD, except for two who had biopsy-confirmed SjD (patients 10, 15). Of the 15 patients, anti-Ro/SSA antibodies were found in 13 (86.7%), SSB antibodies in 7 (46.7%), and rheumatoid factor (RF) positivity in 13 (86.7%). Anti-SSB positivity was observed in seven patients (46.7%), with antibody levels showing greater variation than anti-SSA, ranging from just above the positivity threshold to mildly elevated titres. Notably, patient 7 initially tested negative for anti-SSB (La) antibodies but later showed mild positivity at 33 U/mL (laboratory cutoff < 15 U/mL). Importantly, all ANA-positive patients exhibited significantly elevated anti-SSA antibody levels (>200 U/mL; laboratory reference < 15 U/mL). In all 13 patients, RF levels were markedly elevated at presentation, ranging from 3- to 22-fold above the upper limit of normal.
All patients had serum IgG concentrations above the age-adjusted laboratory reference ranges (upper limit 14.4–16.7 g/L) at initial evaluation and throughout follow-up. Moreover, five patients (33.3%) displayed markedly elevated levels exceeding 20 g/L.
C3 complement levels remained within normal laboratory reference ranges in all patients throughout the entire follow-up period. In patient 1, C4 was significantly decreased at diagnosis (3.8 mg/dL; normal 10–40 mg/dL), with negative cryoglobulins and normal C3 levels. C4 values gradually normalised over the subsequent five months of therapy.
Cryoglobulinaemia was negative in all patients who were tested (8/15).
Lymphopenia was a common finding in this cohort, present in all patients either at diagnosis or emerging during the course of the disease and remained persistent in some cases.
Patient 6 had persistently elevated creatine kinase levels (between 400 and 650 U/L) without clinical signs of myopathy and with no evidence of muscle inflammation on MRI. She was subsequently diagnosed with type I macro-CK syndrome after identifying a high macro-CK complex (3.2%) through CK isoenzyme electrophoresis.
Specifically, patient 8, who presented with the unusual initial manifestation of pericarditis, demonstrated a serological profile highly suggestive of SjD, including a high-titre ANA (1:640), positivity for anti-SSA, anti-SSB, and anti-Ro52 antibodies, elevated RF (173 IU/mL), and increased serum IgG levels (2253 mg/dL), with negative anti-dsDNA and anti-Sm antibodies.
3.4. Salivary Gland Imaging
All individuals showed SGUS findings (parotid, submandibular or lacrimal) consistent with cSjD, which supported and ultimately confirmed the diagnosis based on expert clinical judgement (100%).
For 11 (73.3%) patients, archived ultrasound images or video clips were available for re-evaluation using the new OMERACT semiquantitative scoring system (Figure 2). At diagnosis, one patient (6.7%) had an OMERACT score of 1, seven (46.7%) had a score of 2, and three (20%) had a score of 3.
Lacrimal gland ultrasound (LGUS) was performed at initial presentation or during follow-up, and all but one showed no abnormalities in echotexture, vascularity, or gland size, despite significant changes on parotid and submandibular gland assessment.
On the contrary, patient 12, who presented with left lacrimal gland swelling and ocular dryness, had US and MRI abnormalities of the lacrimal glands (increased left-sided gland volume compared to the contralateral side, hypervascularity, and a heterogeneous echotexture). Notably, her SGUS examination was normal. In this patient, a lacrimal gland biopsy and the presence of specific autoantibodies confirmed the diagnosis.
MRI was performed at diagnosis in only three patients with atypical presentations: patient 10, who had persistent submandibular gland enlargement and negative ANA; patient 12, with lacrimal gland swelling and a normal ultrasound appearance of the parotids and submandibular glands; and patient 15, who had a younger age of disease onset and negative ANA (Figure 3). No MRI showed punctate sialectasis or a “salt-and-pepper” appearance. Furthermore, due to persistently elevated creatine kinase levels, patient 6 underwent an MRI to evaluate possible muscular involvement.
3.5. Histopathological Findings
An MSGB was recommended for 6 of the 15 children in our cohort (40%), and 4 underwent the procedure. The remaining two had postponed the biopsy at the time this report was prepared. In these cases, the attending physician requested a biopsy when atypical features were present, autoantibodies were negative, ultrasound examination of the gland did not reveal characteristic findings, or diagnostic confirmation was required for any reason.
For the other nine patients (60%), whose clinical presentation, serology, and ultrasound findings were already supportive of the diagnosis, biopsy was considered to have no added value and was not pursued.
Among the four patients who underwent MSGB or lacrimal gland biopsy (LSGB), three exhibited diagnostic histopathology (focus score ≥ 1), while the other showed dense lymphocytic infiltration but with a focus score < 1. In patient 7, the initial MSGB was non-diagnostic, displaying a diffuse lymphocytic infiltrate. A diagnostic specimen obtained from the repeat biopsy demonstrated focal lymphocytic sialadenitis (Chisholm–Mason grade 3–4) with multiple focal aggregates (>50 lymphocytes per focus) (Figure 4). The girl (patient 12) who presented with lacrimal enlargement underwent an LSGB, which showed lymphocytic dacryoadenitis, consistent with the histopathologic features of Sjögren’s disease. The specimen revealed no granulomas, necrosis, vasculitis, lupus- or MALT-type changes, or IgG4-related features, thereby supporting the diagnosis of cSjD.
In patient 1, who experienced lower-leg vasculitis (Figure 1) characterised by burning sensations and oedema, investigations were undertaken to clarify the underlying mechanism. Laboratory evaluation revealed hypergammaglobulinaemia (elevated IgG, 3493 mg/dL, IgM 300 mg/dL), high rheumatoid factor activity (423 IU/mL), C4 hypocomplementaemia, positive ANA, and anti-Ro/SSA and anti-La/SSB autoantibodies, but without detectable cryoglobulins in serum. Histopathology from a skin biopsy was compatible with hypergammaglobulinaemic vasculitis.
3.6. Diagnostic Misclassifications Before cSjD
Prior diagnostic misattributions before the definitive diagnosis of cSjD included childhood-onset systemic lupus erythematosus (SLE) in five patients (patients 2, 5, 6, 8, and 14; 33.3%). The differential diagnosis with SLE was based on clinical manifestations, detailed immunological and serological profiles, and organ involvement, further supported by characteristic exocrine glandular findings (Figure 5). Additionally, patient 1 was initially diagnosed with systemic vasculitis, while two other patients were misdiagnosed with juvenile idiopathic arthritis.
3.7. Treatment and Outcomes
3.7.1. Therapeutic Interventions Initiated at Diagnosis
Eight (53.3%) patients were started on hydroxychloroquine (HCQ) at diagnosis, based on the clinician’s judgement, primarily for systemic manifestations of cSjD and serological activity (notably significant hypergammaglobulinaemia and high rheumatoid factor titres), as well as for glandular involvement. Several patients (46.7%) required more than one therapeutic trial of HCQ for 6–12 months by the time of the final assessment. A single 6-month course of HCQ was sufficient to achieve remission of disease manifestations in three patients (20%).
A short course of non-steroidal anti-inflammatory drugs (NSAIDs) (3–5 days) was used for acute episodes of parotitis. In our cohort, two patients used only NSAIDs during the follow-up period.
Systemic corticosteroids were used in three patients to manage acute episodes of parotitis in selected situations, specifically in the presence of severe pain or prolonged flares that did not respond to NSAIDs (patients 3, 6, 7). Oral prednisone was also used in patient 8 for a severe episode of pericarditis with cardiac tamponade, and in patients 10 and 12, where treatment had been initiated before their presentation to our service by the paediatrician because of marked glandular manifestations.
Conventional synthetic DMARDs, such as azathioprine (AZA), were used in five patients—typically in combination with corticosteroids—as a steroid-sparing strategy.
In addition, colchicine was initiated in patient 8 for pericarditis, alongside systemic corticosteroids, and was continued through the last study assessment.
For dry eye symptoms, all patients received lubricating eye drops at diagnosis and subsequently received short courses of topical corticosteroids under ophthalmologic supervision during episodes of significant keratoconjunctival inflammation. Inflammatory skin rashes were managed with topical corticosteroids for the shortest duration necessary.
In patients with xerostomia, recommendations included the use of artificial saliva, routine brushing with fluoride toothpaste, preventive dental care, and xylitol-containing products to minimise dental caries.
3.7.2. Cumulative Treatment up to the Final Follow-Up
Overall, at the time of the last follow-up, seven of the 15 patients (46.7%) had received short courses of prednisone; 8 (53.3%) had been treated with HCQ, 5 (33.3%) with AZA, 1 (6.7%) with mycophenolate mofetil (MMF), and 1 (6.7%) with Colchicine. None received Rituximab or any biologic drug.
MMF at 900–1000 mg/m^2^ per day, divided into two doses, was started to treat hypergammaglobulinemic vasculitis in patient 1 after an inadequate response to HCQ combined with AZA. This therapy successfully controlled the patient’s extraglandular cutaneous manifestations, lowered serum IgG levels, and normalised complement concentrations.
To facilitate clinical application, we propose a table summarising practical treatment considerations for cSjD, based on currently available expert guidelines and our group’s clinical experience with paediatric patients (Table 2).
3.7.3. Disease Complications During Follow-Up and Outcomes at the Last Assessment
Disease duration at the last assessment ranged from 2 to 56 months (median 24; IQR 6–38). At the final follow-up, the median ESSPRI score was 0 (range 0–2.67; IQR 0–1.33), and the median ESSDAI score was 1 (range 0–8; IQR 0–1), indicating mild subjective symptom burden and low disease activity, with residual activity mainly seen in two patients in the glandular, articular, or biological domains.
Chronic musculoskeletal pain and dryness have been most frequently reported by our patients at the last assessment, affecting up to 5/15 (33.3%) in each of these ESSPRI domains. Dryness was clinically significant in only one patient, with a score of 5/10 on a visual analogue scale. Fatigue was reported by three patients at the last assessment, with significant fatigue only in patient 9 (5/10 cm on a visual analogue scale).
Only one girl (patient 3) showed irreversible ocular damage during our study, including impaired tear flow, with an SSDSI score of 1, confirmed by an ophthalmology specialist who observed a clear decrease in tear secretion (abnormal Schirmer test). Two cases developed chronic keratoconjunctivitis. No cases of lymphoma or major chronic organ involvement were observed in our cohort during follow-up. Patient 5, who initially presented with proteinuria, was closely monitored by a nephrologist. After the initial episode, no further urinary protein losses or other signs of renal involvement have been documented to date.
Patient 8 experienced a recurrence of pericarditis with cardiac tamponade following an abrupt tapering of corticosteroids and colchicine, and pericardiocentesis was performed. Upon reinitiation of treatment, the pericarditis resolved and has remained controlled to the present time. Other potential causes of pericarditis were excluded in this case.
No abnormalities in serum amylase or lipase levels, nor any structural pancreatic changes, were detected.
Parotitis episodes improved during follow-up in six patients [40%], mainly with treatment involving NSAIDs, HCQ, and/or corticosteroids. In patients who did not exhibit these features at diagnosis, sicca symptoms and recurrent parotitis appeared later in the disease course in four and five cases, respectively.
Lymphopenia was common during the disease course in our patients, with or without systemic medications (70%).
3.8. Characteristics and Final Diagnoses of Non-Sjögren Cases
Final diagnoses among patients referred with suspected cSjD most commonly included juvenile recurrent idiopathic parotitis, false-positive SSA or SSB results on qualitative serological assays, chronic fibrosing sialadenitis, idiopathic orbital myositis, autoimmune thyroiditis, and IgG4-related disease.
4. Discussion
Our findings are closely in accordance with those reported in recent high-quality paediatric cohorts from various international workgroups, all of which highlight the key clinical features of cSjD [1,2,3,4,5,6,7,8]. Additionally, our cohort offers novel observations, including previously unreported presentations such as pericarditis complicated by cardiac tamponade and co-occurrence with type I macro–creatine kinase syndrome. We also confirm the existence of rare disease phenotypes characterised by isolated lacrimal gland involvement without major salivary gland disease. Furthermore, in our cohort, extraglandular presentation was more common and posed a greater diagnostic challenge. Overall, these findings further emphasise the heterogeneity of the clinical spectrum in children and reinforce the significant diagnostic challenges associated with cSjD.
This single-centre cohort from a 10-year observational study showed a clear female predominance and a wide age range at onset, including very early disease presentation as young as 3 years old, findings consistent with those reported in the literature [1,2,3,9,10]. Notably, nearly all patients in our cohort were diagnosed within the past four years, raising the question of whether the incidence of cSjD has increased in recent years. To date, no published data support a true temporal rise in disease incidence. Although other authors have similarly noted a recent increase in published paediatric case series and cohort studies [15], this trend may more plausibly reflect improved disease recognition, heightened awareness, and increased reporting rather than a genuine increase in occurrence. Nonetheless, delayed diagnosis and under-recognition likely persist, and additional undiagnosed cases may remain.
Recurrent swelling of the parotid gland is a key diagnostic feature of cSjD and is considered one of the most common manifestations at onset, often preceding sicca symptoms or other systemic signs, and reported in approximately 33–91% of paediatric cases [1,3,5,6,7,8]. Parotitis inversely correlates with age, being less common in adult-onset SjD, which highlights its diagnostic significance in paediatric populations [3]. In our series, recurrent parotitis was present at initial presentation in only five patients (33%), but developed later in another five patients, thus supporting the diagnosis over time. Similarly, sicca symptoms were uncommon early in the disease and appeared after 2–3 years of progression in about one-third of patients.
Children presenting with extraglandular manifestations pose the greatest diagnostic challenge, as their clinical presentation is highly heterogeneous and may be more severe. Recently published diagnostic algorithms for children with cSjD-like presentations indicate that the extraglandular pathway is the least sensitive for establishing a diagnosis of cSjD [15]. Patients in this group often have few or no sicca symptoms and may lack obvious inflammation of the salivary or lacrimal glands. Notably, in our cohort, this type of initial presentation was more frequent than presentations dominated by parotitis (53.3%). Lupus-like clinical features were present at disease onset in seven patients, manifesting as facial vasculitic purpura with or without associated arthralgia; some were initially considered by the general paediatrician to have SLE. The presence of positive ANA (fine-speckled pattern), moderate- to high-titre SSA ± SSB, the absence of specific lupus antibodies (anti-dsDNA, anti-Sm), high titres of RF, and suggestive SGUS features at presentation were key elements in considering cSjD. According to current paediatric diagnostic algorithms [15], these patients meet criteria for probable cSjD, and a minor salivary gland biopsy is recommended to confirm the diagnosis. In our cases, MSGB was not performed as serology and ultrasound were suggestive of cSjD at that time. Moreover, five of them developed typical glandular manifestations during the course of the disease.
Additionally, both SLE and SjD may overlap in paediatric populations and have been previously documented [8,27]. The presence of specific lupus antibodies, hypocomplementemia, pancytopenia, or specific lupus organ manifestations (such as lupus nephritis) should raise awareness of this potential association, as these cases are uncommon and often associated with increased mortality and a poorer prognosis than in adults [28].
One patient in our cohort experienced recurrent episodes of vasculitis in the lower legs during the first four years of the disease as the initial symptom. These episodes occurred without other systemic features or renal involvement and preceded other classic signs of SjD, tending to be more aggressive and persistent over time. This manifestation is recognised in children diagnosed with SjD, most commonly presenting as palpable purpura caused by small vessel leukocytoclastic vasculitis. Differentiating cryoglobulinemic vasculitis from other vasculitis subtypes (e.g., hypergammaglobulinemic or urticarial vasculitis) is important for prognosis and management. In her case, histopathology from a skin biopsy was compatible with hypergammaglobulinemic vasculitis. The patient continued to experience recurrent vasculitic flares despite receiving AZA and prednisone. MMF was therefore introduced. Recently published guidelines for the management of SjD recommend using DMARDs only for selected systemic complications; however, MMF is not specifically endorsed due to limited evidence supporting its use in this context. Furthermore, MMF is not recommended in either the Japanese or North American guidelines [22,23,24,25,26]. In our case, MMF resulted in effective control of cutaneous involvement and was associated with reductions in serum IgG levels and increases in complement concentrations over the course of the disease, consistent with previous reports [29]. MMF may therefore be considered in selected cases, as it is generally well tolerated and associated with fewer and less severe adverse effects compared to Rituximab.
Rare but illustrative presentations, such as reported cases of bilateral ranulas [30,31,32] and pulmonary haemorrhage [33,34], underscore the broad clinical spectrum of cSjD and highlight the need for heightened diagnostic awareness. In our series, one patient presented with symptomatic acute exudative pericarditis as the initial disease manifestation. His immunological evaluation revealed findings strongly supportive of SjD, and bilateral parotid ultrasound abnormalities were consistent with the diagnosis, despite the absence of objective ocular or oral dryness. Application of paediatric diagnostic algorithms via the extraglandular pathway classified the case as probable cSjD [15]. Accordingly, an MSGB was recommended to confirm the diagnosis, as in the other patient with extraglandular presentation in our cohort; however, the family deferred the procedure at the time of reporting. During follow-up, the patient also developed objective parotitis.
To our knowledge, no cases of clinically significant, symptomatic acute exudative pericarditis complicated by cardiac tamponade have been reported in children with cSjD in the paediatric literature to date. A single case of cSjD has been described with an initial presentation of haemolytic uremic syndrome and pericarditis, followed by a fatal course due to pulmonary haemorrhage; however, the constellation of severe associated systemic manifestations in that report may have contributed to the occurrence of pericarditis and limits direct comparison [34]. In contrast, pericardial involvement, although uncommon, has been documented in adult-onset SjD, most frequently presenting as a subclinical pericardial effusion or acute exudative pericarditis, and, more rarely, as chronic constrictive pericarditis [35,36].
The association with macro-creatine kinase was another distinctive feature of our series. Macro-creatine kinase type I, recognised as a benign cause of elevated CK, often seen in autoimmune conditions, especially in females, is most often linked to other autoimmune conditions; however, its co-occurrence with primary SjD has not been documented in published case series or reviews to date [37,38].
Anti-SSA (anti-Ro) and anti-SSB (anti-La) autoantibodies, together with RF, demonstrate high diagnostic specificity for cSjD (94.7%, 96.0%, and 95.7%, respectively), but only moderate sensitivity (55.6%, 14.8%, and 33.3%, respectively) [39]. Both the paediatric diagnostic criteria proposed by Bartůňková et al. [13] and more recent diagnostic algorithms [15] allow either anti-SSA or anti-SSB positivity to fulfil the serological component of diagnosis, given that the true prevalence and diagnostic value of anti-SSB antibodies in children remain uncertain.
In our cohort, anti-SSA antibodies were frequently detected, often at very high titres, with a positivity rate of 86.7%, consistent with findings from major paediatric cohorts [1,3,4,8]. Anti-SSB antibodies were identified in seven patients (46.7%), with titres showing greater variability than those of anti-SSA, ranging from just above the positivity threshold to mildly elevated concentrations. Longitudinal changes in anti-SSB status were observed in one patient, who initially tested negative and subsequently developed low-titre positivity. Importantly, no patient demonstrated isolated anti-SSB positivity in the absence of anti-SSA antibodies. Furthermore, isolated positivity for anti-La/SSB antibodies is not included in the 2016 ACR/EULAR classification criteria, which are currently the most widely used criteria used for SjD [11]. The clinical significance of anti-SSB in cSjD, therefore, remains to be fully elucidated.
Seronegative cSjD is a recognised entity, and diagnosis should not be excluded solely on the basis of negative anti-SSA or anti-SSB serology, as illustrated by two patients in our cohort and supported by recent reports [2,40]. Conversely, the widespread availability of qualitative autoantibody assays, when applied without an appropriate clinical context, has contributed to an increasing number of referrals for suspected autoimmune disease in children lacking compatible clinical features, often driven by heightened parental anxiety. Limiting immunological testing to cases supported by careful clinical assessment may improve diagnostic accuracy and substantially reduce unnecessary investigations and parental distress.
Parotid and submandibular ultrasound has become a commonly used first-line imaging modality for assessing salivary gland inflammation, is inexpensive, easily repeatable, and non-invasive, with additional advantages in paediatric assessment [15,41]. SGUS supported the diagnosis of cSjD in all our patients, based on characteristic features of SjD that were evident from the initial presentation. It is an operator-dependent technique, but standardised protocols such as the OMERACT scoring system [21] can significantly enhance its reliability and reproducibility. Based on our experience, SGUS proved highly informative, particularly in patients with extraglandular manifestations at presentation, and was consistent with the results of other studies [5,6]. In addition to its diagnostic role, SGUS may aid in monitoring for complications, including lymphoma [15,41].
LGUS may be particularly valuable for identifying rare disease phenotypes characterised by isolated lacrimal gland involvement in the absence of major salivary gland disease, as observed in one patient from our cohort. However, because the diagnostic performance of LGUS is generally lower and less standardised than that of SGUS [42,43,44], additional investigations—including MRI and tissue biopsy—were required to confirm the diagnosis of cSjD. Notably, in other patients with ultrasound findings suggestive of SjD affecting the major salivary glands, LGUS revealed no abnormalities of the lacrimal glands.
MSGB is a key diagnostic tool in cSjD, particularly when clinical and serological findings are insufficient or atypical, and it demonstrates good diagnostic sensitivity and specificity [45]. However, given its invasive nature and the lack of normative paediatric histopathological data [15], it is not routinely recommended. Repeat biopsy may be considered when initial results are negative or equivocal and clinical suspicion remains high, as in our patient. Such decisions should be individualised, carefully balancing procedural risk against potential diagnostic benefit, especially in the context of evolving clinical features or serological changes.
5. Conclusions
Our findings highlight the marked heterogeneity of childhood-onset SjD, with extraglandular manifestations frequently predominating and often occurring in the absence of sicca or glandular symptoms, thereby posing significant diagnostic challenges. Lupus-like systemic features—including facial vasculitic purpura, with or without arthralgia, and occasional pericarditis, as observed in our cohort—may contribute to frequent initial diagnostic misattribution to SLE. Early use of salivary gland ultrasonography, comprehensive autoantibody profiling, and selective salivary gland biopsy in atypical cases is essential for timely and accurate diagnosis, underscoring the urgent need for paediatric-specific classification criteria.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ramos-Casals M. Acar-Denizli N. Vissink A. Brito-Zerón P. Li X. Carubbi F. Priori R. Toplak N. Baldini C. Faugier-Fuentes E. Childhood-Onset of Primary Sjögren′s Syndrome: Phenotypic Characterization at Diagnosis of 158 Children Rheumatology 2021604558456710.1093/rheumatology/keab 03233493333 · doi ↗ · pubmed ↗
- 2KılbaşG. Ayduran S. Şener S. Coşkuner T. Ulu K. Kısaoğlu H. Aslan E. Könte E.K. Arslanaoğlu C. Aydin T. Results of a Nationwide Multicenter Study in Childhood Sjögren Disease J. Rheumatol.2025521141115010.3899/jrheum.2024-104840664482 · doi ↗ · pubmed ↗
- 3Basiaga M.L. Stern S.M. Mehta J.J. Edens C. Randell R.L. Pomorska A. Irga-Jaworska N. Ibarra M.F. Bracaglia C. Nicolai R. Childhood Sjögren Syndrome: Features of an International Cohort and Application of the 2016 ACR/EULAR Classification Criteria Rheumatology 2021603144315510.1093/rheumatology/keaa 75733280020 PMC 8487648 · doi ↗ · pubmed ↗
- 4Marino A. Romano M. Giani T. Gaggiano C. Costi S. Singh R. Mehta J.J. Liebermaln S.M. Cimaz R. Childhood Sjogren′s Syndrome: An Italian Case Series and a Literature Review-Based Cohort Semin. Arthritis Rheum.20215190391010.1016/j.semarthrit.2020.11.00433261821 · doi ↗ · pubmed ↗
- 5Legger G.E. Erdtsieck M.B. de Wolff L. Stel A.J. Los L.I. Verstappen G.M. Spijkervet F.K. Vissink A. van der Vegt B. Kroese F.G. Differences in Presentation Between Paediatric- and Adult-Onset Primary Sjögren′s Syndrome Patients Clin. Exp. Rheumatol.202139859210.55563/clinexprheumatol/vxe 6h 034796855 · doi ↗ · pubmed ↗
- 6Hammenfors D.S. Valim V. Bica B.E.R.G. Pasoto S.G. Lilleby V. Nieto-González J.C. Silva C.A. Mossel E. Pereira R.M.R. Coelho A. Juvenile Sjögren′s Syndrome: Clinical Characteristics with Focus on Salivary Gland Ultrasonography Arthritis Care Res.202072788710.1002/acr.23839 PMC 697260430697959 · doi ↗ · pubmed ↗
- 7Virdee S. Greenan-Barrett J. Ciurtin C. A Systematic Review of Primary Sjögren′s Syndrome in Male and Paediatric Populations Clin. Rheumatol.2017362225223610.1007/s 10067-017-3745-z 28735431 PMC 5596040 · doi ↗ · pubmed ↗
- 8Ciurtin C. Peng J. Taylor-Gotch R. Peckham H. Wilson R. Al Obaidi M. Jury E.C. P Re S Childhood Sjögren′s Interest Group Clinical phenotypes, classification, and long-term outcomes of childhood-onset Sjögren′s disease into adulthood: A single-centre cohort study Lancet. Rheumatol.20258 e 204e 21610.1016/S 2665-9913(25)00283-841274306 · doi ↗ · pubmed ↗