Broadening the Phenotypic Spectrum of MAFB-Related Disease: Renal, Auricular, Ocular, and Nervous System Involvement
Aviva Eliyahu, Danit Atias-Varon, Ortal Barel, Yulia Khavkin, Elon Pras, Haike Reznik-Wolf, Odelia Chorin, Tomer Poleg, Ari Biller, Pazit Beckerman, Nabil Abu-Amer, Tamara Wygnanski-Jaffe, Lior Greenbaum, Asaf Vivante, Irit Krause

TL;DR
This paper identifies new symptoms linked to MAFB gene variants, expanding the known effects of this genetic condition to include kidney, ear, eye, and brain issues.
Contribution
The study reports novel clinical features associated with MAFB-related disease, including auricular anomalies and neurodevelopmental delay.
Findings
A MAFB variant (c.797T>C; p.Leu266Pro) was found in multiple family members with diverse symptoms.
New features like auricular anomalies, hearing loss, and neurodevelopmental delay were observed in MAFB-related disease.
Structural modeling confirmed the functional impact of the identified MAFB variant.
Abstract
Background: Focal segmental glomerulosclerosis (FSGS) is a leading cause of renal disease presenting with steroid-resistant nephrotic syndrome (SNRS) and variable stages of chronic kidney disease (CKD). Monogenic etiologies for FSGS are increasingly recognized, particularly in pediatric and familial cases. Missense variants in the MAF BZIP Transcription Factor B (MAFB) gene cause a dominantly inherited condition with variable phenotype, ranging from isolated ocular or renal manifestations to syndromic FSGS. Methods: Detailed clinical and genetic investigations were conducted in an extended family presenting with a spectrum of renal and extra-renal manifestations. Results: Using Exome Sequencing (ES), a heterozygous variant, c.797T>C; p.(Leu266Pro) in the MAFB gene was identified in multiple affected family members. Variant segregation confirmed its presence in additional family members.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
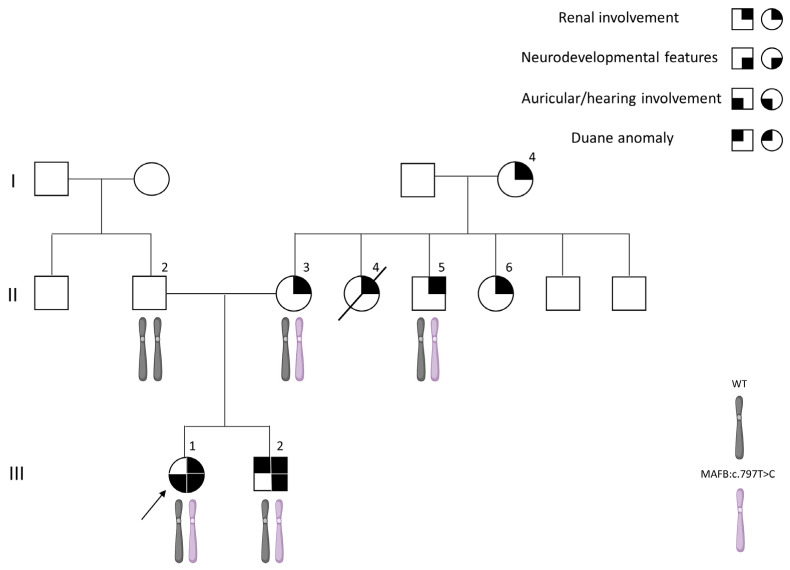 Figure 1
Figure 1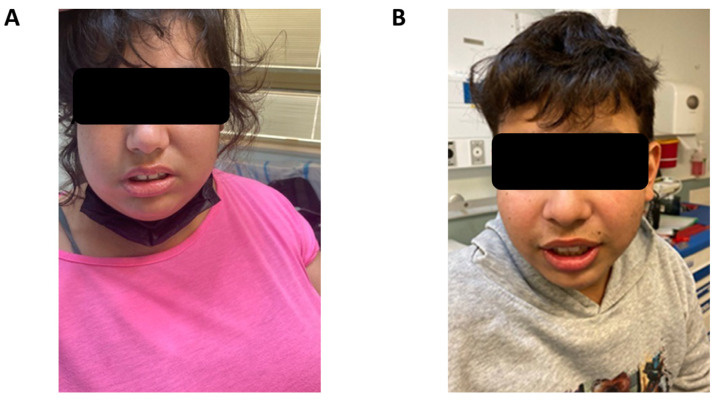 Figure 2
Figure 2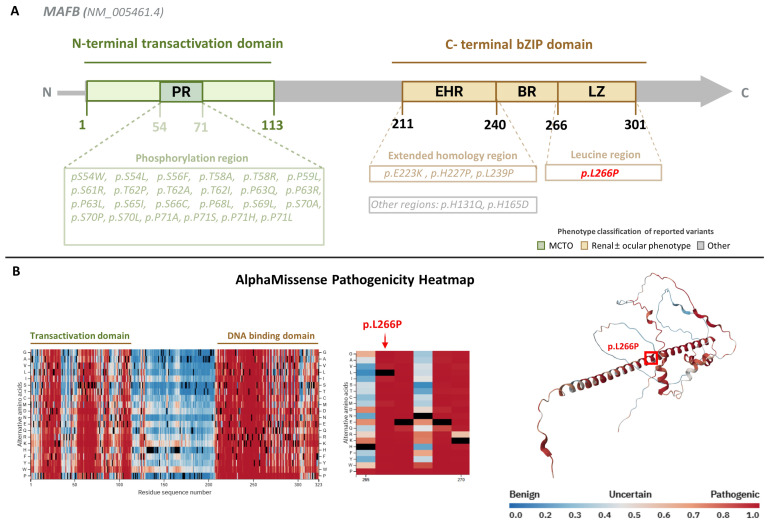 Figure 3
Figure 3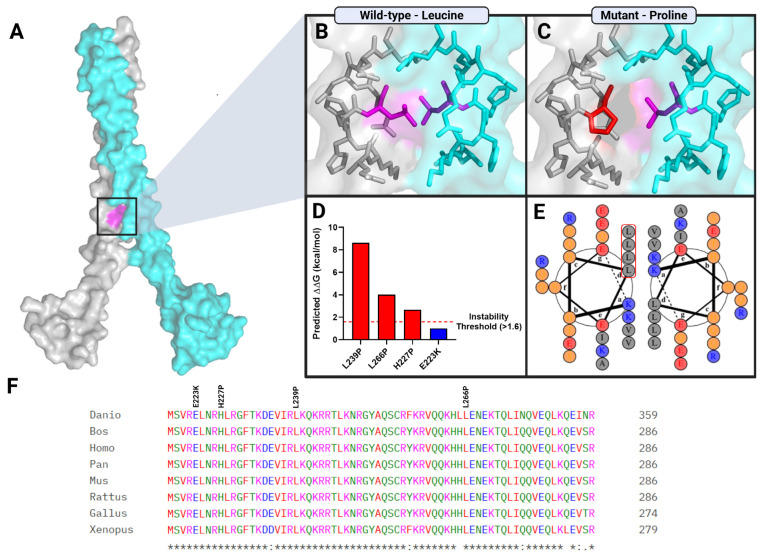 Figure 4
Figure 4- —ISF—Clinical Scientist Grant
- —ERC starting grant
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal Diseases and Glomerulopathies · Chronic Kidney Disease and Diabetes · Genomics and Rare Diseases
1. Introduction
Focal segmental glomerulosclerosis (FSGS) is a major cause of steroid-resistant nephrotic syndrome (SRNS) and a leading contributor to chronic kidney disease (CKD) in both children and adults [1]. The etiology of FSGS is heterogeneous, involving a complex interplay between genetic predisposition and environmental factors [2]. Over the past decade, however, monogenic causes of FSGS have been increasingly recognized, particularly in pediatric and familial cases [3,4]
To date, pathogenic variants in more than 50 genes have been implicated in monogenic forms of FSGS [5,6]. These genes encode proteins critical for slit diaphragm integrity, cytoskeletal organization, foot process architecture, and the maintenance of podocyte homeostasis [5].
Although FSGS most commonly presents as an isolated renal disorder, a subset of affected individuals exhibits syndromic manifestations, which involve extra-renal organ systems and reflect the broader biological roles of genes essential for podocyte function.
Pathogenic variants in MAF BZIP Transcription Factor B (MAFB) (OMIM 608968) are primarily associated with two Mendelian phenotypes: multicentric carpotarsal osteolysis syndrome (MCTO; OMIM #166300), characterized by progressive skeletal osteolysis with possible renal involvement, and Duane retraction syndrome 3 (DRS3; OMIM #617041), a congenital ocular cranial dysinnervation disorder that may co-occur with proteinuric kidney disease.
Recently, heterozygous missense variants in the MAFB gene were reported in four individuals from two unrelated families presenting with FSGS in association with DRS, characterized by restricted horizontal eye movements resulting from congenital abnormal cranial nerve development. Additionally, one affected individual exhibited hearing impairment [7]. These observations suggested a role for MAFB in the coordinated development and function of renal and neuro-ocular systems.
MAFB is a member of the large Maf family of basic leucine zipper (bZIP) transcription factors [8]. It is expressed in multiple cell types, including macrophages, glomerular podocytes, and osteoclasts, where it plays essential roles in cellular differentiation and tissue-specific gene regulation [9,10,11]. Structurally, MAFB consists of an N-terminal transactivation domain and a conserved C-terminal bZIP domain. The bZIP region comprises a basic segment responsible for sequence-specific DNA binding and an adjacent leucine zipper that mediates dimerization, enabling MAFB to function as a homo- or heterodimer [11].
The biological role of MAFB has been further elucidated through studies in animal models, which provide insight into its developmental functions. Beyond renal development, vertebrate models have demonstrated conserved roles for MAFB orthologs in hindbrain and cranial motor neuron development. Recently, Jurgens et al. (2025) showed that disruption of the zebrafish MAFB ortholog, mafba, results in severe malformation or absence of the abducens cranial nerve (CN6) motor nucleus, supporting its evolutionarily conserved role in cranial motor neuron specification [12].
Experimental studies in Mafb-deficient mouse models have demonstrated that MAFB is indispensable for podocyte differentiation, foot process formation, and maintenance of the glomerular filtration barrier, underscoring its critical role in renal development and function [13]. A knock-in mouse model harboring the Mafb p.Leu239Pro variant within the DNA-binding domain recapitulated multiple developmental abnormalities, including defects of the inner ear, kidney, parathyroid, pancreas, and macrophage lineages, with phenotypic features closely resembling Mafb-null mice, further supporting the essential functional role of the bZIP DNA-binding domain [14].
Notably, all missense MAFB variants previously associated with FSGS and DRS cluster within or adjacent to the DNA-binding region of the bZIP domain, supporting the hypothesis that impaired transcriptional regulation underlies the combined renal and neuro-ocular phenotype [7]. In contrast, pathogenic variants clustering within the N-terminal transactivation domain cause MCTO, a skeletal-dominant disorder characterized by progressive osteolysis of the carpal and tarsal bones and variable renal involvement, but lacking the characteristic ocular motility defects observed in DRS, thereby highlighting domain-specific genotype–phenotype correlations [15]
In the present study, we describe a family with multiple affected individuals harboring a rare heterozygous variant c.797T>C; p.(Leu266Pro) in MAFB, presenting with variably expressed renal involvement, accompanied by incomplete penetrant extra-renal features. This report further expands the phenotypic spectrum associated with MAFB-related disease and emphasizes the marked clinical heterogeneity and diagnostic challenges associated with MAFB variation.
2. Materials and Methods
2.1. Clinical Evaluation and Patient Recruitment
Family members were referred for a genetic evaluation and workup at the Nephrogenetic Clinic at the Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Israel. The referral indication was a family history of CKD, which affected multiple family members.
Demographic and clinical data were collected, including results of previous blood tests and imaging. Blood samples were obtained for DNA extraction and genetic analysis. The study has been approved by the Sheba Medical Center Institutional Review Board. Informed, written consent was obtained by the study participants or by their legal guardians, for genetic analysis and for publication of patients’ photographs.
2.2. Exome Sequencing and Bioinformatics Analysis
ES libraries were prepared using the Twist Human Core Exome Plus Kit (Twist Bioscience, San Francisco, CA, USA) and sequenced on an Illumina NovaSeq 6000 platform (San Diego, CA, USA), generating paired-end reads of 150 bp. Sequence alignment to the human reference genome (hg38) was performed using the Illumina DRAGEN Bio-IT Platform (version 3.8), which applies a Smith–Waterman-based alignment algorithm [16]. Variant calling was conducted using the GATK variant caller (version 3.7) [17], with additional variant calling using FreeBayes (version 1.2.0) [18]. Copy number variation analysis (CNV) was performed using DRAGEN pipeline. Annotation of identified variants was carried out using KGG-Seq (version 1.2) [19], followed by further in-house annotation and filtering tools. Variant interpretation and classification were performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines [20].
Sanger sequencing and next-generation sequencing were performed for additional family members as part of a commercial sequencing service.
2.3. Variant Curation and Domain Classification
Previously reported MAFB missense variants were identified through a systematic search of the Human Gene Mutation Database (HGMD^®^ Professional, version 2025.4) and cross-referenced with the primary literature [21]. Variants were categorized according to their location within functional protein domains (N-terminal transactivation domain versus C-terminal basic leucine zipper [bZIP] domain) and their associated clinical phenotype.
Variants located within the N-terminal transactivation domain and previously associated with multicentric carpotarsal osteolysis (MCTO) were included for comparative domain analysis. Variants within the C-terminal bZIP domain associated with renal and/or ocular phenotypes were further evaluated by structural modeling to assess potential effects on DNA binding or dimerization. Variants lacking a consistent genotype–phenotype correlation were excluded from structural modeling.
2.4. Protein/Mutation Modeling Methods
Three-dimensional coordinates of the wild-type protein were obtained from the Protein Data Bank using the crystal structure PDB ID: 2WTY. AlphaMissense software (https://alphamissense.hegelab.org, 10 December 2025) [22] was employed to assess the pathogenicity of the variant. Protein visualization was performed using PyMOL Molecular Graphics System (version 3.1.6) [23]. Predicted change in Gibbs free energy (ΔΔG) was calculated with FoldX5 [24]. Multiple sequence alignments of the MAFB sequences were performed using Clustal Omega (https://www.ebi.ac.uk/jdispatcher/msa/clustalo, 10 December 2025) [25]. To visualize and analyze the coiled-coil domains within MAFB, helical wheel diagrams were generated using DrawCoil 1.0 (https://grigoryanlab.org/drawcoil/, 10 December 2025).
3. Results
3.1. Clinical Characteristics
3.1.1. Patient III-1
The proband is a 20-year-old female of Kurdish-Persian Jewish descent (Figure 1). Her medical history is notable for global developmental delay with severe cognitive impairment, features consistent with autism spectrum disorder (ASD), and profound pre-lingual bilateral hearing impairment diagnosed at seven months of age. Brain magnetic resonance imaging (MRI) at one year revealed cochlear aplasia with a bilateral common cavity configuration, without additional intracranial abnormalities. She subsequently underwent bilateral cochlear implantation. Ophthalmological examination was not feasible due to limited patient cooperation related to behavioral and emotional difficulties. Additional medical history included obesity (BMI 47) and pre-diabetes. Physical examination revealed facial dysmorphic features, including a round face, malar flattening, small palpebral fissures, a prominent nose, short philtrum, and small ears (Figure 2A). Laboratory evaluation at age 20 demonstrated abnormal renal function and nephrotic-range proteinuria. Renal ultrasonography showed bilaterally small kidneys with cortical thinning. A kidney biopsy was not performed. Within months, the patient progressed to end-stage kidney disease (ESKD), requiring initiation of maintenance hemodialysis, followed by renal transplantation.
3.1.2. Patient III-2
The proband’s brother is a 17-year-old male who was evaluated for proteinuria due to a family history of CKD. He had nephrotic-range proteinuria (6.7 g/24 h) with a preserved estimated glomerular filtration rate (eGFR). Renal ultrasonography and audiological assessment were unremarkable, and a kidney biopsy was not performed.
The patient’s developmental history included early developmental delay and attention-deficit/hyperactivity disorder (ADHD), and they had attended a special education program. Physical examination revealed obesity (BMI 32) and facial dysmorphic features similar to those seen in his sister. Ophthalmological evaluation demonstrated a severe bilateral abduction deficit, consistent with DRS type 1 (Figure 2B).
3.1.3. Patient II-3
Patient II-3, the 47-year-old mother of Patients III-1 and III-2, was born to consanguineous parents. She was diagnosed with CKD and proteinuria (1200 mg/24 h) at 16 years of age. A kidney biopsy at that time was consistent with FSGS. She was treated with angiotensin-converting enzyme (ACE) inhibitors for several years, during which her kidney function remained stable, with an eGFR of 110 mL/min/1.73 m^2^. This patient denied a history of hearing impairment, ocular abnormalities, or neurodevelopmental disorders.
Two siblings of patient II-3 (Patients II-4 and II-5) were also diagnosed with FSGS and subsequently progressed to ESKD, as described below.
3.1.4. Patient II-4
Patient II-4 developed ESKD at 23 years of age and initiated renal replacement therapy. She underwent successful cadaveric kidney transplantation at age 29. Her neurodevelopmental history was reported as normal. She passed away at the age of 42 years due to cardiac failure. Genetic testing was not performed.
3.1.5. Patient II-5
Patient II-5 progressed to ESKD at 41 years of age and was treated with hemodialysis. At age 43, he received a successful kidney transplant from a healthy sibling (Patient II-6).
3.1.6. Patient II-6
Patient II-6 exhibited low-grade proteinuria but maintained normal kidney function. She served as the living kidney donor for Patient II-5.
3.1.7. Patient I-4
The mother of Patient II-3 (grandmother of Patients III-1 and III-2) was reportedly diagnosed with multiple myeloma later in life, followed by progressive clinical deterioration that included renal failure requiring dialysis at the age of 75 years. Medical records regarding her renal disease or other systemic findings were not available.
3.2. Genetic Analysis
ES performed on the index patient, her brother and mother (Patients III-1, III-2 and II-3) identified a heterozygous missense variant in MAFB (NM_005461.5): c.797T>C; p.(Leu266Pro). This variant was reported in our previous work summarizing the diagnostic yield of genetic testing within the Nephrogenetic Clinic cohort [26]. However, the initial report only provided a description of the variant with minimal data about its clinical presentation, notably lacking detailed phenotypic characterization and comprehensive familial genetic and clinical investigation.
This c.797T>C; p.(Leu266Pro) variant is absent from population databases (gnomAD frequency -0), and in silico prediction tools uniformly predict a deleterious effect of this substitution (CADD 32, REVEL 0.984). Segregation analysis using Sanger sequencing and/or next generation sequence (NGS) analysis demonstrated maternal inheritance of the variant, which was shared by both affected siblings and by an additional maternal aunt (Patient II-6).
This variant was classified as likely pathogenic variant according to the American College of Medical Genetics and Genomics (ACMG) guidelines [20] (criteria PP1, PM2, PP3, PP4). Although the z score of MAFB is moderate, missense variation is a known mechanism of disease with multiple reports of disease-causing missense variants within this gene.
A summary of the clinical, genetic, and segregation findings is provided in Table 1.
Unfortunately, additional samples from maternal family members were not available for further testing.
Given the neurodevelopmental features observed in Patients III-1 and III-2, chromosomal microarray analysis (CMA) was performed and with benign results in both. Molecular testing for CGG trinucleotide repeat expansion at the FMR1 locus was negative.
No additional pathogenic variants were identified on ES of Patients II-3, III-1, and III-2 that could account for their clinical presentation.
An additional variant in the ACTN gene was reported by ES in patients II-3 and III-2 and in segregation analysis in patient II-6 as well. This variant (NM_004924.6): c.448A>G, p.(Ile150Val) is absent from population databases, and in silico prediction tools predict a deleterious effect of this substitution (CADD 25, REVEL 0.83). However, this variant did not segregate within the family and was absent from the most severely affected family member (Patient III-1). According to the ACMG guidelines [20] this variant was classified as a variant of unknown significance (criteria PM2, PP3, PP2, BS4).
Previously reported MAFB missense variants are clustered according to protein domain and associated phenotype. Variants associated with MCTO are located within the N-terminal transactivation domain, whereas variants associated with renal and/or ocular phenotypes are localized predominantly to the C-terminal bZIP domain. Consistently with the domain-specific clustering, the p.(Leu266Pro) variant identified in the present family is localized within this C-terminal region (Figure 3A).
3.3. Structural Modeling and Stability Prediction
We next examined the structural consequences of p.(Leu266Pro) in the context of the C-terminal bZIP domain. AlphaMissense pathogenicity mapping demonstrated two major constraint hotspots corresponding to the N-terminal transactivation domain and the C-terminal DNA-binding domain, with residue 266 located within a region of highly predicted pathogenicity (Figure 3B).
Structural modeling localized Leu266 to the leucine zipper (LZ) segment of the bZIP domain at the MAFB dimer interface (Figure 3A and Figure 4A). In the wild-type configuration, Leu266 participates in the hydrophobic core of the coiled-coil structure, contributing to interhelical packing stability (Figure 4B). Substitution with proline introduces a helix-disrupting residue lacking the aliphatic side chain required for hydrophobic packing, resulting in distortion of local α-helical geometry at the dimer interface (Figure 4C).
Stability prediction analysis further supported this structural perturbation, yielding a ΔΔG of approximately +4.0 kcal/mol for L266P, well above the predefined instability threshold of >1.6 kcal/mol. This indicates a marked predicted destabilization relative to wild type (Figure 4D). Helical wheel projection confirmed the positioning of Leu266 within the hydrophobic heptad repeat core of the leucine zipper (Figure 4E), and cross-species alignment demonstrated strong evolutionary conservation of this residue across vertebrates (Figure 4F), supporting its structural and functional importance. Notably, other residues affected by disease-associated C-terminal variants also exhibit high evolutionary conservation, underscoring the structural and functional constraint of this domain.
4. Discussion
In this study, we provide a comprehensive clinical description of a MAFB variant, c.797T>C; p.(Leu266Pro), associated with familial FSGS and CKD accompanied by DRS, auricular abnormalities, and neurodevelopmental involvement. Overall, affected family members demonstrated variable clinical expressivity and evidence of reduced penetrance, consistent with previously reported MAFB-related phenotypes [7,27]. In patient II-6, a presentation of low range proteinuria is the only reported manifestation. Moreover, since additional family members did not undergo clinical or genetic evaluation, the accuracy of the phenotypic spectrum description is limited.
To date, approximately 30 disease-causing variants in MAFB have been reported, several within cohorts presenting with combined renal and ocular abnormalities. MAFB is one of multiple transcription factors essential for podocyte differentiation, particularly in the terminal stages of maturation, leading to the formation of a functional glomerulus [28]. Podocyte-specific Mafb knockout studies have shown reduced expression of key podocyte genes—including Nphs1, Magi2, and Tcf21—all required for maintaining the integrity of the slit diaphragm barrier. Variants in these genes are known to cause monogenic FSGS. These findings suggest that in addition to its direct role in podocyte differentiation, MAFB may regulate a broader transcriptional network critical for glomerular protein barrier maintenance [13].
The ocular findings observed in our cohort are consistent with the established role of MAFB in cranial motor neuron development and the pathogenesis of ocular congenital cranial dysinnervation disorders, as supported by prior murine models and recent functional work by Jürgens et al. [12,29].
MAFB protein also plays a role in ear development. Mafb knockout mice demonstrate loss of dorsal otic structures, cochlea and sensory organs. Perturbation of the MAFB–hindbrain pathway influences expression of critical otic genes, including Gbx2, Dlx5, Wnt2b, and Otx2 [30].
Park et al. (2016) proposed a threshold model in which partial reduction in MAFB function leads to isolated DRS, whereas lower levels (e.g., homozygous knockout or dominant-negative effect) result in combined DRS and inner ear abnormalities, indicating tissue-specific sensitivity [31].
A prior report by Pascollini et al. [27] described a family with MAFB-associated disease without renal involvement, including one individual with neurodevelopmental delay [27]. In the present study, comprehensive genetic evaluation of multiple family members with neurologic involvement did not identify an alternative cause for the neurodevelopmental phenotype. We therefore postulate that this variant may contribute to the observed neurodevelopmental abnormalities and suggest that such features could represent part of the expanding spectrum of MAFB-related disease. However, further studies will be required to establish a definitive association between this gene and the neurodevelopmental phenotype. Although limited information exists regarding the role of MAFB in the human brain, experimental evidence demonstrates its involvement in neuronal maturation and microglial function. Loss of MAFB expression in macrophage-lineage cells leads to impaired microglial programming and disruption of brain homeostasis [32], and additional work shows that MAFB promotes maturation of cortical interneurons [33].
These experimental findings underscore the pleiotropic and tissue-specific functions of MAFB in neural development and homeostasis. In this context, accumulating evidence supports a protein domain–dependent model of MAFB pathogenicity. MAFB variants affecting distinct functional domains cause clinically discrete yet partially overlapping phenotypes. Missense variants within the N-terminal transactivation domain cause MCTO, a skeletal-dominant disorder that may include craniofacial anomalies and renal involvement but lacks the characteristic ocular motility defects seen in DRS [15]. In contrast, variants associated with ocular cranial dysinnervation and proteinuric kidney disease, including the variant described here, localize to the C-terminal DNA-binding (bZIP) domain (Figure 3A,B).
The p.(Leu266Pro) variant resides within the leucine zipper region of this domain, a critical segment required for dimerization and transcriptional regulation. In silico pathogenicity analysis using AlphaMissense provides structural support for domain-specific functional vulnerability within MAFB. Structural modeling showed that substitution of leucine with proline is predicted to disrupt hydrophobic packing within the heptad repeat core and introduce conformational constraints incompatible with α-helical stability, resulting in marked destabilization of the bZIP dimer interface, as reflected by a ΔΔG value exceeding the predefined instability threshold (Figure 4B–D).
Notably, prior structural analysis of the nearby MAFB p.Leu239Pro variant—also located within the DNA-binding region—similarly predicted destabilization of the C-terminal domain [7]. Furthermore, a knock-in mouse model harboring the corresponding Mafb p.Leu239Pro substitution exhibited a phenotype closely resembling Mafb-null animals, indicating profound functional impairment [14]. Together, these observations support the concept that structural destabilization of the C-terminal bZIP domain represents a shared pathogenic mechanism among non-MCTO MAFB variants.
Although our in silico structural modeling predicts only modest destabilization for MAFB p.(Glu223Lys), Jürgens et al. demonstrated reduced DNA-binding capacity of this variant using protein-binding microarrays, suggesting that functional impairment may arise from altered transcription factor–DNA interactions.
Conservation analysis further demonstrates strong evolutionary constraint across the C-terminal domain, particularly at residues affected by disease-associated variants (Figure 4F). Collectively, these findings reinforce the domain-dependent functional vulnerability of MAFB and provide a structural framework for the phenotypic segregation observed across its disease spectrum.
Interestingly, although MCTO typically presents with predominantly skeletal manifestations and renal disease is often delayed, partial phenotypic crossover is observed, indicating that domain-specific effects on MAFB function may intersect with tissue-specific thresholds of dosage and genetic background. At present, the reasons for why some individuals develop combined bone–kidney disease while others show isolated organ involvement are not yet clear. Mechanisms underlying variable expressivity and partial penetrance may include unrecognized modifier genes, epigenetic influences, or environmental factors. Functional studies are needed to identify such modifiers and clarify their role.
Accurate molecular diagnosis could improve the outcome of patients and assist in prediction of disease prognosis, promote optional personalized treatment and avoid unnecessary treatments. In the context of MAFB-related disease Kaimori et al. (2021) described a positive effect of cyclosporine treatment in a patient with FSGS due to a MAFB variant [34]. Cyclosporine has a non-immunological effect on the stabilization of the actin cytoskeleton in kidney podocytes [35]. This could explain the direct effect of cyclosporine on reduction in protein excretion and its influence on synaptopodin, an actin binding protein that is an important regulator of podocyte function.
Given the frequent extra-renal manifestations associated with MAFB variants, multidisciplinary evaluation is essential, for example for early identification of hearing loss or ophthalmologic abnormalities. The molecular diagnosis also has implications for kidney transplantation. Whereas idiopathic FSGS is associated with a high risk of recurrence post-transplant, hereditary forms recur infrequently, and preemptive immunosuppression aimed at recurrence prevention is not required.
Improving diagnostic yield facilitates family-based testing, which is invaluable for identifying affected individuals and at-risk relatives. Furthermore, molecular diagnosis enables informed family planning, including evaluation of potential kidney donors and provision of reproductive counseling with options such as prenatal and preimplantation genetic testing.
5. Conclusions
In summary, we report a heterozygous MAFB variant, c.797T>C; p.(Leu266Pro), segregating in multiple related individuals from a single family, presenting with proteinuria, CKD, and variable extra-renal manifestations including DRS, auricular abnormalities, and neurodevelopmental involvement. We provide a comprehensive clinical description of a familial non-MCTO MAFB-associated phenotype and further expand the recognized clinical spectrum of MAFB-related disease. Structural modeling supports a domain-dependent pathogenic mechanism involving disruption of the C-terminal bZIP region. Further functional studies are required to elucidate the molecular mechanisms underlying this condition and refine genotype–phenotype correlations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rosenberg A.Z. Kopp J.B. Focal Segmental Glomerulosclerosis Clin. J. Am. Soc. Nephrol.20171250251710.2215/CJN.0596061628242845 PMC 5338705 · doi ↗ · pubmed ↗
- 2Fogo A.B. Causes and pathogenesis of focal segmental glomerulosclerosis Nat. Rev. Nephrol.201511768710.1038/nrneph.2014.21625447132 PMC 4772430 · doi ↗ · pubmed ↗
- 3Santín S. Bullich G. Tazón-Vega B. García-Maset R. Giménez I. Silva I. Clinical Utility of Genetic Testing in Children and Adults with Steroid-Resistant Nephrotic Syndrome Clin. J. Am. Soc. Nephrol.201161139114810.2215/CJN.0526061021415313 PMC 3087781 · doi ↗ · pubmed ↗
- 4Lepori N. Zand L. Sethi S. Fernandez-Juarez G. Fervenza F.C. Clinical and Pathological Phenotype of Genetic Causes of Focal Segmental Glomerulosclerosis in Adults Clin. Kidney J.20181117919010.1093/ckj/sfx 14329644057 PMC 5888331 · doi ↗ · pubmed ↗
- 5Vivante A. Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease Nat. Rev. Nephrol.20161213314610.1038/nrneph.2015.20526750453 PMC 5202482 · doi ↗ · pubmed ↗
- 6Kagan M. Eliyahu A. Ben Moshe Y. Vivante A. The genetic basis of chronic kidney disease in children and young adults Harefuah 202116083984634957723 · pubmed ↗
- 7Sato Y. Tsukaguchi H. Morita H. Higasa K. Tran M.T.N. Hamada M. Usui T. Morito N. Horita S. Hayashi T. A Mutation in Transcription Factor MAFB Causes Focal Segmental Glomerulosclerosis with Duane Retraction Syndrome Kidney Int.20189439640710.1016/j.kint.2018.02.02529779709 · doi ↗ · pubmed ↗
- 8Wang P.W. Eisenbart J.D. Cordes S.P. Barsh G.S. Stoffel M. Le Beau M.M. Human KRML (MAFB): c DNA Cloning, Genomic Structure, and Evaluation as a Candidate Tumor Suppressor Gene in Myeloid Leukemias Genomics 19995927528110.1006/geno.1999.588410444328 · doi ↗ · pubmed ↗