CD109 Deletion Promotes Myofibroblast Differentiation and Smad-Dependent Matrix Accumulation in Skin Fibrosis
Liqin Xu, Setareh Garousi, Adel Batal, Kenneth W. Finnson, Anie Philip

TL;DR
Deleting CD109 in mice worsens skin fibrosis by increasing collagen and fibroblast activity through TGF-β signaling.
Contribution
This study reveals CD109 as a negative regulator of dermal fibrosis through TGF-β/Smad signaling.
Findings
CD109 KO mice showed increased collagen I and fibronectin in bleomycin-induced fibrosis.
CD109 deficiency enhanced Smad1 and Smad2/3 phosphorylation, indicating stronger TGF-β signaling.
In vitro, CD109 KO fibroblasts had increased TGF-β-induced migration and collagen contraction.
Abstract
Skin fibrosis is characterized by excessive extracellular matrix (ECM) deposition, leading to tissue dysfunction and scarring. Transforming growth factor (TGF)-β is a central mediator of fibrosis. We previously identified CD109 as a TGF-β co-receptor and negative regulator of TGF-β signaling and fibrotic responses and showed that its epidermal overexpression reduces dermal fibrosis in vivo. However, the effects of CD109 loss in the dermis remain unclear. The current study investigates the impact of CD109 knockout (KO) on skin fibrosis using a bleomycin-induced fibrosis mouse model. Following bleomycin treatment, CD109 KO mice showed increased collagen I deposition and elevated fibronectin, CCN2, and α–smooth muscle actin expression in the skin, indicating enhanced ECM production and myofibroblast differentiation compared with wild-type mice. Additionally, CD109 KO mice displayed…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
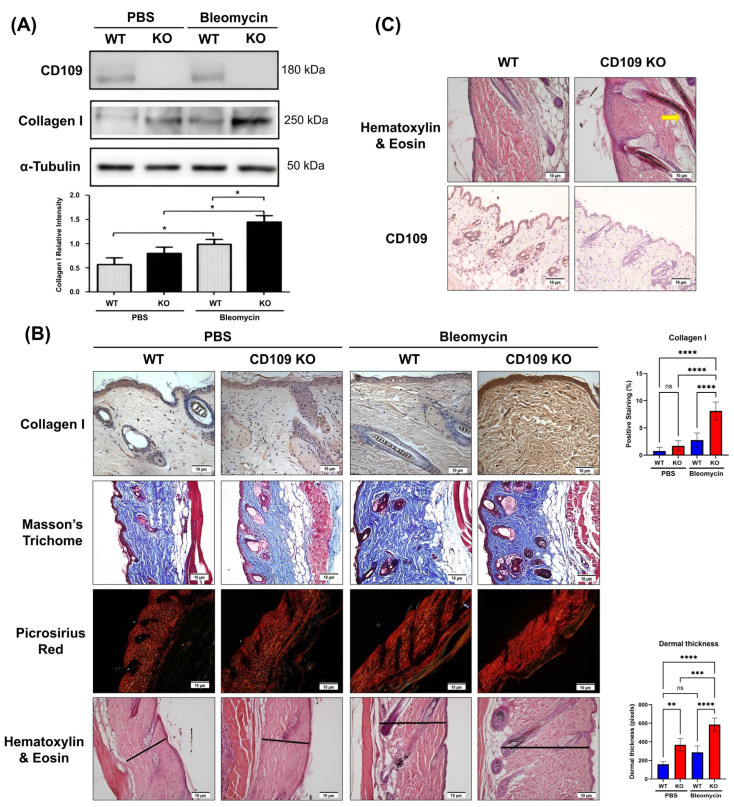 Figure 1
Figure 1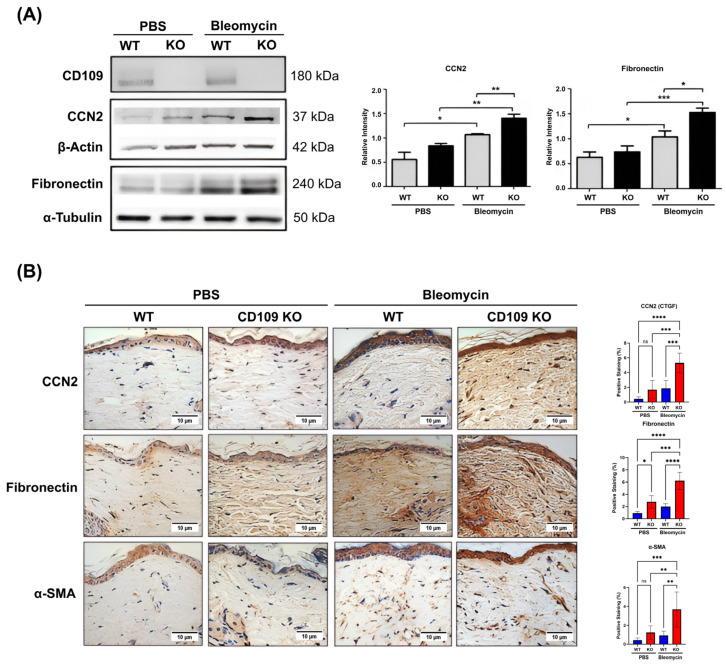 Figure 2
Figure 2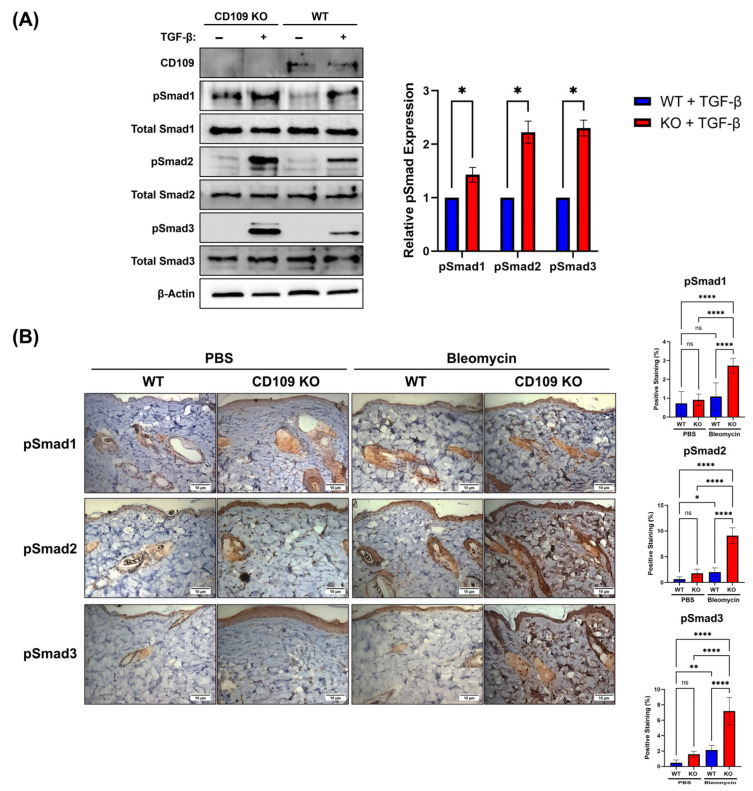 Figure 3
Figure 3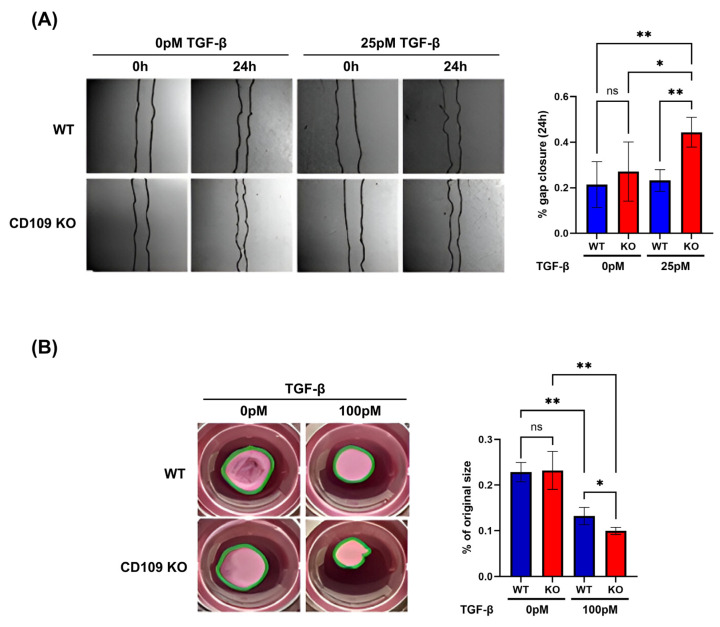 Figure 4
Figure 4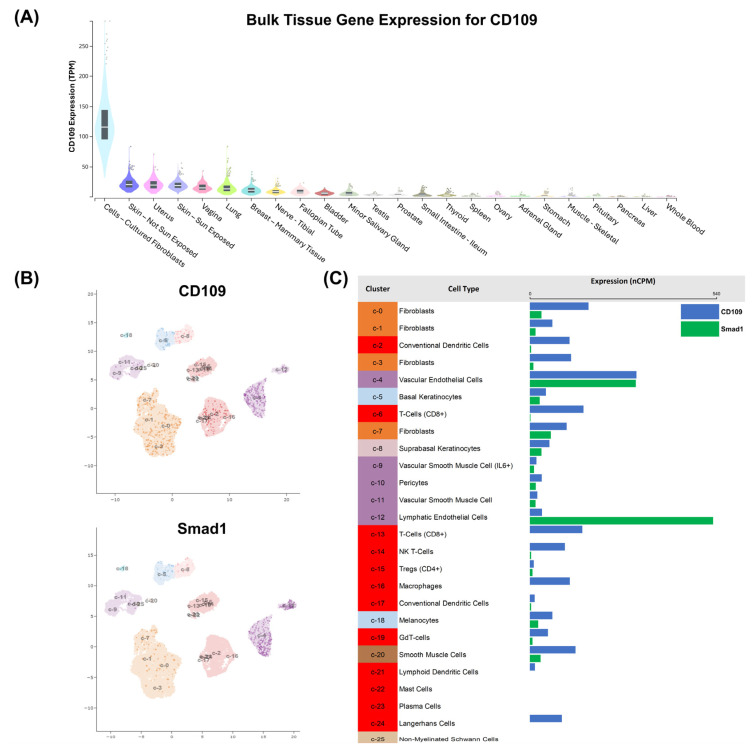 Figure 5
Figure 5- —Canadian Institutes of Health Research (CIHR)
- —Department of Defense Investigator-Initiated Research Award
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective Tissue Growth Factor Research · TGF-β signaling in diseases · Wound Healing and Treatments
1. Introduction
Skin fibrosis, a condition marked by excessive accumulation of extracellular matrix (ECM) proteins like collagen and fibronectin, poses a substantial clinical challenge, especially in disorders such as scleroderma (systemic sclerosis, SSc) and hypertrophic scars [1,2]. This condition is closely associated with impaired wound healing processes and abnormal fibroblast function, which results in increased tissue stiffness, diminished functionality, and cosmetic issues [3]. Central to these processes is transforming growth factor beta (TGF-β), a potent pro-fibrotic cytokine that plays a pivotal role in wound healing and inflammatory responses [1,4].
TGF-β is a pleiotropic cytokine that plays fundamental roles in development and homeostasis by regulating cellular processes including proliferation, differentiation and ECM production [5]. TGF-β canonical signaling is transmitted by a pair of transmembrane serine/threonine kinases known as the TGF-β type II (TβRII) and type I (TβRI, also known as activin receptor-like kinase-5 or ALK5) receptors [6,7]. TGF-β first binds to TβRII, a constitutively active kinase, which in turn phosphorylates ALK5, resulting in its activation [6,7,8]. ALK5 then phosphorylates intracellular Smad2 and Smad3 proteins, which form a complex with Smad4, enter the nucleus and regulate gene expression [9,10]. TGF-β can also signal through a different type I receptor known as activin receptor-like kinase-1 (ALK1), which phosphorylates Smad-1protein [11,12,13,14], leading to different intracellular signaling outcomes including the inhibition of ALK5-Smad2/3 signaling [14,15]. Furthermore, TGF-β is also able to activate so-called ‘non-Smad’ pathways including the mitogen-activated protein (MAP) kinases, phosphatidylinositol-3-kinase and Rho-like GTPases [16,17,18]. The critical role of TGF-β signaling in the development of fibrosis through the activation of fibroblasts and the synthesis of ECM proteins is well documented [19,20].
CD109 is a glycosylphosphatidylinositol (GPI)-anchored protein and a member of the α2-macroglobulin (α2M)/C3, C4, C5 complement family of thioester-containing proteins [21,22,23]. We were the first to demonstrate that CD109 functions as a TGF-β co-receptor [21], negatively regulating TGF-β signaling and inhibiting responses such as extracellular matrix (ECM) synthesis and skin fibrosis [21,24,25,26,27,28,29]. We have shown that CD109 is a critical regulator of TGF-β receptor levels and signaling by promoting TGF-β receptor internalization and Smurf2-mediated degradation via the caveolar pathway [30,31,32]. We found that transgenic (TG) mice overexpressing CD109 in the epidermis exhibit reduced inflammatory cell (neutrophil and macrophage) infiltration, granulation tissue formation, and improved collagen fiber organization during wound healing, when compared to WT littermates [27]. We also showed that epidermal overexpression of CD109 leads to a decrease in bleomycin-induced skin fibrosis, as evidenced by a significant reduction in dermal thickness, collagen cross-linking, collagen and fibronectin content and phospho-Smad2/3 levels, as compared to their WT littermates [28]. Under basal conditions, the TG mice with epidermal CD109 overexpression exhibit decreased ALK5-Smad2/3 signaling and enhanced ALK1-Smad1/5 activation. The decreased ECM production in the dermis displayed by these mice was shown to be due to a paracrine mechanism that likely involves the secretion of soluble factors from epidermal keratinocytes acting on the underlying dermal fibroblasts [24].
The Global CD109 KO mice have been characterized by others previously and have been shown to display epidermal hyperplasia, transient impairment of hair growth and infiltration of immune cells (neutrophils, T and B lymphocytes) in the dermis, but no difference in skin wound closure rate was noted as compared to WT littermates [33]. Also, in a later study, this group has reported that these mice exhibit osteopenia with an osteoporosis-like phenotype in vivo [34]. Another study using these mice has shown that CD109 deletion suppresses chemically induced skin tumorigenesis [35]. Whether CD109 deficiency affects experimental skin fibrosis has not been investigated. Here, we show that CD109 KO mice display enhanced bleomycin-induced skin fibrosis with significantly increased collagen deposition, ECM production, and myofibroblast differentiation as compared to WT mice. Additionally, CD109 KO mice display enhanced phosphorylation of Smad2 and Smad3 proteins during bleomycin-induced skin fibrosis, indicating heightened TGF-β signaling. In vitro, CD109 KO fibroblasts exhibit increased basal proliferation, as well as TGF-β1-induced cell migration and collagen contraction compared to WT fibroblasts. These findings suggest that CD109 deficiency worsens dermal fibrosis by enhancing TGF-β/Smad signaling and myofibroblast activity, highlighting CD109 as an important regulator of skin homeostasis and a potential therapeutic target in fibrotic skin diseases like scleroderma.
2. Results
CD109 KO mice display enhanced dermal collagen deposition as compared to WT mice, during bleomycin-induced skin fibrosis.
The levels of collagen 1 in skin tissue extracts of CD109 KO versus WT mice did not show significant differences under basal conditions. However, during bleomycin-induced skin fibrosis, CD109 KO mice exhibited a significantly greater increase in collagen I levels in their skin as compared to WT mice (p < 0.05) (Figure 1A). Further validation of this finding was obtained through immunohistochemical analysis, which showed that tissue sections obtained from bleomycin-induced fibrotic skin of CD109 KO mice displayed significantly enhanced staining for collagen I as compared to those of WT mice (p < 0.0001) (Figure 1B). Additionally, consistent with these findings, Masson’s Trichrome (Figure 1B) and Picrosirius Red staining (Figure 1B) revealed that collagen was more compactly arranged in tissue sections of bleomycin-induced fibrotic skin of CD109 KO mice, when compared to those of WT mice. Moreover, CD109 KO mice display increased bleomycin-induced dermal thickening as compared to WT mice (p < 0.0001) (Figure 1B). Finally, CD109 KO mice exhibit epidermal hyperplasia and abnormal epidermal appendages, consistent with findings by [30] (Figure 1C).
CD109 KO mice display increased dermal ECM production and myofibroblast differentiation as compared to WT mice, during bleomycin-induced skin fibrosis.
Our results reveal that during bleomycin-induced skin fibrosis, CD109 KO mice skin exhibits significantly higher levels of fibronectin (p < 0.05) and connective tissue growth factor (CTGF, CCN2) (p < 0.01) protein expression when compared to WT mice skin, as evidenced by Western blot analysis (Figure 2A). Results from IHC staining for fibronectin (p < 0.0001) and CCN2 (p < 0.001) provided further validation of those results (Figure 2B). The differentiation of fibroblasts into myofibroblasts is characterized by excessive ECM synthesis and increased α-SMA expression. Our results using IHC staining demonstrate that α-SMA levels are higher in the skin of CD109 KO mice than in WT mice following bleomycin treatment (p < 0.01), pointing to increased myofibroblast differentiation in CD109-deficient mice during skin fibrosis (Figure 2B).
CD109 KO mice display increased phosphorylation of Smad1, Smad2, and Smad3 as compared to WT mice.
We next investigated the impact of CD109 deletion on activation of Smad2/3 and Smad1 pathways in vitro using dermal fibroblasts treated with TGF-β and in vivo during bleomycin-induced skin fibrosis. Using Western blot and IHC, we examined the levels of phosphorylated Smad1, Smad2, and Smad3 proteins in dermal fibroblasts. Western blot analysis highlights increased pSmad1 (p < 0.05), pSmad2 (p < 0.05), and pSmad3 (p < 0.05) phosphorylation after TGF-β treatment (Figure 3A). IHC analysis revealed elevated levels of pSmad1 (p < 0.0001), pSmad2 (p < 0.0001), and pSmad3 (p < 0.0001) in CD109 KO mice as compared to WT counterparts after bleomycin treatment (Figure 3B).
CD109 KO fibroblasts showed enhanced TGF-β1-induced cell migration and collagen gel matrix contraction as compared to WT fibroblasts.
Analysis of cell migration using an in vitro wound healing assay (scratch assay) showed that the CD109 KO and WT fibroblasts migrated at similar rates under baseline conditions at 24 h (p = 0.698). However, CD109 KO fibroblasts showed a significantly higher rate of TGF-β1-induced cell migration as compared to WT fibroblasts (p < 0.01) (Figure 4A). Stimulation with 25 pM TGF-β1 of CD109 KO fibroblasts showed a wound gap closure of 44.35 ± 6.5% at 24 h versus 23.16 ± 4.8% in WT fibroblasts (p < 0.01) (Figure 4A). In addition, CD109 KO fibroblasts showed a significantly greater increase in TGF-β1-induced collagen gel contraction as compared to WT fibroblasts (p < 0.05) (Figure 4B).
CD109 is widely expressed in human tissues (Figure 5A) and it is differentially expressed in subpopulations of skin-associated cells (Figure 5B upper panel and Figure 5C).
To examine CD109 expression in the human body, we analyzed publicly available bulk tissue RNA expression data from the Genotype-Tissue Expression Project (GTEx) database, which contains measurements from many different human tissues. CD109 expression, reported as transcripts per million (TPM), was compared across a wide range of tissues using visualization tools available on the GTEx Portal.
Bulk transcriptomic analysis demonstrated that CD109 is highly expressed in fibroblast-rich tissues, including skin, while showing minimal expression in most internal organs and circulating blood, indicating preferential enrichment within mesenchymal compartments (Figure 5A). To further explore gene expression at the single-cell level in human skin, we used single-cell RNA sequencing datasets from the Human Protein Atlas single-cell database to identify gene expression in individual skin cell populations. Expression levels for CD109 and Smad1 were summarized as normalized counts per million (nCPM) and visualized as bar charts corresponding to clusters of cells identified by single-cell RNA sequencing analysis and mapped using Uniform Manifold Approximation and Projection (UMAP) plots. These results revealed that CD109 expression is enriched in fibroblast clusters and vascular endothelial cells (Figure 5B,C). In contrast, Smad1 displayed a broader distribution but was reduced in CD109-high fibroblast clusters, showing an inverse correlation with CD109 expression, while higher levels were observed in endothelial and lymphatic endothelial cells (Figure 5B,C).
3. Discussion
Skin fibrosis, characterized by excessive ECM deposition and tissue remodeling, is the hallmark of conditions such as hypertrophic scarring and keloid formation, where wound healing is dysregulated [1], and of diseases like scleroderma (systemic sclerosis, SSc), where fibrosis extends beyond the skin to internal organs [2]. We have shown previously that CD109 protein levels are increased in SSc patient skin as compared to normal healthy skin [29] and decreased in psoriasis patient skin as compared to adjacent uninvolved skin [32], suggesting that dysregulation of CD109 expression may play a pathological role in these conditions. We have also shown that TG mice overexpressing CD109 in the epidermis show improvement in wound healing and fibrosis parameters in experimental models in vivo [27,28]. Altering CD109 expression in the dermal fibroblasts has not been investigated. In the current study, using a global CD109 KO mouse model, we show that CD109-deficient mice exhibit enhanced bleomycin-induced skin fibrosis, as characterized by increased dermal thickening, ECM production and myofibroblast differentiation when compared to WT mice. Although collagen I levels did not differ significantly between WT and CD109 KO skin under basal conditions, Western blot analysis revealed a consistent trend toward increased collagen I in CD109-deficient mice. While not statistically significant, this observation raises the possibility that CD109 contributes to the regulation of basal dermal collagen homeostasis. Subtle alterations in ECM set-points under resting conditions may predispose tissue to exaggerated fibrotic responses upon injury or challenge, and future studies will be required to directly address this possibility. This enhanced skin fibrosis is associated with heightened activation of Smad1, Smad2, and Smad3 signaling, confirming that CD109 functions as a negative regulator of TGF-β-driven fibrotic responses in the skin. Additionally, isolated CD109 KO mouse fibroblasts demonstrate increased TGF-β1-induced migration and collagen gel matrix contraction, further underscoring the role of endogenous CD109 in controlling fibroblast activity and fibrosis progression.
CD109 expression is elevated in scleroderma skin, likely reflecting a compensatory response to chronic TGF-β activation, but reduced in psoriatic lesions, where loss of CD109-mediated inhibition of TGF-β contributes to keratinocyte hyperproliferation and aberrant ECM remodeling [29,32,36]. These opposing patterns suggest distinct, cell-type-specific roles for CD109, restraining TGF-β-driven fibrosis in dermal fibroblasts and maintaining epidermal homeostasis in keratinocytes [37]. Further, understanding how CD109 interacts with immune and endothelial cells within the dermal microenvironment may reveal additional mechanisms underlying its anti-fibrotic activity in skin [38].
While our finding that CD109 deletion leads to increased phosphorylation of Smad2 and Smad3 proteins in the current study is consistent with our previous report that epidermal overexpression of CD109 diminishes phosphorylation of Smad2 and Smad3 [24], our observation that Smad1 phosphorylation is upregulated in both the CD109 deletion (current study) and overexpression [24] models is intriguing. The analysis of the gene bulk expression and single-cell databases demonstrated an inverse expression pattern in skin fibroblast cell populations with elevated CD109 expression associated with lower Smad1 expression (Figure 5). This inverse relationship supports the concept that CD109-rich fibroblast populations are associated with reduced Smad1 transcriptional output, consistent with a modulatory role for CD109 in shaping Smad-dependent signaling landscapes within mesenchymal cells relevant to fibrotic regulation. However, the possibility that the increased Smad 1 phosphorylation is bone morphogenetic protein (BMP)-mediated (and not TGF-β-induced) cannot be ruled out. The critical role of Smad3 pathway activation in mediating fibrosis is well established. For example, targeting Smad3 can attenuate fibrosis [39], and disrupting Smad3 signaling reduces collagen synthesis, accumulation, and transcription, leading to less fibrosis [40]. The role of Smad2 and Smad1 in mediating skin fibrosis is less well understood. The relative significance of Smad3, Smad2, Smad1 and MAP kinases in mediating the antifibrotic effects of CD109 is an interesting question to pursue.
While the present study focused on defined components of the TGF-β signaling pathway and established fibrotic markers, the CD109 KO mouse model offers a powerful platform for broader discovery-based analyses. Unbiased approaches such as global transcriptomic or proteomic profiling of WT and CD109-deficient skin, both at baseline and following fibrotic challenge, may reveal additional signaling pathways, regulatory networks, and previously unrecognized mediators contributing to altered skin homeostasis and fibrosis susceptibility. Such studies could further clarify cell-type-specific and non-canonical mechanisms regulated by CD109 and expand our understanding of its role beyond the classical TGF-β axis.
A limitation of this study is the use of the bleomycin-induced mouse model, which, although widely used [39], does not fully reflect the complexity of human fibrotic skin disorders such as scleroderma [41]. In addition, the use of a global CD109 KO mouse model precludes cell-type-specific interpretation of the observed effects. Consequently, potential contributions from non-fibroblast populations cannot be excluded, and these may have influenced the fibrotic phenotype independently of fibroblast-intrinsic mechanisms. A further limitation is that we did not directly quantify TGF-β receptor protein levels (e.g., TβRII/ALK5/ALK1) in WT vs. CD109 KO skin; however, our pathway readouts (pSmads) and fibrosis endpoints robustly indicate heightened signaling in CD109-deficient skin. Furthermore, in previous studies, we have demonstrated that CD109 is an important regulator of TGF-β receptor levels [30,31,32]. Future studies using fibroblast-specific CD109 knockout mice and patient-derived skin biopsies will clarify CD109’s role in fibrosis and inflammation. Integrating genetic models with human data will better define their cell-specific functions and therapeutic potential in fibrotic skin disease.
In summary, this study identifies CD109 as a critical negative regulator of dermal fibrosis. Loss of CD109 enhanced fibrotic responses in vivo, as reflected by increased ECM deposition, myofibroblast differentiation, and Smad2/3 activation in bleomycin-treated CD109 KO mice relative to wild-type controls. Consistently, CD109-deficient fibroblasts displayed enhanced migration and collagen contraction in vitro, indicative of a profibrotic phenotype. Collectively, these findings position CD109 as a key regulator of skin homeostasis and underscore its potential as a therapeutic target in fibrotic skin disease.
4. Materials and Methods
Generation of Mouse Colony: Frozen sperm from CD109 heterozygotic C57BL/6 mice was obtained from Dr. Masahide (Department of Pathology, Nagoya University Graduate School of Medicine, Nagoya, Japan) [33] and used to expand the colony. For experiments, sixteen CD109 KO and sixteen WT male mice, aged 4–6 weeks, were selected from third-generation offspring.
Genotyping of CD109 Mice: Genomic DNA was extracted from mouse tail samples using the REDExtract-N-Amp™ Tissue PCR Kit (XNAT, Sigma-Aldrich, St. Louis, MO, USA). PCR was performed with specific primers to identify CD109 KO and WT alleles. Genotyping of mice was conducted using PCR with four specific primers: P1 (forward) 5′-GTCCCGCTTTCTGGTGACAG-3′, P2 (reverse) 5′-GTGTGACTGTTAGACAGTGCAG-3′, P3 (forward) 5′-CCATCGCCATCTGCTGCACG-3′, and P4 (reverse) 5′-ACGATCCTGAGACTTCCACAC-3′. PCR amplification was carried out under the following conditions: initial denaturation at 96 °C for 2 min, followed by 32 cycles of denaturation at 94 °C for 30 s, annealing at 65 °C for 30 s, and extension at 72 °C for 30 s. The expected amplicon sizes were 205 bp for the wild-type allele and 603 bp for the targeted allele [33].
In Vivo Bleomycin Treatment: Thirty-two mice, comprising sixteen CD109 KO and sixteen WT mice, were divided into groups receiving either bleomycin dissolved in phosphate-buffered saline (PBS) or PBS alone. Under anesthesia with isoflurane, their dorsal skin was shaved, and a 1 × 1 cm area for injection was outlined. The mice were subjected to treatment every other day, receiving either a 100 μL subcutaneous injection at a single site of bleomycin sulfate (15 μg, Wisent Bioproducts) in PBS or an equivalent volume of PBS alone. This regimen was maintained over a span of 28 days. Mice were euthanized on day 29 for sample collection.
Histology and Immunohistochemistry (IHC): Treated skin areas were processed for Hematoxylin and Eosin, Masson’s trichrome, or Picrosirius Red staining. Dermal thickness was quantified from the basement membrane to the hypodermis across five high-power fields for each section, with two distinct sections evaluated per specimen. This analysis was conducted using Image Pro Plus 6 software (Media Cybernetics, Bethesda, MD, USA). IHC was carried out at 4 °C overnight using specific antibodies to evaluate the expression levels of fibrosis markers such as α-smooth muscle actin (α-SMA), type I collagen, connective tissue growth factor (CTGF, CCN2), and fibronectin, compared to IgG negative controls. Additionally, the activation of TGF-β downstream signaling proteins, including phospho-Smad1 (pSmad1), phospho-Smad2 (pSmad2), and phospho-Smad3 (pSmad3), was assessed in a similar manner. The quantification of these markers was achieved by measuring the optical density of the calibrated images, which were restricted to the fibroblast-rich dermal compartment, using Fiji ImageJ 1.54g software [42]. The statistical significance was calculated using an ordinary one-way ANOVA.
Western Blot and Densitometry: Full-thickness skin tissue was homogenized in RIPA buffer supplemented with protease inhibitor cocktail, sodium orthovanadate and phenylmethylsulphonyl fluoride (PMSF). Tissue homogenate was clarified at 12,000× g for 10 min. The supernatant was collected for protein quantification using the Bio-Rad Lowry Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA). Samples were resolved by 7.5% or 10% SDS-PAGE and transferred to a nitrocellulose membrane. Measurement of markers of fibrosis (α-SMA, fibronectin, CTGF, collagen I) was performed using primary and HRP-conjugated secondary antibodies. Each experiment was performed in at least quadruplicate, and densitometry of immunoblots was measured using ImageJ 1.54g software (NIH) [43]. Student’s t-test was used for the calculation of statistical significance.
Isolation of Dermal Fibroblasts: CD109 KO mice and their WT mice, aged 0 to 5 days, were euthanized, the treated skin area was collected and fibroblasts were isolated. The procedure involved cleaning the bodies with 10% Poviodine-Iodine solution (Laboratoire Atlas Inc., Montreal, QC, Canada), rinsing with sterile water, and harvesting the skin under sterile conditions. The skin was minced and digested in a 0.1% collagenase-containing medium. After incubation and stirring for up to 2 h, the mixture was centrifuged at 180× g for 10 min to pellet the cells, which were then washed and suspended in fresh medium (DMEM). The cell culture was maintained at 37 °C with 5% CO_2_, with a medium change after 24 h and subsequently as needed.
Fibroblast Cell Count and Proliferation: Mouse fibroblasts were prepared at a concentration of approximately 1 × 10^6^ cells/mL. For viability assessment, cells were stained using a 0.4% trypan blue solution and allowed to sit for 5 min at room temperature. A 20 μL sample of the cell-trypan blue mixture was then applied to a hemocytometer to count both viable and non-viable cells. In total, 0.3 × 10^5^ CD109 KO and WT fibroblasts were seeded in each well of six-well plates and cultured under previously described conditions for dermal fibroblast isolation. Upon reaching confluence, fibroblasts were detached and recounted using the trypan blue exclusion method to compare viable cell counts between KO and WT cell populations.
In Vitro Wound Healing Scratch Assay: Third-passage CD109 KO and WT mouse fibroblasts (0.1 × 10^6^ cells) were cultured to confluence, serum-starved for 6.5 h, and then treated with mitomycin C (to inhibit proliferation) and with 0 pM, 25 pM, or 100 pM of TGF-β1 (Sanofi Genzyme), respectively. A scratch assay was performed on the cell monolayer using a 200 μL pipette tip, and gap closure was monitored over 24 h using serial photography. The extent of cell migration was quantified using ImageJ 1.54 g software [43] based on the closure of the scratched area. The statistical significance was calculated using an ordinary one-way ANOVA.
Collagen Contraction Assay: A total of 10 mg of rat tail collagen (11179179001, Sigma-Aldrich) was dissolved in 2.5 mL of 0.2% mM acetic acid solution at 4 °C, according to the manufacturer’s protocol, for a final concentration of 4 mg/mL. This collagen solution was mixed with DMEM containing 10% fetal bovine serum, NaOH and third passage CD109 KO and WT mouse fibroblasts to create a uniform gel mixture. This mixture was aliquoted into a 24-well plate and allowed to solidify at room temperature for 30 min. After solidification, 800 μL of supplemented medium was added, and the plate was incubated at 37 °C with 5% CO_2_ overnight. Post-incubation, fibroblasts were serum-starved for 6–8 h and treated with TGF-β1. Photographic documentation was made at 0 and 24 h to assess collagen contraction, which was quantified using ImageJ 1.54g software (NIH) [43] based on changes in gel size. The statistical significance was calculated using an ordinary one-way ANOVA.
Bulk Tissue Gene Expression and Single-Cell RNA Sequencing Analysis: Bulk tissue RNA expression data for CD109 (ENSG00000156535.15) were obtained from the Genotype-Tissue Expression (GTEx) Project, Analysis Release V10 (dbGaP accession phs000424.v10.p2), and reported as transcripts per million (TPM) across a broad range of human tissues using GTEx-provided visualization tools (https://www.gtexportal.org/home/gene/CD109/geneExpressionTab; accessed on 21 January 2026). Single-cell RNA sequencing data for CD109 and Smad1 were obtained from the Human Protein Atlas (HPA) (https://www.proteinatlas.org/; accessed 21 January 2026) single-cell database, which integrates curated human single-cell transcriptomic datasets. Cluster-level gene expression was summarized using normalized counts per million (nCPM) and visualized as bar charts linked to the corresponding UMAP clusters.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hameedi S.G. Saulsbery A. Olutoye O.O. The pathophysiology and management of pathologic scarring-a contemporary review Adv. Wound Care 202514486410.1089/wound.2023.018538545753 PMC 11839539 · doi ↗ · pubmed ↗
- 2Son H.H. Moon S.J. Pathogenesis of systemic sclerosis: An integrative review of recent advances J. Rheum. Dis.2025328910410.4078/jrd.2024.012940134549 PMC 11931279 · doi ↗ · pubmed ↗
- 3Bainbridge P. Wound healing and the role of fibroblasts J. Wound Care 201322407–408, 410–41210.12968/jowc.2013.22.8.40723924840 · doi ↗ · pubmed ↗
- 4Tie Y. Tang F. Peng D. Zhang Y. Shi H. TGF-β signal transduction: Biology, function and therapy for diseases Mol. Biomed.202234510.1186/s 43556-022-00109-936534225 PMC 9761655 · doi ↗ · pubmed ↗
- 5Morikawa M. Derynck R. Miyazono K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology Cold Spring Harb. Perspect. Biol.20168 a 02187310.1101/cshperspect.a 02187327141051 PMC 4852809 · doi ↗ · pubmed ↗
- 6Liu T. Feng X.H. Regulation of TGF-β signalling by protein phosphatases Biochem. J.201043019119810.1042/BJ 2010042720704570 PMC 3154754 · doi ↗ · pubmed ↗
- 7Wrighton K.H. Lin X. Feng X.-H. Phospho-control of TGF-β superfamily signaling Cell Res.20091982010.1038/cr.2008.32719114991 PMC 2929013 · doi ↗ · pubmed ↗
- 8Thatcher J.D. The TGF-β signal transduction pathway Sci. Signal.20103 tr 410.1126/scisignal.3119 tr 420424268 · doi ↗ · pubmed ↗