Integrative Multi-Omics Analysis Identifies NUP205 as a Candidate Prognostic Biomarker in Liver Hepatocellular Carcinoma
Eun-A Jeong, Jae-Ho Lee, Jongwan Kim

TL;DR
This study identifies NUP205 as a potential biomarker for liver cancer prognosis and drug resistance, based on multi-omics analysis and in vitro experiments.
Contribution
The novel contribution is the comprehensive investigation of NUP205's role in liver hepatocellular carcinoma, including its epigenetic regulation and drug sensitivity associations.
Findings
NUP205 expression is elevated in liver hepatocellular carcinoma and linked to poor prognosis and advanced clinicopathological features.
Promoter hypomethylation and miRNA interactions regulate NUP205 expression in liver cancer.
NUP205 knockdown reduces mRNA levels and increases sensitivity to doxorubicin in liver cancer cells.
Abstract
Patients with Liver Hepatocellular carcinoma (LIHC) have a poor prognosis due to late-stage diagnosis and the limited efficacy of drug treatments. Dysregulation of nuclear pore complex (NPC) components, particularly nucleoporins (NUPs), may play a role in tumor progression. However, the specific role of NUP205 in LIHC has not been comprehensively investigated. We evaluated the expression, prognostic significance, epigenetic regulation, microRNA(miRNA) interactions, drug sensitivity, and biological functions of NUP205 in LIHC. Comprehensive bioinformatics analyses were performed using publicly available databases and web-based analysis platforms, including The Cancer Genome Atlas (TCGA), UALCAN, and the Kaplan–Meier Plotter (KM Plotter), among others. In vitro validation was performed using small interfering RNA (siRNA)-mediated knockdown of NUP205 in HepG2 cells, followed by…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
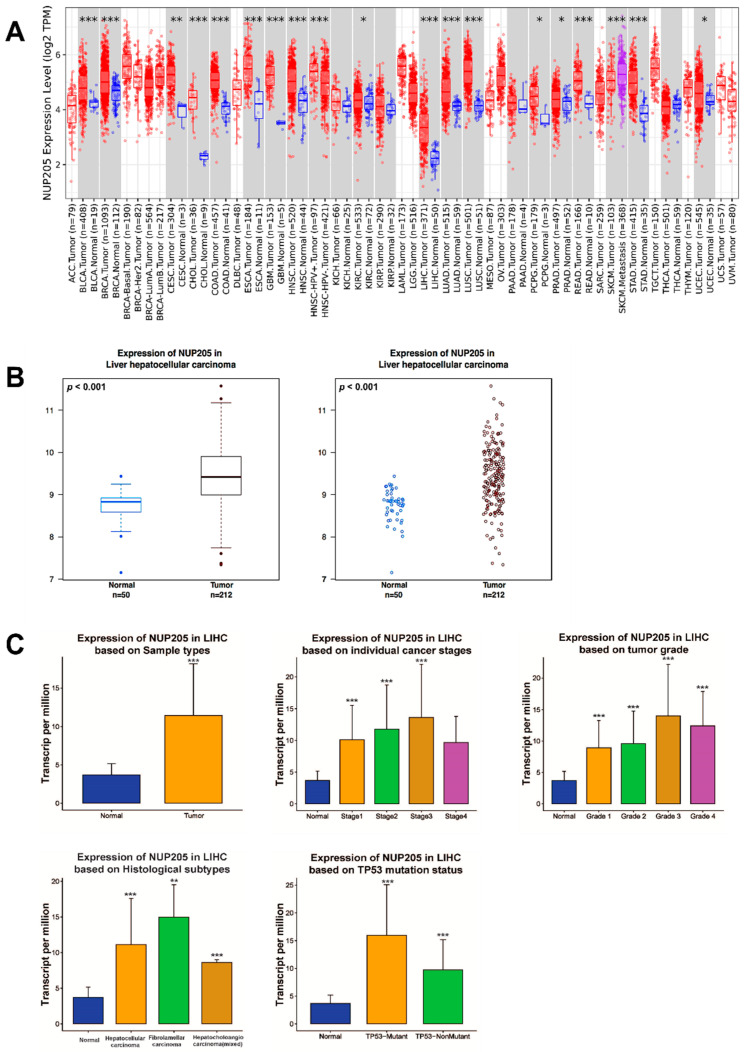 Figure 1
Figure 1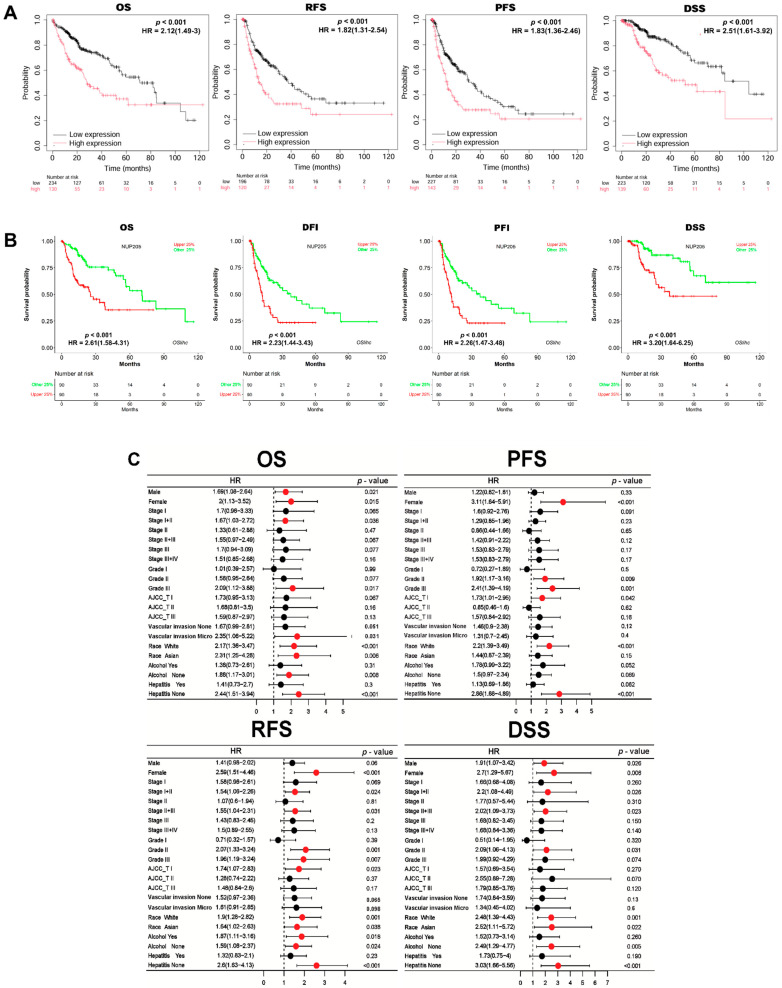 Figure 2
Figure 2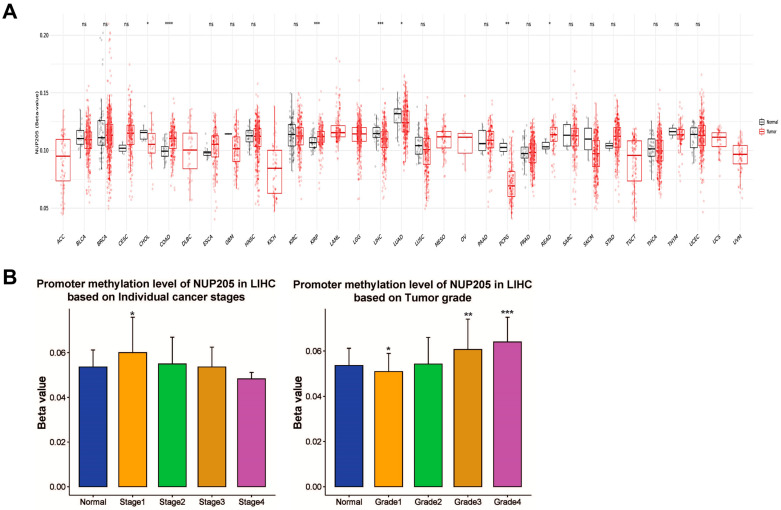 Figure 3
Figure 3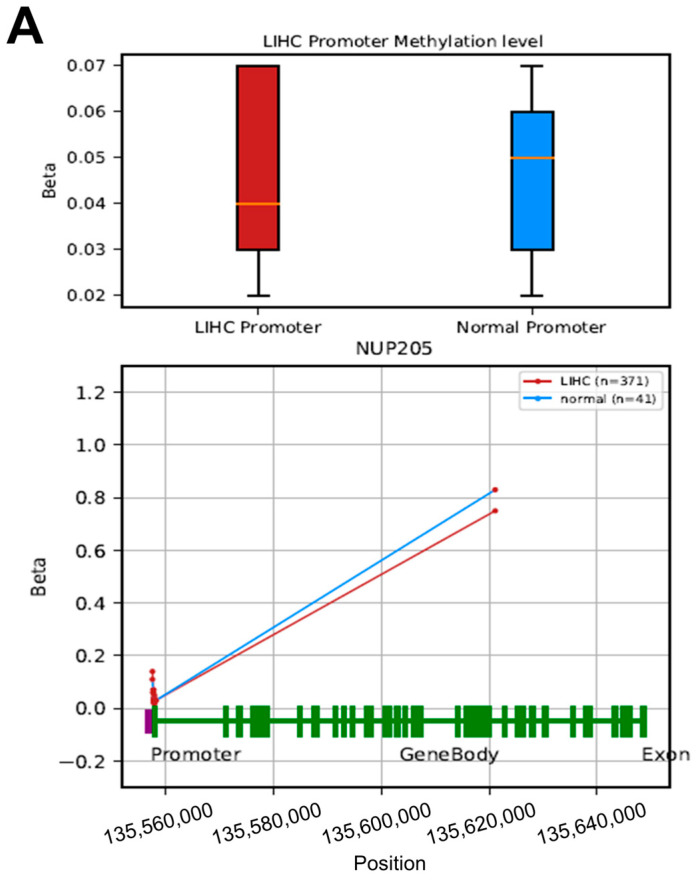 Figure 4
Figure 4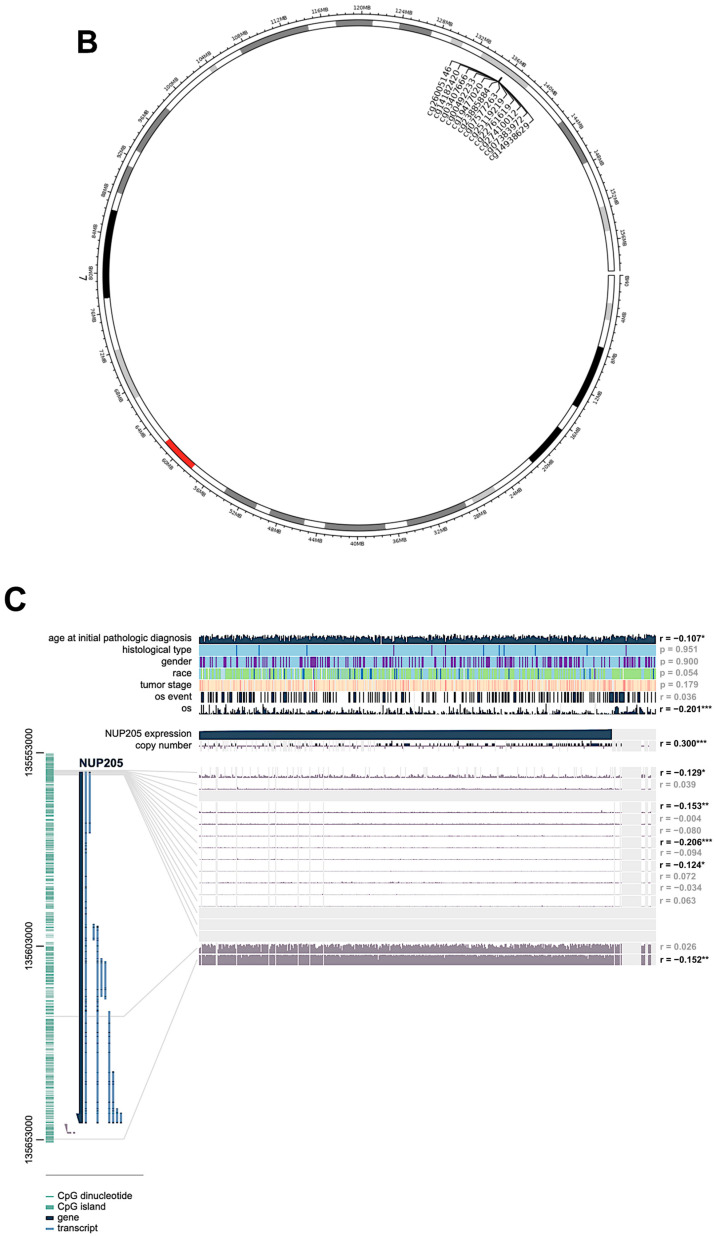 Figure 5
Figure 5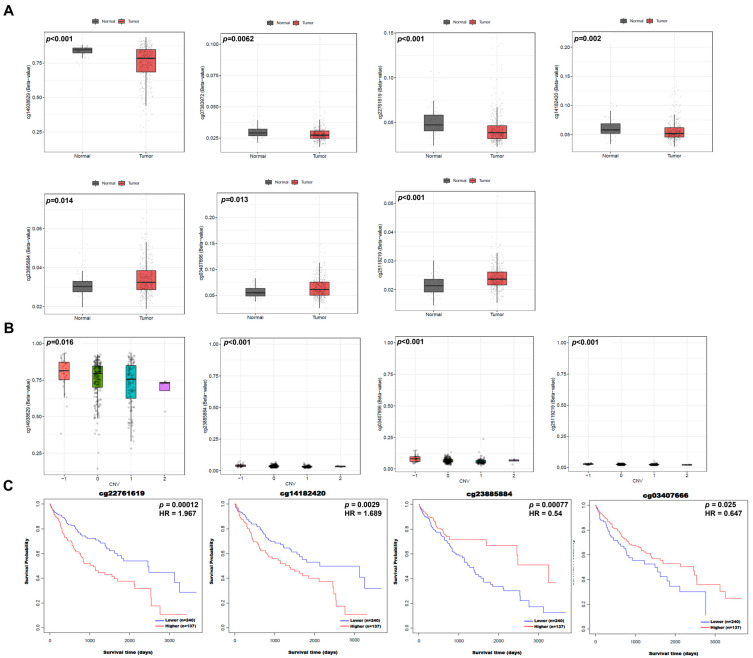 Figure 6
Figure 6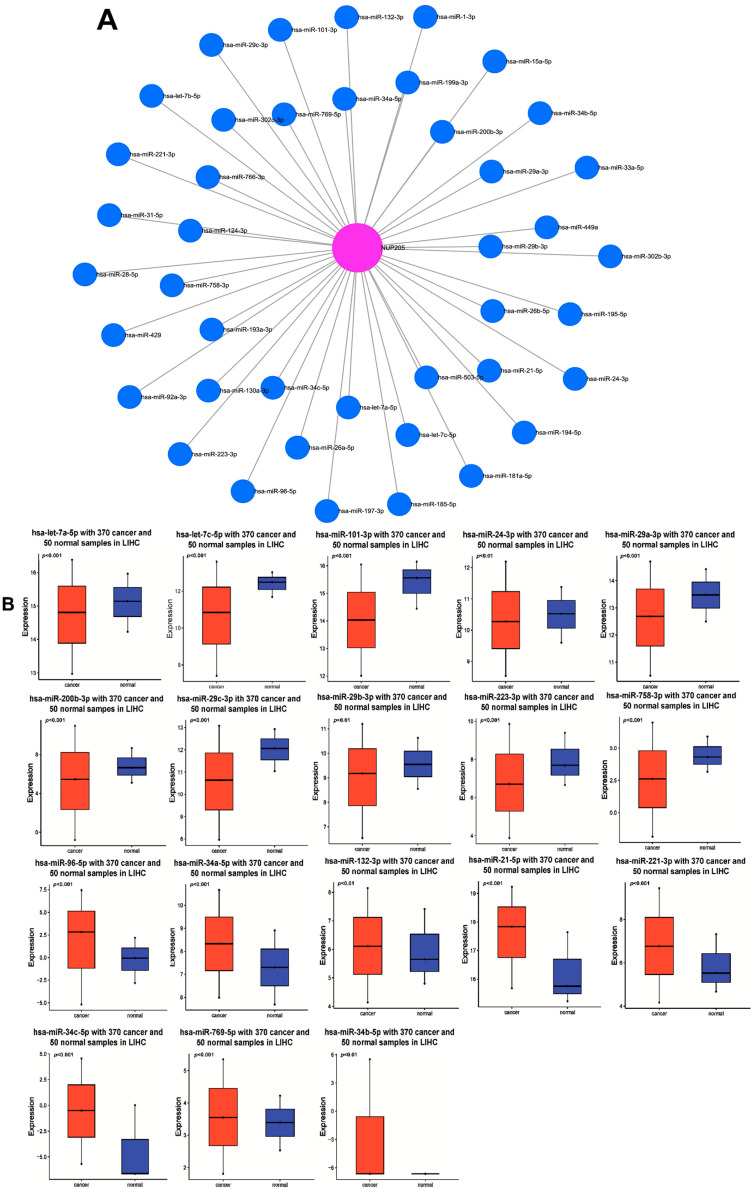 Figure 7
Figure 7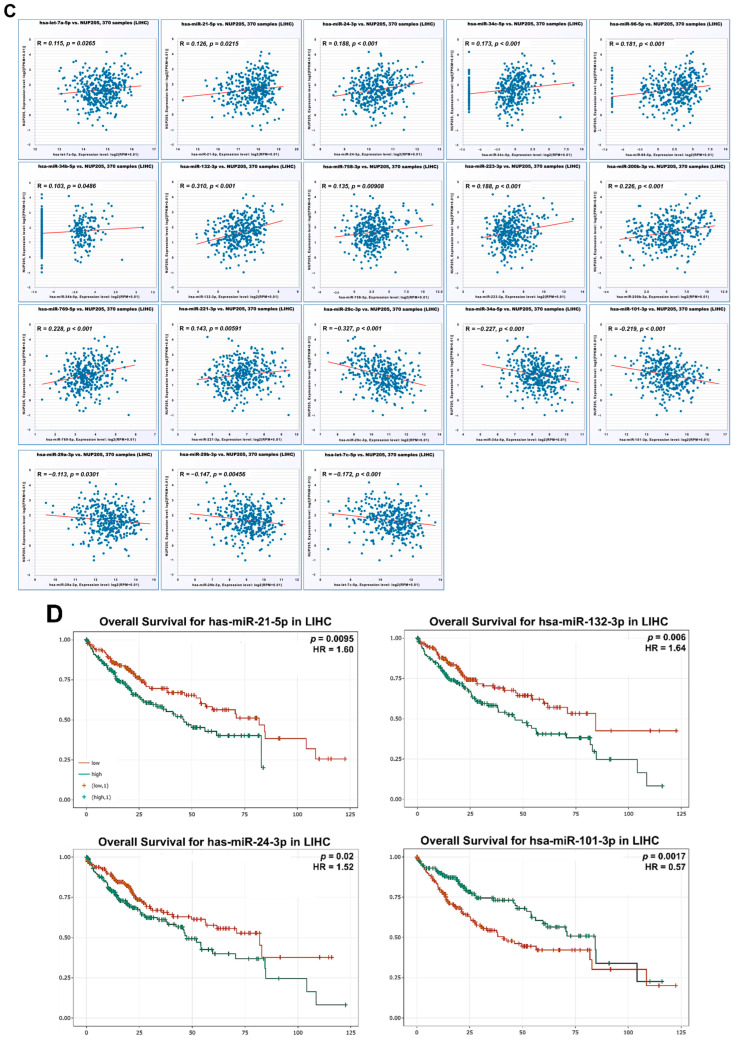 Figure 8
Figure 8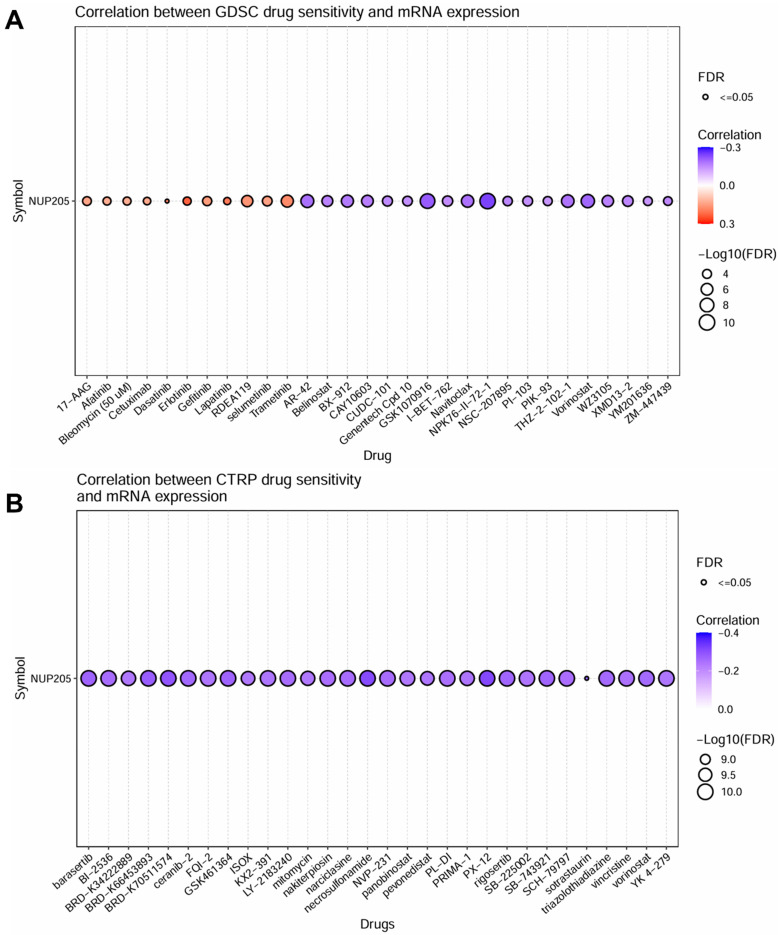 Figure 9
Figure 9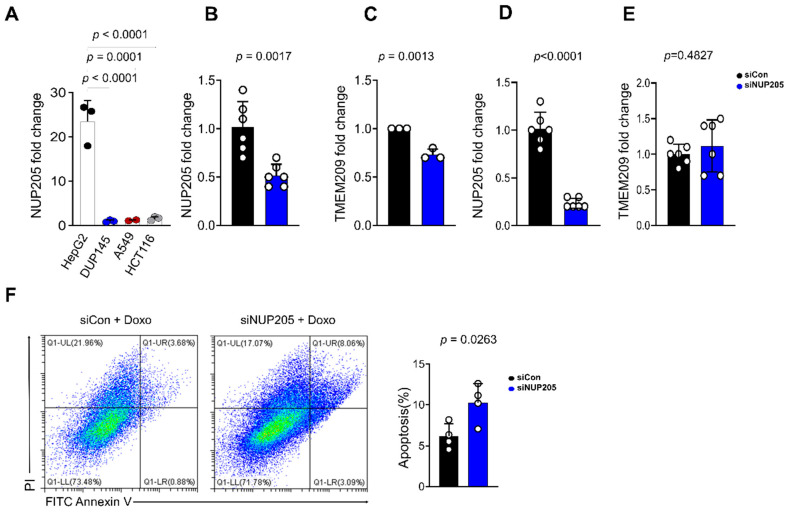 Figure 10
Figure 10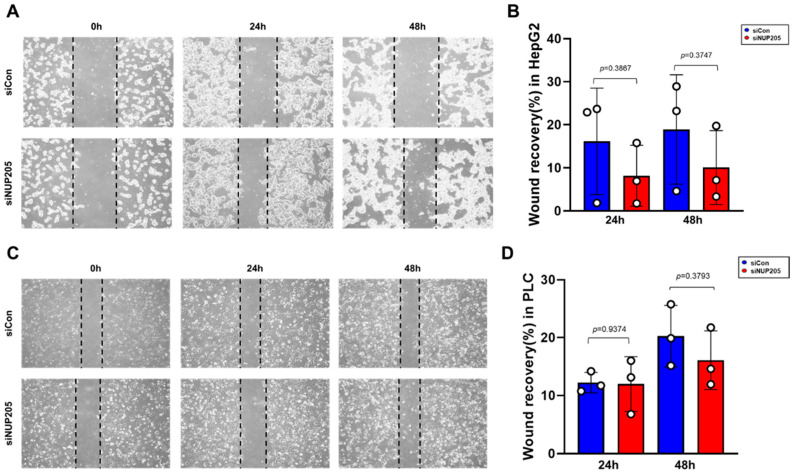 Figure 11
Figure 11Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNuclear Structure and Function · RNA Research and Splicing · Ferroptosis and cancer prognosis
1. Introduction
Liver Hepatocellular carcinoma (LIHC), the most common form of primary liver cancer, represents a significant global health challenge characterized by its increasing incidence and high mortality rates. In 2020, approximately 905,700 new cases of liver cancer were diagnosed worldwide, accompanied by 830,200 deaths, highlighting the persistently poor prognosis [1,2,3]. The principal etiological risk factors for LIHC include chronic infections with hepatitis B and C viruses, excessive alcohol consumption, and metabolic-associated fatty liver diseases, such as non-alcoholic fatty liver disease (NAFLD), which frequently occur in the context of cirrhosis [4,5]. Although advances in vaccination and antiviral therapies have reduced the incidence of virus-related LIHC in certain regions, the overall global incidence continues to increase, partly driven by the NAFLD epidemic and other lifestyle-related factors [5]. Prognosis remains unfavorable, and early-stage LIHC amenable to curative resection or liver transplantation can achieve 5-year survival rates exceeding 70%; the majority of patients present with advanced disease, for which 5-year survival rates fall below 20% [6,7,8]. These clinical challenges underscore the urgent need to identify robust prognostic biomarkers and novel molecular targets for LIHC.
Nucleoporin 205 (NUP205) has recently attracted attention as a potential oncogenic factor in several malignancies. NUP205 is a core structural component of the nuclear pore complex (NPC), which is essential for nucleocytoplasmic transport and maintenance of cellular homeostasis [9,10]. Dysregulation of NPC components has been increasingly linked to tumorigenesis and immune evasion [11]. Among nucleoporins, NUP205 was prioritized in this study based on preliminary The Cancer Genome Atlas (TCGA) analyses showing consistent upregulation in LIHC and significant associations with adverse clinical outcomes. While several nucleoporins have been implicated in cancer development, the functional and clinical relevance of NUP205 in LIHC remains insufficiently characterized.
Aberrant overexpression of NUP205 has been reported in LIHC and other malignancies, including colorectal, bladder, and lung cancers, where it is associated with enhanced tumor progression and poor prognosis [12,13,14]. Functional studies have further suggested that NUP205 promotes tumor cell proliferation in LIHC models [15]. However, the molecular mechanisms responsible for NUP205 dysregulation and its contribution to LIHC progression remain largely unclear.
Accumulating evidence indicates that gene dysregulation in LIHC is driven by multiple interacting molecular mechanisms, including epigenetic modifications, genomic alterations, and post-transcriptional regulation. DNA methylation abnormalities, copy-number variations, and microRNA (miRNA)-mediated regulation have each been shown to influence oncogene activation, tumor suppressor silencing, and therapeutic resistance in LIHC [16,17,18,19,20,21]. Importantly, these regulatory layers do not operate independently but instead form interconnected networks that collectively shape tumor behavior. Consequently, integrative multi-omics approaches are essential to fully elucidate the complex regulatory landscape of potential drivers like NUP205, capturing complementary alterations that single-omics analyses may miss [22].
The dysregulation of oncogenes in LIHC arises from complex and interdependent molecular mechanisms operating across multiple regulatory layers, including transcriptional, epigenetic, and post-transcriptional processes. Single-omics approaches are often insufficient to fully capture this complexity, as alterations at one molecular level may be compensated or modulated by regulatory events at another. Therefore, integrative multi-omics analyses provide a biologically grounded framework for generating robust hypotheses by revealing coordinated dysregulation across complementary molecular domains [22,23].
In this study, we aimed to elucidate the role of NUP205 in LIHC through cross-validation using multiple public databases, complemented by validation via in-house experimental assays. Our analyses consistently revealed significant upregulation of NUP205 expression in LIHC tissues, which was further confirmed at both the messenger RNA (mRNA) and protein levels through laboratory experiments. Elevated NUP205 expression was strongly associated with poor prognosis, underscoring its potential as a prognostic biomarker for LIHC. Furthermore, we investigated the mechanisms underlying NUP205 overexpression by examining its relationship with DNA methylation and miRNA regulation. These findings provide an integrative multi-omics-based framework suggesting that NUP205 dysregulation in LIHC may be influenced by epigenetic and post-transcriptional regulatory mechanisms. This framework offers a rationale for further experimental investigation of NUP205 as a candidate biomarker and regulatory factor in LIHC rather than definitive mechanistic or therapeutic evidence.
2. Results
2.1. NUP205 mRNA Expression Is Upregulated in LIHC
NUP205 mRNA expression was markedly elevated in a variety of cancer types relative to normal tissues, including LIHC, bladder urothelial carcinoma, breast invasive carcinoma, cervical squamous cell carcinoma and endocervical adenocarcinoma, cholangiocarcinoma, colon adenocarcinoma, esophageal carcinoma, glioblastoma multiforme, head and neck squamous cell carcinoma (HNSC), Human Papillomavirus (HPV)-positive HNSC, kidney renal clear cell carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, pancreatic adenocarcinoma, pheochromocytoma and paraganglioma, prostate adenocarcinoma, skin cutaneous melanoma–metastasis, stomach adenocarcinoma, and uterine corpus endometrial carcinoma (Figure 1A). To validate these findings in LIHC, NUP205 expression was further examined using the Wanderer database, which independently confirmed significant upregulation of NUP205 in LIHC tissues compared to normal tissues (p = 0.001) (Figure 1B). NUP205 expression was assessed across various clinicopathological subgroups within LIHC using the UALCAN database. This analysis demonstrated significant associations between NUP205 expression and multiple clinical subgroups, including primary tumor, tumor stage (I–III), tumor grade (I–IV), and histological subtype (Figure 1C). The consistent findings across multiple independent datasets underscore the potential utility of NUP205 as a pan-cancer biomarker and highlight its diagnostic and prognostic relevance, particularly for LIHC.
2.2. Upregulated NUP205 Is Associated with Poor Prognosis in LIHC
To evaluate the prognostic value of NUP205 expression in LIHC, we performed survival analyses using the Kaplan–Meier Plotter (KM Plotter) and Online consensus Survival for liver hepatocellular carcinoma (OSlihc) databases. Elevated NUP205 expression was significantly associated with poor prognosis in LIHC, as demonstrated by worse Overall Survival (OS) (Hazard Ratio [HR]: 2.12, p < 0.001), Recurrence-Free Survival (RFS) (HR: 1.82, p < 0.001), Progression-Free Survival (PFS) (HR: 1.83, p < 0.001), and Disease-Specific Survival (DSS) (HR: 2.51, p < 0.001) (Figure 2A). Subsequent analyses confirmed that upregulated NUP205 expression was significantly associated with poorer prognosis in OS (HR: 2.6081, p < 0.001), Disease-Free Interval (DFI) (HR: 2.2252, p < 0.001), PFS (HR: 2.26, p < 0.001), and DSS (HR: 3.20, p < 0.001) (Figure 2B). Further investigation revealed a consistent association between elevated NUP205 levels and an unfavorable prognosis in patients with LIHC. The relationship between NUP205 expression and clinicopathological features was also examined, revealing that higher NUP205 expression was correlated with worse prognosis across various subgroups. Specifically, for OS, increased NUP205 expression was associated with poorer outcomes in male (HR: 1.69, p = 0.021), female (HR: 2.0, p = 0.015), stage I+II (HR: 1.67, p = 0.036), grade III (HR: 2.09, p = 0.017), vascular invasion (micro, HR: 2.35, p = 0.031), race (white, HR: 2.17, p < 0.001), race (asian, HR: 2.31, p = 0.006), alcohol (none, HR: 1.88, p = 0.008), and hepatitis (none, HR: 2.44, p < 0.001). For PFS, increased NUP205 expression was associated with poorer outcomes in female (HR: 3.11, p < 0.001), grade II (HR: 1.92, p = 0.009), grade III (HR: 2.41, p = 0.001), American Joint Committee on Cancer (AJCC)_T I (HR: 1.73, p = 0.042), race (white, HR: 2.2, p < 0.001), and hepatitis (none, HR: 2.86, p < 0.001). For RFS, increased NUP205 expression was associated with poorer outcomes in female (HR: 2.59, p < 0.001), stage I+II (HR: 1.54, p = 0.024), stage II+III (HR: 1.55, p = 0.031), grade II (HR: 2.07, p = 0.001), grade III (HR: 1.96, p = 0.007), AJCC_T I (HR: 1.74, p = 0.023), race (white, HR: 1.9, p = 0.001), race (asian, HR: 1.64, p = 0.038), alcohol (yes, HR: 1.87, p = 0.018), alcohol (none, HR: 1.59, p = 0.024), and hepatitis (none, HR: 2.6, p < 0.001). For DSS, increased NUP205 expression was associated with poorer outcomes in male (HR: 1.91, p = 0.026), female (HR: 2.7, p = 0.006), stage I+II (HR: 2.2, p = 0.066), stage II+III (HR: 2.02, p = 0.023), grade II (HR: 2.09, p = 0.031), race (white, HR: 2.48, p = 0.001), race (asian, HR: 2.52, p = 0.022), alcohol (none, HR: 2.49, p = 0.005), and hepatitis (none, HR: 3.03, p < 0.001) (Figure 2C). These subgroup analyses should be interpreted as exploratory. Collectively, these results suggest that elevated NUP205 expression is a robust indicator of poor prognosis in LIHC, underscoring its potential utility as a prognostic biomarker.
2.3. NUP205 Is Associated with DNA Methylation in LIHC
To elucidate the epigenetic regulation of NUP205 in LIHC, DNA methylation analyses were conducted using multiple public databases, including UALCAN, OncoDB, SMART, and MEXPRESS. DNA methylation, primarily occurring at cytosine–phosphate–guanine (CpG) sites and catalyzed by DNA methyltransferases (DNMTs), is a key epigenetic mechanism regulating gene expression without altering the DNA sequence. In general, promoter hypermethylation is associated with transcriptional repression, whereas promoter hypomethylation facilitates transcriptional activation. Therefore, the hypomethylation of the NUP205 promoter observed in this study may contribute to its increased expression in LIHC and may be associated with poor clinical outcomes. In these analyses, promoter regions were defined according to standardized transcription start site (TSS)-centered annotations provided by each TCGA-based platform. These findings indicated significant hypomethylation of the NUP205 promoter region across various cancer types, including LIHC (Figure 3A). Further investigation revealed that DNA methylation of NUP205 was significantly associated with clinicopathological characteristics such as pathological stage I and histological grades I, III, and IV in LIHC (Figure 3B). NUP205 is associated with hypomethylation in LIHC compared to normal tissue and has 12 probes (Figure 4A,B). Correlation analysis demonstrated that methylation at these NUP205-associated probes was negatively correlated with DNA methylation levels at several CpG sites, including cg26005146 (R = −0.129, p < 0.05), cg03407666 (R = −0.153, p < 0.01), cg23885884 (R = −0.206, p < 0.001), cg25119219 (R = −0.124, p < 0.05), and cg16572020 (R = −0.152, p < 0.01) (Figure 4C). Furthermore, probes associated with NUP205 exhibited hypomethylation in LIHC tissues compared with normal tissues. Several probes, including cg14938629 (p < 0.001), cg07383972 (p = 0.0062), cg22761619 (p < 0.001), and cg19477020 (p = 0.002), showed significant hypomethylation in LIHC. In contrast, other NUP205-associated probes, such as cg23885884 (p = 0.014), cg03407666 (p = 0.013), and cg25119219 (p < 0.001), exhibited relative hypermethylation in LIHC compared to normal controls (Figure 5A). Analysis of the relationship between these probes and copy-number variations (CNVs) in LIHC revealed that several probes, including cg14938629 (p = 0.016), cg23885884 (p < 0.001), cg03407666 (p < 0.001), and cg25119219 (p < 0.001), were significantly associated with CNV alterations (Figure 5B). Moreover, evaluation of the prognostic relevance of NUP205-associated probes indicated that cg22761619 (HR: 1.967, p < 0.001), cg14182420 (HR: 1.689, p = 0.0029), cg23885884 (HR: 0.54, p < 0.001), and cg03407666 (HR: 0.647, p = 0.0025) were significantly associated with OS, suggesting that hypomethylation at these loci is associated with poorer prognosis (Figure 5C). These findings underscore the potential of NUP205 as an epigenetic biomarker and suggest its utility in risk stratification, molecular subtyping, and the development of precise epigenetic therapies for LIHC.
2.4. NUP205 Is Associated with miRNAs in LIHC
To identify miRNAs potentially targeting NUP205, predictive interaction analysis was performed using the miRNet database, a comprehensive platform for exploring miRNA–target gene networks. The resulting network comprised 41 miRNAs predicted to interact with NUP205, which was positioned as the central node (magenta), while each miRNA was represented as a peripheral node (blue circles) (Figure 6A). To assess the functional impact of NUP205-targeting miRNAs on LIHC, candidate miRNAs were analyzed for their expression levels, correlation with NUP205, and prognostic significance. To investigate the regulatory relationship between NUP205 and its target miRNAs in LIHC, we analyzed their differential expression patterns. Several NUP205-targeting miRNAs were significantly downregulated in LIHC tumor tissues compared to normal liver tissues, including hsa-let-7a-5p (p < 0.001), hsa-let-7c-5p (p < 0.001), hsa-miR-101-3p (p < 0.001), hsa-miR-24-3p (p = 0.0073), hsa-miR-29a-3p (p < 0.001), hsa-miR-200b-3p (p < 0.001), hsa-miR-29c-3p (p < 0.001), hsa-miR-29b-3p (p = 0.0085), hsa-miR-223-3p (p < 0.001), and hsa-miR-758-3p (p < 0.001). Several NUP205-associated miRNAs were significantly upregulated in LIHC, including hsa-miR-96-5p (p < 0.001), hsa-miR-34a-5p (p < 0.001), hsa-miR-132-3p (p = 0.0017), hsa-miR-21-5p (p < 0.001), hsa-miR-221-3p (p < 0.001), hsa-miR-34c-5p (p < 0.001), hsa-miR-769-5p (p = 0.031), and hsa-miR-34b-5p (p = 0.0036) (Figure 6B). Correlation analysis was performed to assess the association between NUP205 expression and these miRNAs. Positive correlations were observed between NUP205 and several miRNAs, including hsa-let-7a-5p (R = 0.115, p = 0.0265), hsa-miR-21-5p (R = 0.126, p = 0.0215), hsa-miR-24-3p (R = 0.188, p < 0.001), hsa-miR-34c-5p (R = 0.173, p < 0.001), hsa-miR-96-5p (R = 0.181, p < 0.001), hsa-miR-34b-5p (R = 0.103, p = 0.0486), hsa-miR-132-3p (R = 0.301, p < 0.001), hsa-miR-758-3p (R = 0.135, p = 0.00908), hsa-miR-223-3p (R = 0.188, p < 0.001), hsa-miR-200b-3p (R = 0.226, p < 0.001), hsa-miR-769-5p (R = 0.228, p < 0.001), and hsa-miR-221-3p (R = 0.143, p = 0.00591). In contrast, negative correlations were identified between NUP205 and hsa-miR-29c-3p (R = −0.327, p < 0.001), hsa-miR-34a-5p (R = −0.227, p < 0.001), hsa-miR-101-3p (R = −0.219, p < 0.001), hsa-miR-29a-3p (R = −0.113, p = 0.0301), hsa-miR-29b-3p (R = −0.147, p = 0.00456), and hsa-let-7c-5p (R = −0.172, p < 0.001) (Figure 6C). To investigate the prognostic value of NUP205-targeting miRNAs in LIHC, survival analyses were conducted based on miRNA expression levels. Elevated expression of several miRNAs, including hsa-miR-21-5p (HR: 1.60, p = 0.0095), hsa-miR-132-3p (HR: 1.64, p = 0.006), and hsa-miR-24-3p (HR: 1.52, p = 0.02), was significantly associated with poorer prognosis. Conversely, reduced expression of hsa-miR-101-3p was associated with poor prognosis (HR: 0.57, p = 0.0017) (Figure 6D). Collectively, these findings suggest that NUP205 is intricately regulated by a network of miRNAs in LIHC, some of which exhibit prognostic significance and may represent potential candidates for therapeutic interventions in miRNA-based treatment strategies.
2.5. NUP205 Is Associated with Drug Sensitivity in LIHC
To explore the potential therapeutic implications of NUP205 in LIHC, we investigated the correlation between NUP205 mRNA expression and drug sensitivity using data from the Genomics of Drug Sensitivity in Cancer (GDSC) and Cancer Therapeutics Response Portal (CTRP) database. The GDSC-based drug sensitivity analysis is shown in Figure 7A. NUP205 expression was positively correlated with the half-maximal inhibitory concentration (IC_50_) values for agents, including 17-AAG, Afatinib, Bleomycin, Cetuximab, Dasatinib, Erlotinib, Gefitinib, Lapatinib, RDEA119, selumetinib, and Trametinib. This positive correlation indicates that a higher NUP205 expression may contribute to drug resistance. In contrast, negative correlations were observed between NUP205 expression and sensitivity to drugs, such as AR-42, Belinostat, BX-912, CAY10603, CUDC-101, Genentech cpd 10, GSK1070916, I-BET-762, Navitoclax, NPK76-II-72-1, NSC-207895, PI-103, PIK-93, THZ-2-102-1, Vorinostat, WZ3105, XMD13-2, YM201636, and ZM-447439. This negative correlation indicates that a higher NUP205 expression may increase drug sensitivity. These findings suggest that elevated NUP205 expression may contribute to resistance or sensitivity to distinct classes of targeted therapies, including kinase inhibitors and DNA-damaging agents. The CTRP-based drug sensitivity analysis, depicted in Figure 7B, revealed a significant negative correlation between NUP205 and sensitivity to a broad range of anticancer compounds, including Barasertib, BI-2536, BRD-K34222889, BRD-K66453893, BRD-K70511574, Ceranib-2, FQI-2, GSK461364, ISOX, KX2-391, LY-2183240, Mitomycin, Nakiterpiosin, Narciclasine, Necrosulfonamide, NVP-231, Panobinostat, Pevonedistat, PL-DI, PRIMA-1, PX-12, Rigosertib, SB-225002, SB-743921, SCH-79797, Sotrastaurin, Triazolothiadiazine, Vincristine, Vorinostat, and YK4-279. This negative correlation indicates that a higher NUP205 expression may lead to increased drug sensitivity. These findings suggest that elevated NUP205 expression is associated with increased drug sensitivity in LIHC, particularly to epigenetic modulators, heat shock protein inhibitors, and cyclin-dependent kinase inhibitors. While some of these agents are not currently indicated as primary treatments for LIHC, these findings suggest that NUP205 may influence drug responsiveness across multiple therapeutic classes, highlighting its potential relevance as a predictive biomarker and a target for further investigation. Collectively, these drug sensitivity analyses indicated that NUP205 expression may modulate responsiveness to multiple anticancer agents, underscoring its potential role as a predictive biomarker for therapeutic response.
2.6. NUP205 Is Associated with Chemicals in LIHC
To investigate the regulatory effects of the chemical compounds on NUP205 expression, a chemical–gene interaction analysis was conducted using data from the Comparative Toxicogenomics Database (CTD). This analysis identified 66 chemical compounds associated with the upregulation of NUP205 expression (Table S1), suggesting that they are pharmacological modulators of this gene. Furthermore, the top 21 gene-chemical interaction profiles involving NUP205 were characterized, revealing a significant overlap with several genes exhibiting similar chemical responsiveness. Notably, nucleoporin 93 (NUP93) demonstrated the highest similarity to NUP205, with a similarity index of 0.3400 and 51 shared interacting chemicals. Other genes with elevated similarity indices included RNA Binding Motif protein 28 (RBM28) (0.3306; 41 chemicals), Testis Expressed 2 (TEX2) (0.3206; 42 chemicals), Isoleucyl-tRNA Synthetase 1 (IARS1) (0.3118; 53 chemicals), and Timeless Circadian Clock Protein (TIMELESS) (0.3099; 53 chemicals). Interestingly, multiple components of the NPC (NUP93 and NUP155) and chromatin-associated regulators Chromatin Assembly Factor 1 Subunit A (CHAF1A), Cytoskeleton-Associated Protein 2 Like (CKAP2L), and Inner Centromere Protein (INCENP) were among the highest-ranked genes, suggesting that NUP205 may functionally interact with or be co-regulated by similar chemical agents implicated in nuclear transport and chromatin remodeling. Additionally, genes such as Cytoskeleton Associated Protein 4 (CKAP4) and FAST Kinase Domains 2 (FASTKD2) exhibited substantial chemical interaction overlap (52 and 41 chemicals, respectively), indicating a potentially coordinated transcriptional response to pharmacological perturbations (Table S2). Collectively, these findings delineate a network of chemically coregulated genes associated with NUP205 and may inform future investigations into shared druggable vulnerabilities within NUP205-related molecular pathways.
2.7. Gene–Gene Interaction (GGI) Network Analysis of NUP205
Next, a GGI network analysis was performed to elucidate the functional landscape associated with NUP205. As shown in Figure S1, the resulting network revealed a highly interconnected regulatory architecture comprising physical interactions, co-expression, co-localization, predicted associations, genetic interactions, shared protein domains, and pathway-level connectivity. NUP205 occupies a central hub position within this network and interacts directly or indirectly with multiple components of the NPC, including NUP93, NUP98, NUP107, NUP153, NUP155, and NUP50. These associations suggest a coordinated functional role in nucleocytoplasmic transport and the structural organization of the nuclear pore. Furthermore, NUP205 is connected to key transport-related regulators such as nuclear RNA export factor 1 (NXF1), karyopherin subunit beta 1 (KPNB1), karyopherin subunit alpha 1 (KPNA1), and RAN binding protein 1 (RANBP1), indicating its involvement in both mRNA transport and non-coding RNA (ncRNA) export from the nucleus. Functional enrichment analysis revealed that NUP205 and its interacting partners are significantly associated with mRNA transport, ncRNA export, regulation of gene silencing, and RNA- and miRNA-mediated gene silencing. Collectively, these findings suggest that NUP205 is embedded within a complex regulatory framework that integrates RNA processing, nuclear transport, and post-transcriptional gene silencing, thereby positioning it as a potential nodal regulator at the interface between transcriptional control and nucleocytoplasmic communication.
2.8. Protein–Protein Interaction (PPI) and Functional Enrichment Analysis of NUP205
Next, Protein–Protein Interaction (PPI) network analysis was performed to elucidate the mechanistic role of NUP205 in biological processes. As shown in Figure S2 (top panel), the STRING-based PPI network identified a cluster of highly interconnected proteins that were functionally associated with NUP205. This cluster included NUP93, NUP98, NUP107, NUP133, NUP155, NUP160, NUP85, NUP153, SEH1-like nucleoporin (SEH1L), and NUP214, which are core components of the NPC. To further assess the functional significance of these interacting proteins, Gene Ontology (GO) enrichment analyses were performed across three categories: biological process (BP), cellular components (CC), and molecular functions (MF). In GO-BP, NUP205-associated proteins were significantly enriched in RNA- and transport-related processes, including mRNA transport, protein import into the nucleus, nucleocytoplasmic transport, RNA export from the nucleus, and NPC assembly. These findings highlight the involvement of NUP205 in nucleocytoplasmic trafficking, nuclear organization, and post-transcriptional gene regulation (Figure S2B). GO-CC analysis revealed the predominant localization of these proteins to the nuclear pore, nuclear membrane, nuclear pore inner and outer rings, and central transport channel, consistent with the established structural and transport functions of NPC-associated proteins within the nuclear envelope (Figure S2C). GO-MF analysis revealed the structural constituents of the nuclear pore and the structural molecule activity. Collectively, these results suggest that NUP205 and its interactors play critical roles in maintaining the integrity and function of the NPC, supporting the hypothesis that NUP205 functions as a structural scaffold and regulatory mediator of the nucleocytoplasmic transport machinery (Figure S2D).
2.9. NUP205 Expression Is Elevated in HepG2 Cells Compared to Other Carcinoma Cell Lines
To evaluate the cancer-type specificity of NUP205 expression, we analyzed its mRNA levels in various carcinoma cell lines, including those derived from prostate, lung, colon, and liver cancers. Notably, NUP205 expression was significantly higher in HepG2 cells than in other cancer cell lines, such as prostate cancer, lung cancer, and colon cancer (Figure 8A). These findings indicate a potential LIHC-specific role for NUP205 in tumor biology.
2.10. Knockdown Using Small Interfering RNA (siRNA)-NUP205 Reduces Transmembrane Protein 209 (TMEM209) Expression
To elucidate the functional significance of NUP205 expression in HepG2 cells, we knockdown NUP205 by transfection with small interfering RNA (siRNA)-NUP205 and then quantified the transcript level using quantitative reverse transcription PCR (RT-qPCR). Transfection with NUP205-specific siRNA resulted in a significant reduction in NUP205 mRNA expression relative to the control group in HepG2 cells (p < 0.01; Figure 8B). Reduces transmembrane protein 209 (TMEM209) promotes proliferation, migration, invasion, and epithelial–mesenchymal transition in LIHC cell lines in vitro. TMEM209 enhances proliferation and metastasis of LIHC cells via a karyopherin-β1 (KPNB1)-dependent mechanism. Accordingly, the TMEM209/KPNB1/Wnt/β-catenin signaling axis has been identified as a critical pathway in LIHC progression [24]. Furthermore, TMEM209 overexpression and TMEM209–NUP205 interaction between NUP205 have been implicated as key drivers of lung cancer proliferation [12]. Notably, our data showed that NUP205 knockdown significantly downregulated TMEM209 expression (p = 0.0013; Figure 8C). Additionally, transfection with NUP205-specific siRNA resulted in a significant reduction in NUP205 mRNA expression compared to the control group in PLC/PRF/5 cells (p < 0.0001; Figure 8D). However, NUP205 knockdown does not downregulate TMEM209 expression (p = 0.4827; Figure 8E). These results suggest that NUP205 may contribute to LIHC progression, at least in part, by modulating TMEM209 expression.
2.11. NUP205 Knockdown Enhances Doxorubicin (DOX)-Induced Apoptosis in HepG2 Cells
To investigate the effect of NUP205 expression on chemosensitivity, doxorubicin (DOX), a widely used chemotherapeutic agent for various cancers [25,26], was used. Our study demonstrated that the downregulation of NUP205 in HepG2 cells significantly increased apoptosis (Figure 8F). These findings suggest that NUP205 plays a role in the drug response mechanisms of LIHC and that its inhibition enhances the apoptotic effect of DOX. Therefore, targeting NUP205 may represent a promising therapeutic strategy to improve the efficacy of chemotherapy in patients with LIHC. In addition, cell recovery was examined using a wound healing assay. A slight reduction in cell migration was observed in siNUP205-treated cells compared with the control group. However, this difference did not reach statistical significance (Figure 9A–D).
3. Discussion
In this study, we comprehensively characterized the oncogenic role of NUP205 in LIHC by integrating multi-omics analyses to elucidate its prognostic significance, epigenetic regulation, miRNA networks, drug sensitivity profiles, and functional interactions. Our results revealed that NUP205 was significantly upregulated in LIHC, correlated with advanced clinicopathological features, and predicted poor prognosis. Furthermore, mechanistic investigations indicated that the upregulation of NUP205 is driven by epigenetic alterations and miRNA interactions, which also influence drug sensitivity in LIHC. Moreover, functional validation using in vitro assays demonstrated that NUP205 knockdown was associated with reduced TMEM209 mRNA expression in HepG2 cells. While this observation suggests a potential regulatory relationship, direct protein-level interactions or causal mechanisms were not assessed in this study.
Based on integrative multi-omics analyses complemented by limited in vitro functional assays, we propose a conceptual regulatory framework in which NUP205 overexpression in LIHC is associated with epigenetic alterations and dysregulated miRNA networks. Rather than establishing a definitive mechanistic model, this framework highlights coordinated molecular associations that may contribute to tumor progression and altered drug responsiveness, which require further validation at the protein and functional levels.
NUP205 is a key component of the NPC and plays a vital role in nucleocytoplasmic transport and the maintenance of nuclear architecture. Increasing evidence has linked the abnormal overexpression of NPC components to tumorigenesis and immune evasion in various cancer types [11]. Previous studies have identified NUP205 as an oncogenic factor in several cancers, including colorectal, lung, and nasopharyngeal cancers, where its upregulation is associated with increased proliferation and poor clinical outcomes [12,13,14]. In LIHC, earlier research based on limited datasets suggested a prognostic role for NUP205 [15,27]. Our study extends these findings by providing a comprehensive multi-omics validation using multiple public databases and functional experiments. The results of this study suggest epigenetic and post-transcriptional mechanisms underlying NUP205 dysregulation in LIHC, including the identification of DNA methylation patterns and miRNA regulators with prognostic relevance.
Integrative DNA methylation analysis revealed that the upregulation of NUP205 in LIHC was significantly associated with promoter hypomethylation. This is consistent with previous studies that connected the aberrant epigenetic activation of critical genes to LIHC progression [28,29]. Notably, specific CpG site probes showed inverse correlations with NUP205 expression and were independently associated with poor prognosis, indicating their potential as differentially methylated markers (DMMs) for risk stratification. These results support previous reports that DNA hypomethylation, especially in cirrhotic or hepatitis B virus (HBV)-infected liver tissues, promotes oncogenic activation and hepatocarcinogenesis [30,31]. Importantly, the biological relevance of NUP205 upregulation inferred from epigenetic analyses is further supported by our functional experiments, in which NUP205 knockdown suppressed TMEM209 expression and enhanced DOX-induced apoptosis in LIHC cell models. While causality between DNA methylation and these downstream phenotypes was not directly tested, these findings collectively suggest that epigenetically associated NUP205 overexpression contributes to tumor progression and therapeutic response in LIHC.
In addition to epigenetic dysregulation, we identified a network of miRNAs that may regulate NUP205 expression. Several miRNAs were significantly downregulated in LIHC, whereas oncomiRs, such as hsa-miR-21-5p, hsa-miR-24-3p, and hsa-miR-132-3p, were overexpressed and exhibited a positive correlation with NUP205 expression. Notably, elevated expression of hsa-miR-21-5p, hsa-miR-24-3p, and hsa-miR-132-3p was associated with poor patient survival, underscoring their potential as prognostic biomarkers and therapeutic targets. Although miRNA–target interactions were initially identified using miRNet, key miRNAs were further supported by cross-validation with The Encyclopedia of RNA Interactomes (ENCORI), which incorporates cross-linking immunoprecipitation sequencing (CLIP-seq)-supported evidence and TCGA-based expression and survival analyses, thereby reducing potential noise from prediction-only approaches. The relatively modest correlation coefficients observed between NUP205 and several miRNAs are consistent with the multi-layered nature of gene regulation in bulk tumor tissues, where miRNA-mediated effects coexist with epigenetic, transcriptional, and genomic regulatory mechanisms [21,22,32].
Drug therapy remains a significant challenge in enhancing survival outcomes in patients with advanced LIHC [33,34]. Our pharmacogenomic analysis reveals that the upregulation of NUP205 is associated with responsiveness to various targeted therapies and cytotoxic agents, including kinase inhibitors and epigenetic modulators. DOX, an anthracycline antibiotic derived from Streptomyces species, is widely used as an effective chemotherapeutic agent against a range of malignancies, including carcinomas, leukemia, solid tumors, soft-tissue sarcomas, and breast cancer [25,26]. Our findings demonstrate that NUP205 knockdown sensitizes cells to DOX-induced apoptosis, suggesting a contributory role in drug therapy. However, the clinical application of DOX is limited because of its dose-dependent and irreversible cardiotoxicity [35,36]. Moreover, the precise mechanisms underlying the therapeutic effects of DOX remain a subject of ongoing debate.
GGI and PPI analyses suggested potential associations between NUP205 and other NPC components involved in nucleocytoplasmic transport. These interaction networks were derived from in silico prediction-based databases and should therefore be interpreted as hypothesis-generating rather than as evidence of direct PPIs or regulatory activity in LIHC. Enrichment analyses indicated that genes associated with NUP205 were enriched in pathways related to mRNA transport, nuclear export, and miRNA-mediated gene silencing. These findings reflect pathway-level associations based on transcriptomic data and do not establish direct mechanistic roles for NUP205 in these processes.
Despite the advantages of integrating transcriptomic, epigenetic, and functional validation data, this study has several limitations. First, its retrospective design and dependence on publicly available datasets may have introduced a cohort-specific bias. Second, although in vitro experiments provided functional evidence, further in vivo validation is necessary to confirm the role of NUP205 in tumor growth and response to drug therapy. In addition, because HepG2 cells are derived from hepatoblastoma rather than hepatocellular carcinoma, findings obtained from this cell line should be interpreted as mechanistic support for the multi-omics analyses rather than definitive evidence of LIHC-specific behavior. Accordingly, the HepG2 cell line was not employed as a definitive disease model of LIHC but rather as an experimental system to explore general mechanisms of gene activity regulation and nucleocytoplasmic transport associated with NUP205 dysregulation. This clarification is important to avoid overinterpretation of the in vitro findings as LIHC-specific phenotypes.
In this context, publicly available functional dependency data from the Achilles/DepMap project provide additional insight. To our knowledge, NUP205 does not emerge as a strong dependency gene in HepG2 or PLC/PRF/5 cell lines, suggesting that its role may not be universally essential for cell viability. Publicly available dependency datasets indicate that NUP205 does not represent a strong universal survival dependency in commonly used LIHC cell lines. This finding underscores the context-dependent nature of NUP205 function and supports the interpretation that the observed phenotypes in this study reflect regulatory or modulatory effects rather than essential viability dependency.
Finally, the precise molecular mechanisms by which NUP205 regulates TMEM209 or modulates apoptotic pathways remain incompletely understood and warrant further investigation. These limitations reflect the broader analytical scope of the present study. Importantly, the regulatory relationships and biological interpretations proposed in this study are derived from integrative transcriptomic, epigenetic, and in silico analyses, complemented by in vitro functional assays. Together, the findings provide a framework for hypothesis generation and support future experimental studies aimed at further clarifying these regulatory links.
4. Materials and Methods
4.1. Data Sources
We conducted a retrospective in silico analysis of NUP205 expression in patients with LIHC. Publicly available multi-omics datasets and online analytical tools were used to investigate gene expression, epigenetic regulation, prognostic value, and interaction networks. The principal data source was TCGA LIHC cohort, comprising approximately 371 tumor samples and 50 adjacent normal liver tissue samples (RNA-seq expression data) [37].
4.2. Gene Expression Analysis
To evaluate the mRNA expression levels of NUP205 in LIHC, we used multiple publicly available bioinformatics platforms, including TIMER2.0, Wanderer, and UALCAN. TIMER2.0 (http://timer.cistrome.org/; accessed on 7 April 2025) is a comprehensive tool for the systematic analysis of immune infiltrates, gene expression, and survival outcomes, incorporating clinical data across various cancer types [38,39]. Wanderer (http://maplab.cat/wanderer; accessed on 7 April 2025) is a user-friendly, gene-centric web application designed to access and visualize TCGA DNA methylation data along with the corresponding mRNA expression across multiple cancer types [40]. UALCAN (http://ualcan.path.uab.edu; accessed on 8 April 2025) was used to compare mRNA expression levels between tumor and normal tissues and to evaluate expression patterns across clinicopathological subgroups, including tumor stage, grade, and TP53 mutation status [38].
4.3. Survival Analysis
To evaluate the prognostic significance of NUP205 in LIHC, comprehensive survival analyses were performed using multiple publicly accessible bioinformatics platforms, including the KM plotter (http://kmplot.com/analysis/; accessed on 10 April 2025) [38,39] and OSlihc (https://bioinfo.henu.edu.cn/LIHC/LIHCList.jsp; accessed on 12 April 2025) [39]. Both platforms integrate datasets from TCGA, Genotype-Tissue Expression (GTEx), and the Gene Expression Omnibus (GEO). The survival endpoints assessed using the KM plotter included OS, RFS, DSS, and PFS. OSlihc evaluated OS, DFS, RFS, and DSS.
4.4. DNA Methylation Analysis
To examine the DNA methylation status of NUP205 and its prognostic significance in LIHC, several publicly accessible bioinformatics platforms were utilized. The UALCAN platform (methylation module) was used to evaluate the promoter methylation β-values of NUP205 in tumor versus normal tissues, as well as across clinical subgroups. SMART (http://www.bioinfo-zs.com/smartapp/; accessed on 18 April 2025) was used to investigate the inverse correlation between gene expression and DNA methylation across pan-cancer cohorts, facilitating the visualization of expression-methylation relationships by integrating data from TCGA Illumina Human Methylation 450K arrays (Illumina, Inc., San Diego, CA, USA) and RNA-sequencing profiles [41]. OncoDB (https://oncodb.org/; accessed on 22 April 2025) served as an initial screening tool for promoter methylation status in LIHC, enabling the comparison of average β-values between tumors and matched normal tissues [42]. MEXPRESS (https://mexpress.be/; accessed on 28 April 2025) was used to map the methylation levels of CpG sites across the NUP205 gene locus, aligning each probe with TSS and exon/intron structures, allowing direct comparison between tumor and normal samples while integrating relevant clinical metadata [43]. Finally, MethSurv (https://biit.cs.ut.ee/methsurv; accessed on 29 April 2025) was used to perform univariate Cox regression analyses on individual CpG sites to evaluate their prognostic significance, with KM survival curves generated for each site [44]. DNA methylation data were derived from TCGA Illumina Human Methylation 450K arrays and were obtained in the form of preprocessed β-values as provided by each platform. CpG probes with missing β-values were excluded according to platform-specific pre-processing pipelines, and only probes annotated to the NUP205 gene region were retained for downstream analyses. In total, 12 NUP205-associated CpG probes were included in the final methylation and survival analyses.
4.5. Non-Coding RNA Regulatory Network
To investigate the post-transcriptional regulation of NUP205, we examined miRNA interactions using miRNet 2.0 and ENCORI (starBase; https://rnasysu.com/encori/index.php; accessed on 7 June 2025). miRNet 2.0 (https://www.mirnet.ca; accessed on 5 June 2025) is a web-based platform designed for constructing and visualizing miRNA-target interaction networks [45]. (Encyclopedia of RNA Interactomes, also known as starBase v3.0) (https://starbase.sysu.edu.cn; accessed on 9 June 2025) provides TCGA expression data for mRNAs and miRNAs, facilitating correlation analyses to identify miRNAs that are inversely correlated with specific mRNAs across cancer samples. Additionally, ENCORI offers survival analyses for these miRNAs in LIHC. For each miRNA potentially targeting NUP205, we assessed its prognostic significance using ENCORI’s Kaplan–Meier plots comparing the high and low expression groups [46].
4.6. Drug Sensitivity Analysis
The Gene Set Cancer Analysis (GSCA) platform (https://guolab.wchscu.cn/GSCA/#/; accessed on 11 June 2025) integrates TCGA datasets encompassing 33 cancer types and including data on mRNA expression, DNA methylation, somatic mutations, immune infiltration, and drug sensitivity derived from the CTRP and the GDSC [47]. The CTD (http://ctdbase.org; accessed on 12 June 2025) is a manually curated resource that associates genes with chemicals, diseases, and phenotypes, based on evidence extracted from the peer-reviewed literature [48].
4.7. Gene Network and Functional Association (GeneMANIA)
To further investigate the genes associated with NUP205, we utilized GeneMANIA (http://genemania.org; accessed on 14 June 2025) with both the Cytoscape plugin and web interface. GeneMANIA identifies genes that are functionally similar or related to a query gene by analyzing extensive functional association data, including co-expression, co-localization, protein and genetic interactions, pathways, and domain similarities [49]. It visualizes gene–gene functional interaction networks, integrating data on co-expression, physical interactions, co-localization, and pathway associations.
4.8. PPI Network (STRING)
We constructed a PPI network for NUP205 to identify potential functional associations with other proteins. The STRING database (https://string-db.org; accessed on 16 June 2025) was used to construct a PPI network for NUP205. Interacting proteins and enriched GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms were retrieved using the default medium confidence threshold [50]. The combined confidence score integrates experimentally validated, curated, and predicted interactions, and only first-shell interactors of NUP205 were included for downstream functional enrichment analyses.
4.9. Cell Culture and siRNA Transfection
HepG2 cells were cultured at 37 °C in a humidified atmosphere containing 5% CO_2_ in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. 11965092) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Cat. No. 16000044) and 1× penicillin–streptomycin (Welgene, Gyeongsan, Republic of Korea; Cat. No. LS20202). PLC/PRF/5 cells were maintained at identical temperature and CO_2_ conditions in RPMI-1640 medium (Welgene, Gyeongsan, Republic of Korea; Cat. No. LM01103) supplemented with the same concentrations of FBS and the antibiotic mixture. Gene silencing was performed using the Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. 13778150) with NUP205-specific siRNA or a scrambled negative control (Ambion, Thermo Fisher Scientific, Waltham, MA, USA; siRNA IDs s23176 and s23177) following the manufacturer’s protocols. Forty-eight hours post-transfection, total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. 15596018), and RNA concentration and purity were assessed using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The primer sequences used for RT-qPCR analyses are summarized in Table 1.
4.10. Quantitative Real-Time PCR (qRT-PCR)
Complementary DNA (cDNA) was synthesized from total RNA using the ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan; Cat. No. QPK-201) according to the manufacturer’s protocol. The transcript levels of NUP205, TMEM209, and the reference gene, GAPDH, were quantified using the CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The thermal cycling conditions consisted of an initial denaturation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 72 °C for 30 s.
4.11. Annexin V Staining Assay
HepG2 cells were knocked down with siRNA-NUP205 and then treated with 2 μM DOX. After 24 h, both the culture supernatant and adherent cells were harvested using a cell scraper and resuspended in phosphate-buffered saline (PBS). The collected cells were then centrifuged at 1500× g for 5 min, and the resulting pellet was resuspended in 1× binding buffer (BD Biosciences, Franklin Lakes, NJ, USA; Cat. No. 5166121E). Subsequently, the cells were incubated with FITC-conjugated Annexin V and propidium iodide (PI) (BD Biosciences; Cat. No. 556547) at room temperature in the dark. Apoptotic cells were analyzed using a CytoFLEX S flow cytometer (Beckman Coulter, Brea, CA, USA).
4.12. Wound Healing Assay
HepG2 and PLC/PRF/5 cells were seeded in 6-well plates and cultured to 70–80% confluence. Cells were transfected with control siRNA or target-specific siRNA NUP205 using Lipofectamine 2000. After 6 h, the medium was replaced, and the cells were cultured at 37 °C. A scratch was created in the center of each well, washed with PBS, and incubated in appropriate media (R10 for PLC/PRF/5 and D10 for HepG2). Cell recovery was assessed at 0, 24, 48 h. Cell migration was quantified using the iSolution Lite ×64 software (IMT i-Solution Inc., Vancouver, BC, Canada).
4.13. Statistical Analysis
All statistical analyses were performed using integrated tools available on publicly accessible bioinformatics platforms, including TIMER2.0, UALCAN, OSlihc, Kaplan–Meier Plotter, SMART, GSCA, MethSurv, MEXPRESS, ENCORI, miRNet, CTD, GeneMANIA, and STRING. For analyses of gene expression and DNA methylation, either Student’s t-test or Wilcoxon rank-sum test was used, according to the data distribution characteristics and platform-specific defaults. Relationships between continuous variables, such as gene expression and DNA methylation levels or gene–miRNA expression pairs, were evaluated using Pearson correlation coefficients. Survival analyses were performed using the log-rank test, and univariate Cox proportional hazards regression models with HRs and 95% confidence intervals (CIs) were used to evaluate prognostic significance. To account for multiple hypothesis testing, Benjamini–Hochberg false discovery rate (FDR) corrections were applied where applicable. A significance threshold of p < 0.05 or FDR < 0.05 was applied unless otherwise specified.
To minimize potential batch effects, all transcriptomic, methylation, and miRNA analyses were conducted within the same platform-specific datasets (e.g., TCGA-LIHC) rather than through direct cross-platform data merging. Public databases were used primarily for cross-validation of trends and associations rather than for pooled quantitative analyses. As all datasets were obtained from platforms with standardized normalization pipelines, no additional batch-effect correction was applied.
Network-based functional associations were visualized using Cytoscape version 3.10.1. Experimental data were analyzed using GraphPad Prism software version 3.0 (GraphPad Software, San Diego, CA, USA). Data are presented as the mean ± standard error of the mean (SEM) from at least three independent experiments. Differences between the experimental groups were assessed using two-way analysis of variance (ANOVA), followed by Tukey’s post hoc multiple comparison test to adjust for multiple testing. Statistical significance was set at p < 0.05.
5. Conclusions
In this study, we identified overexpression of NUP205 as an important oncogenic driver characterized by adverse prognostic impact and chemoresistance association in LIHC. Integrative analyses demonstrate that NUP205 expression is associated with promoter hypomethylation and miRNA suppression. Furthermore, functional assays confirmed its role in regulating tumor cell survival and drug response. These findings suggest that NUP205 may serve as a valuable prognostic biomarker and therapeutic target. Future in vivo validation and mechanistic investigations are needed to further elucidate its role in LIHC progression and treatment resistance.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Konyn P. Ahmed A. Kim D. Current epidemiology in hepatocellular carcinoma Expert. Rev. Gastroenterol. Hepatol.2021151295130710.1080/17474124.2021.199179234624198 · doi ↗ · pubmed ↗
- 2Rumgay H. Arnold M. Ferlay J. Lesi O. Cabasag C.J. Vignat J. Laversanne M. Mc Glynn K.A. Soerjomataram I. Global burden of primary liver cancer in 2020 and predictions to 2040 J. Hepatol.2022771598160610.1016/j.jhep.2022.08.02136208844 PMC 9670241 · doi ↗ · pubmed ↗
- 3Sung H. Ferlay J. Siegel R.L. Laversanne M. Soerjomataram I. Jemal A. Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20217120924910.3322/caac.2166033538338 · doi ↗ · pubmed ↗
- 4Mauro E. de Castro T. Zeitlhoefler M. Sung M.W. Villanueva A. Mazzaferro V. Llovet J.M. Hepatocellular carcinoma: Epidemiology, diagnosis and treatment JHEP Rep.2025710157110.1016/j.jhepr.2025.10157141244300 PMC 12615749 · doi ↗ · pubmed ↗
- 5Kim H.S. El-Serag H.B. The epidemiology of hepatocellular carcinoma in the USA Curr. Gastroenterol. Rep.2019211710.1007/s 11894-019-0681-x 30976932 · doi ↗ · pubmed ↗
- 6Calderon-Martinez E. Landazuri-Navas S. Vilchez E. Cantu-Hernandez R. Mosquera-Moscoso J. Encalada S. Al Lami Z. Zevallos-Delgado C. Cinicola J. Prognostic scores and survival rates by etiology of hepatocellular carcinoma: A review J. Clin. Med. Res.20231520020710.14740/jocmr 490237187717 PMC 10181349 · doi ↗ · pubmed ↗
- 7Jemal A. Ward E.M. Johnson C.J. Cronin K.A. Ma J. Ryerson B. Mariotto A. Lake A.J. Wilson R. Sherman R.L. Annual report to the nation on the status of cancer, 1975-2014, featuring survival J. Natl. Cancer Inst.2017109 djx 03010.1093/jnci/djx 03028376154 PMC 5409140 · doi ↗ · pubmed ↗
- 8Siegel R.L. Miller K.D. Wagle N.S. Jemal A. Cancer statistics, 2023 CA Cancer J. Clin.202373174810.3322/caac.2176336633525 · doi ↗ · pubmed ↗