Associations of Tumor Somatic Mutations and Genetic Alterations with Survival Outcomes in Melanoma Patients Treated with Ipilimumab
Mohammad Ali Khaksar, Islam Eljilany, Ibrahim Yassine, Xiaoqing Yu, Jamie K. Teer, Jose R. Conejo-Garcia, Maureen Lyons, William LaFramboise, Ahmad A. Tarhini

TL;DR
This study explores how tumor mutations affect survival in melanoma patients treated with ipilimumab, identifying potential genomic markers for treatment response.
Contribution
The study identifies specific tumor mutations associated with survival outcomes in melanoma patients treated with neoadjuvant ipilimumab.
Findings
BRAF and NRAS mutations were detected in 73% of patients and showed mutual exclusivity and concurrence patterns.
Mutations in several genes were linked to shorter survival outcomes, though they did not maintain significance after multiple testing correction.
The study highlights the potential role of tumor genomic alterations in anti-tumor immunity and treatment response.
Abstract
Background: Identifying patients most likely to benefit from immune checkpoint inhibitors (ICIs) remains a significant challenge in advanced melanoma. We evaluated the association between tumor somatic mutations and clinical outcomes, focusing on relapse-free survival (RFS) and overall survival (OS) in locoregionally advanced melanoma patients treated with neoadjuvant ipilimumab. Methods: Tumor specimens and matched peripheral blood samples from 22 patients underwent whole-exome sequencing (WES) to identify non-synonymous somatic mutations. Tumor mutational burden (TMB) was quantified, and specific mutations were analyzed for associations with survival outcomes. Results: The analysis revealed a mutational landscape dominated by single-nucleotide missense mutations with a median TMB of 11.4 mutations/MB. BRAF and NRAS mutations were detected in 73% of patients and exhibited mutual…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
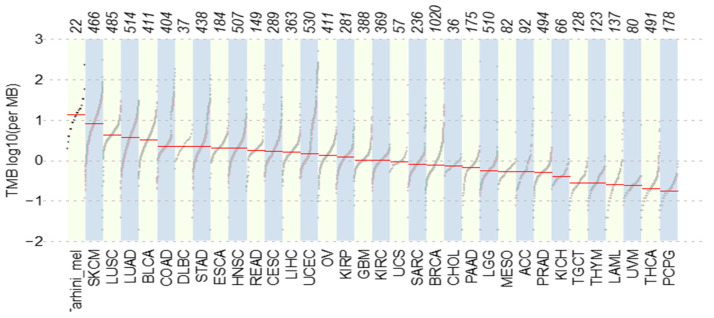 Figure 1
Figure 1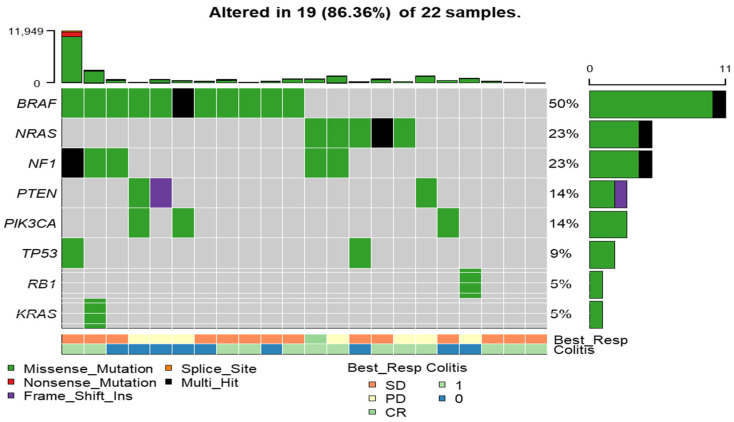 Figure 2
Figure 2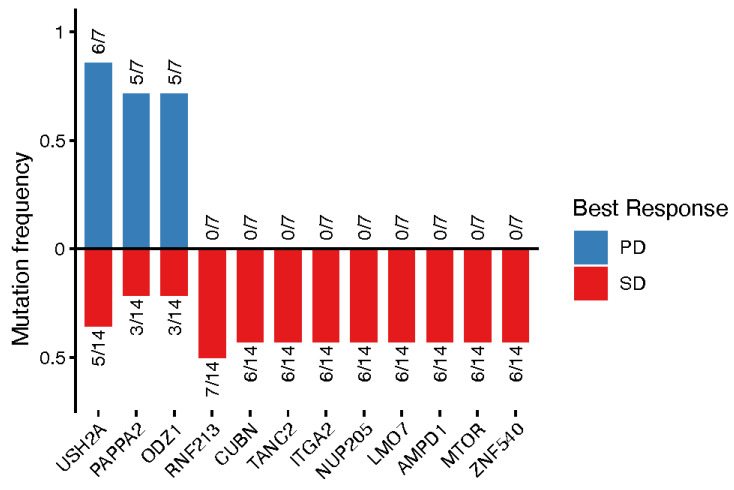 Figure 3
Figure 3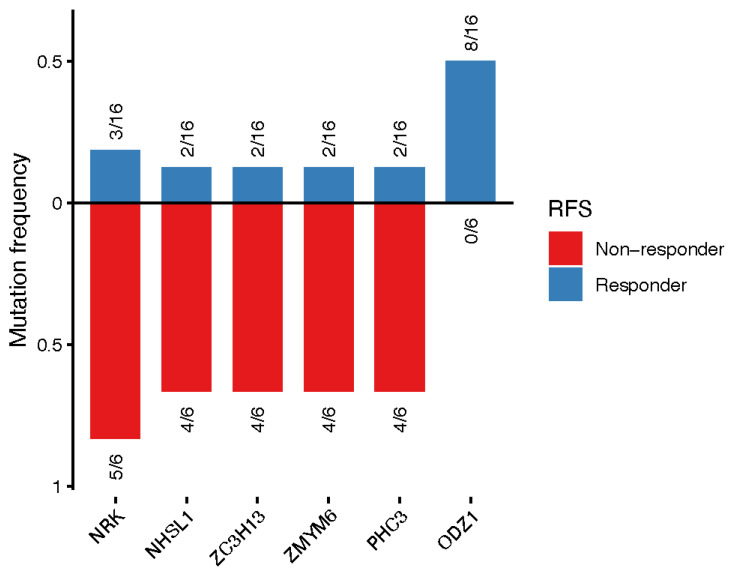 Figure 4
Figure 4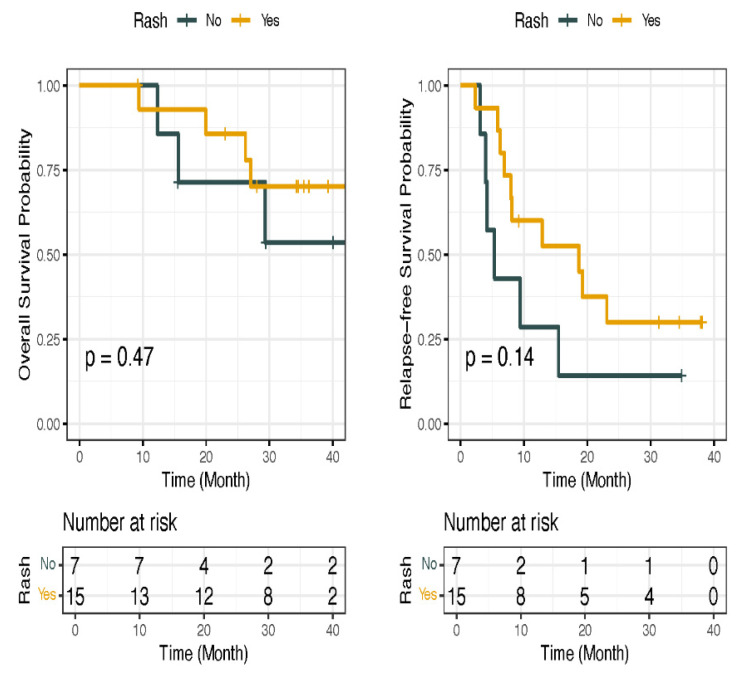 Figure 5
Figure 5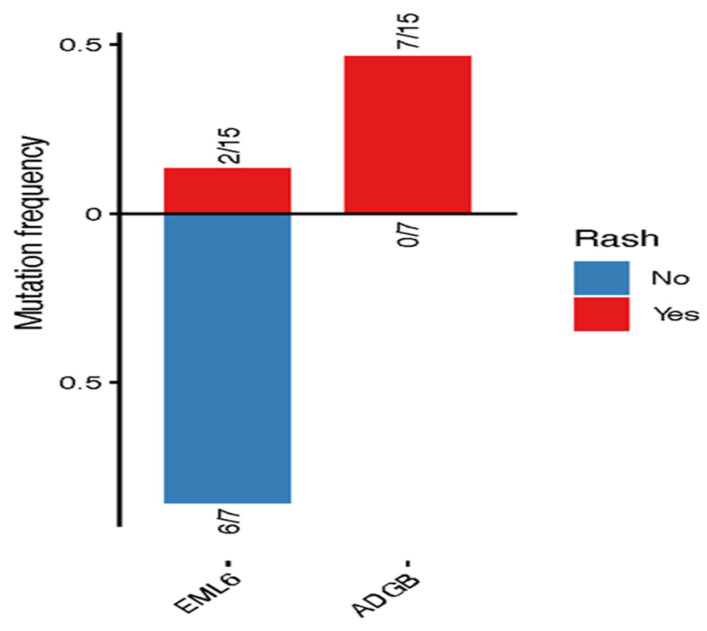 Figure 6
Figure 6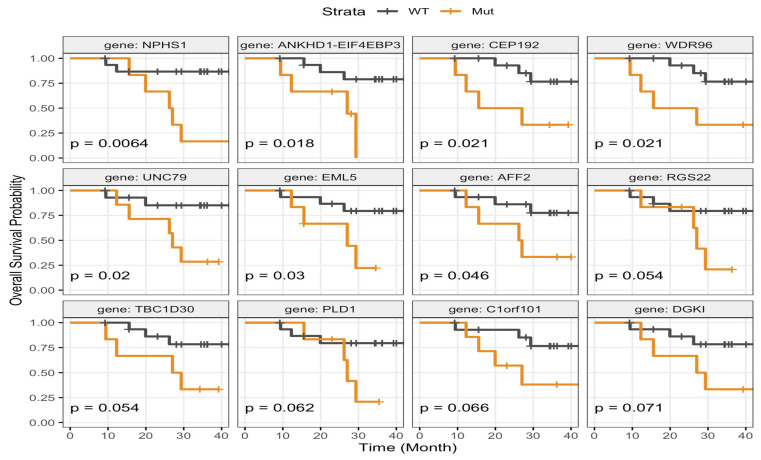 Figure 7
Figure 7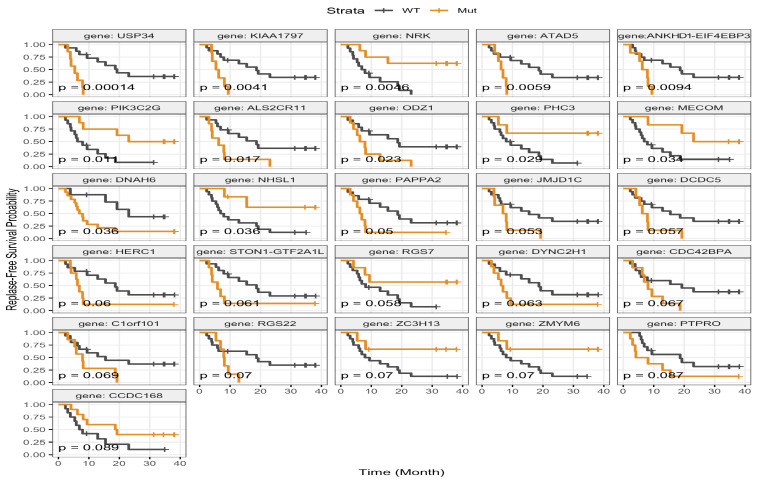 Figure 8
Figure 8- —Bristol-Myers Squibb
- —National Institutes of Health (NIH)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMelanoma and MAPK Pathways · Cancer Immunotherapy and Biomarkers · Cutaneous Melanoma Detection and Management
1. Introduction
The advent of immune checkpoint inhibitors (ICIs) has fundamentally changed the way locoregionally advanced melanoma is treated. By targeting inhibitory immune pathways, including cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) and programmed death-1 (PD-1), ICIs reinvigorate anti-tumor immune responses, thereby enhancing immune surveillance and promoting tumor regression [1,2]. Despite these advances, clinical responses to ICIs remain highly heterogeneous. While a subset of patients achieves durable clinical benefits, others exhibit limited or no response. Accordingly, there is an obvious need to identify reliable predictive biomarkers that can inform treatment decisions [3].
Accumulating evidence suggests that tumor mutational burden (TMB) and specific somatic mutations may serve as valuable biomarkers of response to ICIs. Elevated TMB, reflecting an increased load of somatic mutations within the tumor genome, has been associated with increased neo-antigen generation and improved immune recognition, thereby augmenting therapeutic efficacy [4]. Additionally, mutations in key oncogenes, including BRAF and NRAS, influence tumor biology, modulate the immune microenvironment, and contribute to variability in treatment response and resistance, highlighting the value of comprehensive genomic profiling in melanoma management [5,6,7,8]. Large-scale genomic efforts, including those conducted by The Cancer Genome Atlas (TCGA), have significantly advanced understanding of melanoma biology by delineating recurrent driver mutations and their associations with clinical outcomes. However, these datasets are predominantly derived from retrospective analyses or heterogeneous patient populations, which may limit their applicability to specific clinical contexts [9]. In particular, data describing mutational profiles and clinical correlations in the neoadjuvant setting, especially for ipilimumab, a CTLA-4 inhibitor, remain limited. This knowledge gap is particularly significant as neoadjuvant therapy offers an opportunity to evaluate early tumor responses and their potential impact on long-term survival [10].
To address these critical gaps, the present study characterizes the somatic mutational landscape of locoregionally advanced melanoma patients treated with neoadjuvant ipilimumab. Through whole-exome sequencing (WES) of tumor specimens and matched peripheral blood samples, we evaluate associations between tumor-specific genomic alterations and survival outcomes. By identifying genomic features associated with therapeutic benefit, this study provides exploratory insights that could contribute to the development of biomarker-guided stratification approaches in melanoma.
2. Materials and Methods
2.1. Patient Cohort and Sample Collection
This study (NCT 00972933) included patients diagnosed with locoregionally advanced melanoma who received treatment at the University of Pittsburgh Medical Center (UPMC) previously [11]. Eligibility criteria required availability of tumor tissue and matched peripheral blood mononuclear cell samples (PBMCs) with comprehensive clinical data, including follow-up information regarding disease progression and overall survival. Eligible patients had histologically confirmed locoregionally advanced melanoma and were candidates for neoadjuvant ipilimumab therapy, but had received no prior systemic treatment for advanced disease. The study was approved by the institutional review board at UPMC (IRB# PRO09010033), and all participants in this study (n = 22) provided written informed consent [12].
Tumor samples were obtained at baseline prior to initiation of neoadjuvant immunotherapy and again at the time of definitive surgical resection. Specimens were preserved as either snap-frozen tissue or formalin-fixed, paraffin-embedded (FFPE) samples, and tumor tissue domains were macro-dissected to enrich for tumor cell content. Peripheral blood cell samples were collected at the same time points. Genomic DNA was extracted from tissue and blood using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. For response assessment, modified WHO criteria were used and imaging studies were performed at baseline (before the initiation of ipilimumab), then at 6–8 weeks after initiation of ipilimumab (before surgery), and subsequently at 3-month intervals. Responses were classified as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD). However, radiologic confirmation of responses at 6–8 weeks was not performed due to the planned surgery.
2.2. Whole-Exome Sequencing Techniques
Whole-exome sequencing (WES) was performed on all samples using the SOLiD^®^ 5500 Next-Generation Sequencing platform (Thermo Fisher Scientific, Waltham, MA, USA). DNA concentration was measured using a Qubit™ Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), and DNA purity and quality were assessed by spectrophotometry (NanoDrop ND-1000, Thermo Fisher Scientific) and microcapillary electrophoresis (Bioanalyzer 2100, Agilent Technologies, Santa Clara, CA, USA), respectively. Exome library preparation utilized the SureSelect Human All Exon V5 kit (Agilent Technologies, Santa Clara, CA, USA), capturing approximately 50 Mb of human exonic regions from 5 μg initial DNA substrate. After hybridization to exon-specific probes, the libraries were amplified by polymerase chain reaction (PCR) and paired-end sequencing performed on amplified fragments by sequential oligonucleotide ligation and detection comprising 75 bp reads. Sequencing achieved an average read depth of approximately 100× per exome. Raw sequencing data were processed using SOLiD^®^ BioScope™ software (version 1.3) for base calling and initial quality filtering. Low-quality reads (Phred score < 20) and adapter sequences were removed prior to downstream analysis.
2.3. Genomic Analysis
Bioscope software (version 1.3) was used to generate BAM files by aligning filtered color-space sequencing reads to the reference genome (hg19) converted to color space. Duplicate reads were marked using Picard tools, and variant refinement included insertion/deletion realignment and base quality score recalibration using the Genome Analysis Toolkit (GATK) (version 2.2-16). Somatic single-nucleotide variants (SNVs) were identified using MuTect (version 1.1.4) [13] and Strelka (version 1.0.13) [14], and small insertions/deletions (indels) were identified using Somatic Indel Detector (GATK) (part of GATK Lite, version 2.2-16) and Strelka (version 1.0.13). Functional annotation of variants was carried out using ANNOVAR (version 06212012) to classify coding consequences, including missense, nonsense, and splice-site mutations. Tumor mutational burden (TMB) was defined as the total number of protein-altering somatic mutations per exome divided by the number of targeted bases: 61,91,1501 million base pairs (median TMB 11.4, range 1.6–198.4). Positional clustering analysis was performed using OncodriveCLUST to identify mutation hotspots suggestive of driver events. Pathway enrichment analysis was conducted using the maftools package (version 2.22.0) to identify potentially affected biological pathways.
2.4. Study Outcomes
Clinical outcomes included relapse-free survival (RFS) and overall survival (OS). Survival time was calculated from the initiation of ipilimumab therapy to the date of the event of interest or the last follow-up.
2.5. Statistical Analysis
All statistical analyses were performed using R software (version 4.0.1). Kaplan–Meier survival curves were generated to estimate survival distributions, and differences between groups were compared using the log-rank test. Univariable and multivariable Cox proportional hazards models were utilized to assess associations between somatic mutations across all genes and survival outcomes, including OS and RFS. For each model, hazard ratios (HRs) with 95% confidence intervals (CIs), nominal p-values, and Benjamini–Hochberg (BH)-adjusted p-values were reported. Multivariable models included TMB as a prespecified covariate based on its established biological relevance in melanoma. To assess sensitivity to model specification, we quantified the absolute change in log hazard ratios (ΔlogHR) following adjustment for TMB. Given the limited cohort size (n = 22) and the number of outcome events (7 OS events and 16 RFS events), model complexity was restricted to preserve events-per-variable (EPV) ratios and reduce the risk of overfitting. Univariable models were additionally performed as sensitivity analyses to evaluate the robustness of associations without TMB adjustment. Statistical significance was determined using false discovery rate (FDR)-adjusted p-values (<0.05) as the significance threshold. Patterns of mutually exclusive and co-occurring mutations were evaluated using the CoMEt tool. Data visualization was performed using ggplot2 (version 4.0.0) in R (version 4.4.1). Oncoplots of driver mutations were generated using the maftools package (version 2.22.0), and pathway enrichment results were visualized usingmaftools package (version 2.22.0).
3. Results
3.1. Patient Characteristics and Demographics
This study included 22 patients diagnosed with locoregionally advanced melanoma who were treated with neoadjuvant ipilimumab. Table S1 summarizes the baseline characteristics of the enrolled patients, including age, sex, tumor stage, and Eastern Cooperative Oncology Group (ECOG) performance status, as previously published [12].
3.2. Non-Synonymous Mutational Summary
Comprehensive genomic analysis revealed a median TMB of 11.4 (range 1.6–198.4), which is higher than levels reported in TCGA. Marked inter-patient variability in TMB was observed (Figure 1).
3.3. Co-Occurrence of Selected Genes
As illustrated in Figure 2, alterations in eight genes were identified in 19 of 22 patients (86.36%). Of note, BRAF mutations were detected in 50% of patients and were more frequent among those with stable disease (SD; 57%) compared with patients with progressive disease (PD; 43%). However, this difference was not statistically significant (Fisher’s Exact test p = 0.66). Additionally, Figure S1 shows the alterations in other genes. Mutations in NRAS and NF1 were detected in 23% of patients, reflecting the heterogeneity of oncogenic drivers in melanoma. Additional alterations were observed in PTEN (14%), PIK3CA (14%), TP53 (9%), RB1 (5%), and KRAS (5%), highlighting their roles in cell-cycle regulation and immune evasion. Co-occurrence analysis revealed mutual exclusivity between BRAF and NRAS mutations (p < 0.05), indicating their distinct but overlapping contributions to melanoma oncogenesis. Furthermore, KRAS mutations frequently co-occurred with NF1 alterations, suggesting potential synergistic interactions (Figure S2). Pathway enrichment analysis revealed that RTK-RAS pathway was the most frequently affected oncogenic pathway, followed by NOTCH and WNT pathways (Figure S3). Of note, as shown in Figure S4, positional clustering identified NRAS and SLC35B4 as potential contributors to melanoma.
3.4. Association of Mutations with Clinical Outcomes
As illustrated in Figure 3, mutations in USH2A, PAPPA2, and ODZ1 were enriched in patients with PD compared with those with wild-type tumors (p < 0.05), with mutation rates of 86%, 74%, and 74%, respectively. In contrast, mutations in the remaining genes were more frequently observed in patients with SD, occurring in approximately 50% of cases.
Genes associated with relapse, as assessed by RFS, are shown in Figure 4. Approximately 66% of patients whose tumors harbored mutations in NHSL1, ZC3H13, ZMYM6, and PHC3 and 83% of patients whose tumors harbored mutations in NRK remained relapse-free compared with those with wild-type alleles. In contrast, 50% of patients with ODZ1 mutations experienced disease relapse (p < 0.05).
In the present study (Figure 5), we found that patients who developed rash as an immune-related adverse event (irAE) exhibited longer OS and RFS compared with those who did not develop rash following ipilimumab; however, these differences did not reach statistical significance (OS: p = 0.47; RFS: p = 0.14). Moreover, EML6 mutations were more prevalent among patients who did not develop rash and showed no association with survival outcomes (Figure S5), whereas ADGB mutations were enriched in patients who developed rash following ICI therapy (Figure 6). Additionally, Kaplan–Meier analyses demonstrated that patients harboring ADGB mutations had numerically longer OS and RFS compared with those with wild-type ADGB; however, these differences did not reach statistical significance (Figure S6). Furthermore, similar analyses were performed for colitis irAE and the PTPRO gene, as described in detail in the Supplementary Results (Figures S7 and S8).
Using the log-rank test (p < 0.05 considered statistically significant), alterations in NPHS1 (p = 0.0064), CEP192 and WDR96 (both p = 0.021), UNC79 (p = 0.02), EML5 (p = 0.03), and AFF2 (p = 0.046) were associated with shorter OS, as demonstrated in Figure 7. Moreover, mutations in ANKHD1–EIF4EBP3 (p = 0.018), a transcriptional readthrough transcript, were also associated with shorter OS. In other words, patients whose tumors harbored the wild-type versions of these genes tended to have longer survival.
Associations between genomic alterations and RFS are shown in Figure 8. Wild-type alleles of USP34 (p = 0.00014), KIAA1797 (p = 0.0041), ATAD5 (p = 0.0059), ALS2CR11 (p = 0.017), ODZ1 (p = 0.023), and DNAH6 (p = 0.036) were associated with longer RFS. Additionally, mutations in NRK (p = 0.0046), PIK3C2G (p = 0.011), PHC3 (p = 0.029), MECOM (p = 0.034), and NHSL1 (p = 0.036) were also associated with longer RFS.
3.5. Cox Proportional Hazard Regression Analysis
Univariable and multivariable Cox proportional hazard regression analyses were performed to evaluate the association between genomic alterations in 339 genes and survival outcomes, including RFS and OS, with tumor mutational burden (TMB) included as covariate (Table S2). As shown in Table 1, univariable Cox regression analysis demonstrated that tumors harboring mutations in CEP192 (HR = 4.96, 95% CI 1.10–22.37; p = 0.03), WDR96 (HR = 4.96, 95% CI 1.10–22.37; p = 0.03), UNC79 (HR = 5.65, 95% CI 1.09–29.22; p = 0.03), NPHS1 (HR = 7.23, 95% CI 1.38–37.85; p = 0.02) and EML5 (HR = 4.6, 95% CI 1.01–20.85; p = 0.04) were associated with shorter OS. After adjustment for TMB in multivariable models, CEP192 (HR = 5.311, 95% CI 1.03–27.37; p = 0.04), WDR96 (HR = 5.37, 95% CI 1.03– 27.95; p = 0.04), UNC79 (HR = 5.72, 95% CI 1.02–32.07; p = 0.04), and NPHS1 (HR = 7.16, 95% CI 1.31–39.00; p = 0.02) retained statistical significance. However, after correction for multiple testing using the false discovery rate (FDR) method, none of these associations remained statistically significant. Similar analyses were performed for RFS, which demonstrated that alterations in USP34 (univariable: HR = 9.72, 95% CI 2.4–39.3; p = 0.001; multivariable: HR = 15.79, 95% CI 3.27–76.32; p = 0.0006), KIAA1797 (univariable: HR = 5.03, 95% CI 1.49–16.98; p = 0.009; multivariable: HR = 6.39, 95% CI 1.77–23.06; p = 0.004), ODZ1 (univariable: HR = 3.07, 95% CI 1.11–8.43; p = 0.03; multivariable: HR = 3.13, 95% CI 1.13–8.68; p = 0.03), DNAH6 (univariable: HR = 3.25, 95% CI 1.02–10.33; p = 0.04; multivariable: HR = 3.40, 95% CI 1.04–11.1; p = 0.04), ALS2CR11 (univariable: HR = 3.25, 95% CI 1.18–9; p = 0.02; multivariable: HR = 3.41, 95% CI 1.21–9.6; p = 0.02), and ATAD5 (univariable: HR = 5.24, 95% CI 1.42–19.33; p = 0.01; multivariable: HR = 6.96, 95% CI 1.67–29.04; p = 0.007) genes were associated with shorter RFS in both univariate and multivariate Cox regression models. Conversely, patients with tumors harboring mutations in PIK3C2G (univariable: HR = 0.23, 95% CI 0.07–0.77; p = 0.017; multivariable: HR = 0.15, 95% CI 0.03–0.64; p = 0.01), NRK (univariable: HR = 0.18, 95% CI 0.05–0.67; p = 0.01; multivariable: HR = 0.11, 95% CI 0.02–0.55; p = 0.007), PHC3 (univariable: HR = 0.21, 95% CI 0.05–0.97; p = 0.04; multivariable: HR = 0.07, 95% CI 0.006–0.82; p = 0.03), NHSL1 (univariable: HR = 0.23, 95% CI 0.05–1.02; p = 0.05; multivariable: HR = 0.093, 95% CI 0.009–0.93; p = 0.04), and MECOM (univariable: HR = 0.27, 95% CI 0.07–0.98; p = 0.04; multivariable: HR = 0.15, 95% CI 0.03–0.81; p = 0.03) had improved RFS. However, similar to OS, none of the findings withstood FDR correction. Furthermore, mutations in ANKHD1–EIF4EBP3, a transcriptional readthrough transcript, were associated with poorer RFS (univariable: HR = 4.36, 95% CI 1.3–14.58; p = 0.01; multivariable: HR = 5.22, 95% CI 1.48–18.46; p = 0.01) and OS (univariable: HR = 5.27, 95% CI 1.14–24.4; p = 0.03; multivariable: HR = 5.38, 95% CI 1.06–27.43; p = 0.04). However, these associations did not remain statistically significant after FDR correction. As a final step, to assess sensitivity to model specification, we quantified the absolute change in log hazard ratios (ΔlogHR) following adjustment for TMB. For most genes, the change in effect size was modest (median |ΔlogHR| = 0.28 for OS and 0.08 for RFS), indicating limited impact of TMB adjustment on the estimated associations (Table S2).
4. Discussion
This study identified genomic alterations associated with response to neoadjuvant ipilimumab in locoregionally advanced melanoma, highlighting the complexity of the melanoma genomic landscape and the potential clinical value of comprehensive tumor profiling for risk stratification, outcome prediction, and more tailored immunotherapy approaches in the future. Our findings demonstrated a high prevalence of BRAF mutations among our patients, with moderate frequencies of NRAS and NF1 mutations, reflecting the genomic heterogeneity of melanoma. Pathway enrichment analysis demonstrates predominant involvement of the RTK-RAS signaling pathway (Figure S3), reinforcing its central role in melanoma pathogenesis. BRAF mutations, particularly the V600E substitution, act as oncogenic drivers by activating the MAPK signaling pathway, largely independent of upstream RAS activity, thereby promoting tumor proliferation and disease progression [15]. The higher prevalence of BRAF mutations among patients suggests a potentially nuanced interaction between MAPK pathway and partial immune responses to neoadjuvant ipilimumab. While these findings are consistent with the established role of BRAF mutations in melanoma biology, they raise questions regarding their association with therapeutic outcomes. While BRAF-targeted therapies have shown efficacy in BRAF-mutant melanoma, the enrichment of BRAF mutations among patients with stable disease warrants further investigation into their potential influence on responses to ICIs [16,17]. Furthermore, NRAS mutations, which activate both MAPK and PI3K pathways, alongside NF1 mutations that disrupt RAS pathway regulation, underscore the molecular heterogeneity of melanoma and may modulate antitumor immune responses [18,19,20,21,22]. Of note, the prevalence of BRAF mutations observed in our cohort is consistent with prior reports in melanoma (approximately 40–60%), while the frequencies of NRAS and NF1 mutations fall within the previously reported range of 15–25% [22,23,24]. Although BRAF mutations have been linked to more aggressive disease phenotypes [9,25], a recent study showed that patients with BRAF mutant melanoma have consistently demonstrated improved survival compared to those with BRAF wild-type tumors [26]. While BRAF-targeted therapies are effective in metastatic disease [27], the relationship between BRAF status and ICI responsiveness remains complex and context-dependent, warranting further investigation in larger prospective cohorts.
In this exploratory analysis, several somatic gene alterations were associated with OS and RFS. For instance, alterations in genes such as CEP192, EML5, WDR96, UNC79, NPHS1, and ANKHD1–EIF4EBP3 were consistently associated with poorer OS or alterations in genes like USP34, KIAA1797, ANKHD1–EIF4EBP3, ATAD5, and ODZ1 were consistently associated with poorer RFS across analytical approaches. However, interpretation of these associations requires caution. The limited number of OS events (n = 7) and RFS events (n = 16), combined with a small cohort size (n = 22) and multiple testing burden, increases susceptibility to model instability and false discovery. Notably, none of the observed associations retained statistical significance after FDR correction, reinforcing the exploratory nature of these findings. Collectively, these findings may provide preliminary insights into genomic determinants of recurrence risk, but they remain exploratory and require validation in larger cohorts.
To provide biological context, the genes identified in our analysis can be broadly grouped into functional categories relevant to tumor progression and immune modulation. First, alterations in genes involved in centrosome regulation and cytoskeletal organizations such as CEP192, EML5, and ODZ1 were associated with adverse outcomes. CEP192 is essential for centrosome amplification and has been linked to poor prognosis and immunosuppressive tumor microenvironments in other malignancies [28,29]. EML5 is involved in regulating microtubule organization and cytoskeletal architecture, which are critical for maintaining cellular integrity and intracellular transport. Disruption of these processes may increase cellular adaptability under therapeutic stress, potentially facilitating tumor survival and disease progression. Although EML5 is not classified as a canonical oncogenic driver, its dysregulation may be associated with treatment resistance and unfavorable clinical outcomes [30]. With respect to ODZ1, a previous study showed an inverse association between the number of ODZ1-positive cells in glioblastoma specimens and both overall survival and progression-free survival. Mechanistically, ODZ1 enhances tumor aggressiveness by promoting cytoskeletal reorganization through Myc-mediated activation of the RhoA–ROCK signaling pathway, thereby increasing cellular invasion and motility [31]. Second, alterations affecting proliferative and translational signaling pathways were observed, including mutations in ANKHD1–EIF4EBP3 and PIK3C2G.
ANKHD1 has been identified as a contributor to tumor progression and is considered a potential regulator within the Hippo signaling pathway. In non-small-cell lung cancer, elevated ANKHD1 expression has been linked to advanced disease stage and poor prognosis, where it enhances cellular proliferation and invasion by activating Yes-associated protein (YAP) and suppressing Hippo pathway activity [32]. ANKHD1-EIF4EBP3, also known as MASK-BP3–, arises from the alternative splicing and genomic fusion of two functionally distinct loci involved in proliferative signaling and translational control. Given that both ANKHD1 and EIF4EBP3 biological roles overlap with Ras/MAPK pathway, this fusion protein may integrate proliferative signaling with translational control, thereby facilitating tumor growth and progression, However, its precise oncogenic role remains to be fully defined [33,34,35,36]. Similarly, Gao et al. [37] reported that loss of PIK3C2G expression in A549 cells (a non-small-cell lung cancer cell line) suppressed proliferation, migration, and invasion, while simultaneously promoting apoptosis and disrupting cell cycle progression. Therefore, it is plausible that functionally impairing mutations in PIK3C2G may exert anti-tumor effects. Nevertheless, the functional consequences of the specific mutations identified in our cohort require experimental validation.
Third, alterations in genes implicated in genomic stability and tumor suppression, including ATAD5 and KIAA1797 (FOCAD), were associated with survival outcomes. ATAD5 regulates proliferating nuclear antigen (PCNA) deubiquitination and maintains DNA replication fidelity. Loss of function in ATAD5 has been linked to genomic instability and increased susceptibility to several malignancies [38,39,40]. KIAA1797, also known as Focadhesin (FOCAD), has been implicated in glioblastoma biology. Brockschmidt et al. reported that KIAA1797 is frequently deleted in glioblastoma [41]. They further demonstrated that restoration of wild-type KIAA1797 expression significantly reduced colony formation, migration, and invasion in vitro. With respect to WDR96, also known as CFAP43, and NPHS1, which encodes nephrin, a transmembrane protein expressed in kidney podocytes, there are limited studies exploring their roles in malignancies. WDR96 plays a crucial role in ciliary and flagellar motility, and loss of function alterations in this gene have been linked to male infertility and normal pressure hydrocephalus [42,43]. NPHS1 mutations account for a substantial proportion of congenital nephrotic syndrome [44].
In addition to the findings mentioned above, Li and Shi reported significant upregulation of USP34, a regulator of the Wnt signaling pathway, in acute myeloid leukemia (AML) compared with matched healthy controls and found that elevated expression was associated with adverse clinical outcomes [45,46].
Furthermore, we observed that ADGB mutations were associated with the development of rash and improved survival outcomes. Consistent with prior reports, patients who experienced dermatitis irAEs demonstrated prolonged OS and RFS. The development of rash has been proposed as a clinical indicator of enhanced systemic immune activation, potentially reflecting a more robust antitumor immune response. These findings suggest that tumor-intrinsic genomic features may intersect with host immune activation patterns, linking toxicity and therapeutic benefit.
Collectively, our data suggests that in the neoadjuvant setting, somatic alterations beyond canonical driver mutations may contribute to variability in recurrence risk and immune responsiveness. However, the absence of FDR-significant associations, combined with limited event numbers and lack of functional validation, precludes definitive conclusions. These results should therefore be considered hypothesis-generating.
This study has several limitations. The small, single-center cohort limits statistical power and generalizability. The retrospective design precludes causal inference, and intratumoral heterogeneity was not comprehensively assessed. Sampling from a single tumor region may underestimate the genomic complexity influencing therapeutic response. Furthermore, the functional consequences of the identified alterations were not experimentally validated. Despite these limitations, our findings provide preliminary insights into the genomic architecture of melanoma in the neoadjuvant immunotherapy setting and highlight the importance of integrative genomic and clinical analyses. Future studies incorporating larger, multi-institutional cohorts and functional modeling will be essential to validate these associations and clarify the mechanistic interplay between tumor genomics, immune activation, and clinical outcomes.
5. Conclusions
The present study identified tumor somatic mutations associated with clinical outcomes in advanced melanoma patients treated with neoadjuvant ipilimumab. Validation in larger cohorts and functional studies are warranted to clarify the biological roles of these genes and their potential contribution to responses to ICIs in melanoma and other malignancies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ribas A. Wolchok J.D. Cancer immunotherapy using checkpoint blockade Science 20183591350135510.1126/science.aar 406029567705 PMC 7391259 · doi ↗ · pubmed ↗
- 2Hodi F.S. O’Day S.J. Mc Dermott D.F. Weber R.W. Sosman J.A. Haanen J.B. Gonzalez R. Robert C. Schadendorf D. Hassel J.C. Improved survival with ipilimumab in patients with metastatic melanoma N. Engl. J. Med.201036371172310.1056/NEJ Moa 100346620525992 PMC 3549297 · doi ↗ · pubmed ↗
- 3Ziogas D.C. Theocharopoulos C. Lialios P.P. Foteinou D. Koumprentziotis I.A. Xynos G. Gogas H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment Cancers 202315271810.3390/cancers 1510271837345056 PMC 10216291 · doi ↗ · pubmed ↗
- 4Snyder A. Makarov V. Merghoub T. Yuan J. Zaretsky J.M. Desrichard A. Walsh L.A. Postow M.A. Wong P. Ho T.S. Genetic basis for clinical response to CTLA-4 blockade in melanoma N. Engl. J. Med.20143712189219910.1056/NEJ Moa 140649825409260 PMC 4315319 · doi ↗ · pubmed ↗
- 5Davies H. Bignell G.R. Cox C. Stephens P. Edkins S. Clegg S. Teague J. Woffendin H. Garnett M.J. Bottomley W. Mutations of the BRAF gene in human cancer Nature 200241794995410.1038/nature 0076612068308 · doi ↗ · pubmed ↗
- 6Van Allen E.M. Wagle N. Sucker A. Treacy D.J. Johannessen C.M. Goetz E.M. Place C.S. Taylor-Weiner A. Whittaker S. Kryukov G.V. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma Cancer Discov.201449410910.1158/2159-8290.CD-13-061724265153 PMC 3947264 · doi ↗ · pubmed ↗
- 7Muñoz-Couselo E. Adelantado E.Z. Ortiz C. García J.S. Perez-Garcia J. NRAS-mutant melanoma: Current challenges and future prospect Onco Targets Ther.2017103941394710.2147/OTT.S 11712128860801 PMC 5558581 · doi ↗ · pubmed ↗
- 8Dika E. Veronesi G. Altimari A. Riefolo M. Ravaioli G.M. Piraccini B.M. Lambertini M. Campione E. Gruppioni E. Fiorentino M. BRAF, KIT, and NRAS Mutations of Acral Melanoma in White Patients Am. J. Clin. Pathol.202015366467110.1093/ajcp/aqz 20932017841 · doi ↗ · pubmed ↗