Genome-Wide Analysis of the MADS-Box Gene Family and Expression Pattern Under Abiotic Stresses in Lilium davidii var. unicolor
Xinyi Wang, Yuntao Zhu, Yuwei Nie, Tian Lan, Shuyi Zhang, Yiran Zhao, Jing Wang, Chunli Ma, Hengbin He

TL;DR
This study identifies and analyzes MADS-box genes in Lanzhou lily, revealing their roles in development and responses to environmental stresses like drought and cold.
Contribution
The study provides the first genome-wide analysis of MADS-box genes in Lilium davidii var. unicolor and identifies specific genes involved in abiotic stress responses.
Findings
62 LdMADS genes were identified, with notable expansion in the SOC1 subfamily.
LdMADS4, LdMADS14, LdMADS25, and LdMADS26 responded to multiple abiotic stress conditions.
Tissue-specific expression patterns were observed, with M-type and MIKC* genes active in reproductive tissues.
Abstract
The MADS-box gene family encodes a critical class of transcription factors that regulate diverse developmental processes in plants. However, its role in abiotic stress responses remains poorly characterized in Lilium davidii var. unicolor (Lanzhou lily). In this study, we identified 62 LdMADS genes in the Lanzhou lily genome, classifying them into 17 Type I and 45 Type II members. Notably, the SOC1 subfamily exhibited a pronounced expansion. These LdMADS members were distributed across all twelve chromosomes and displayed considerable structural variation, with some genes harboring exceptionally long introns. Tissue-specific expression profiling revealed that M-type and MIKC* genes were predominantly and specifically expressed in ovaries and anthers, whereas MIKCC members exhibited complex and diverse expression patterns across multiple tissues. The selection of candidate LdMADS genes…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
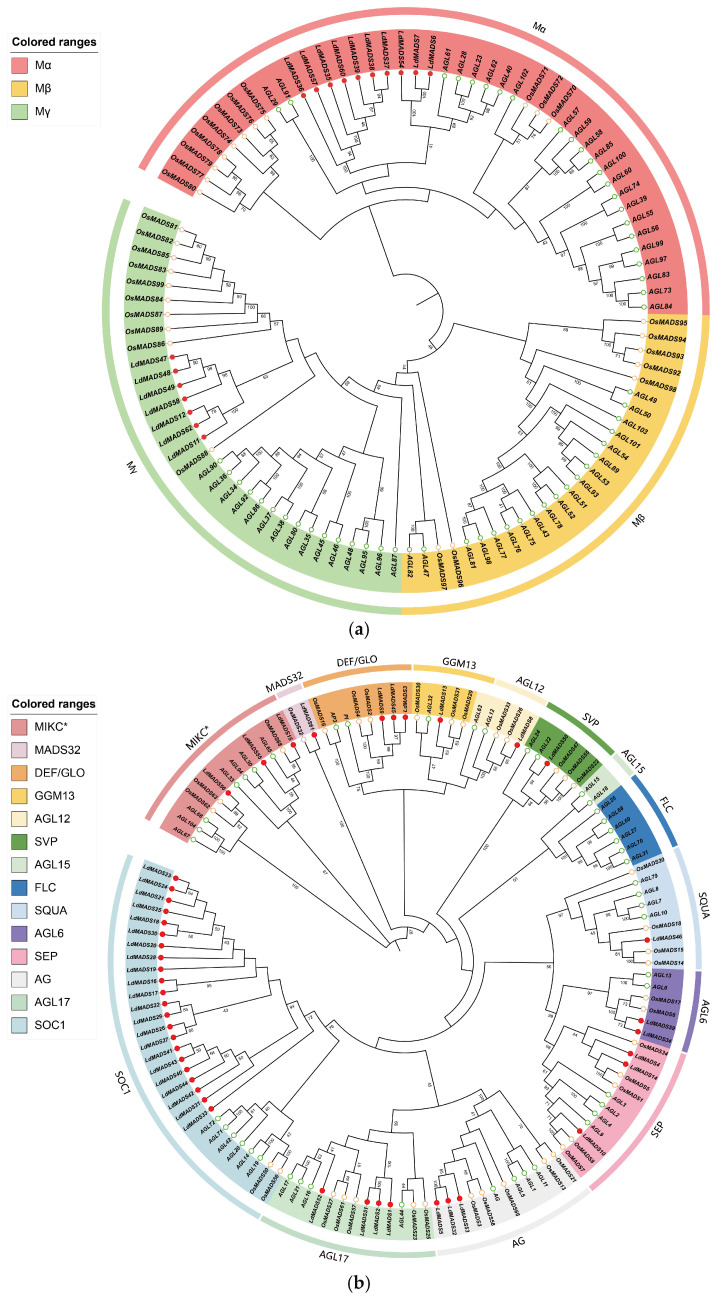 Figure 1
Figure 1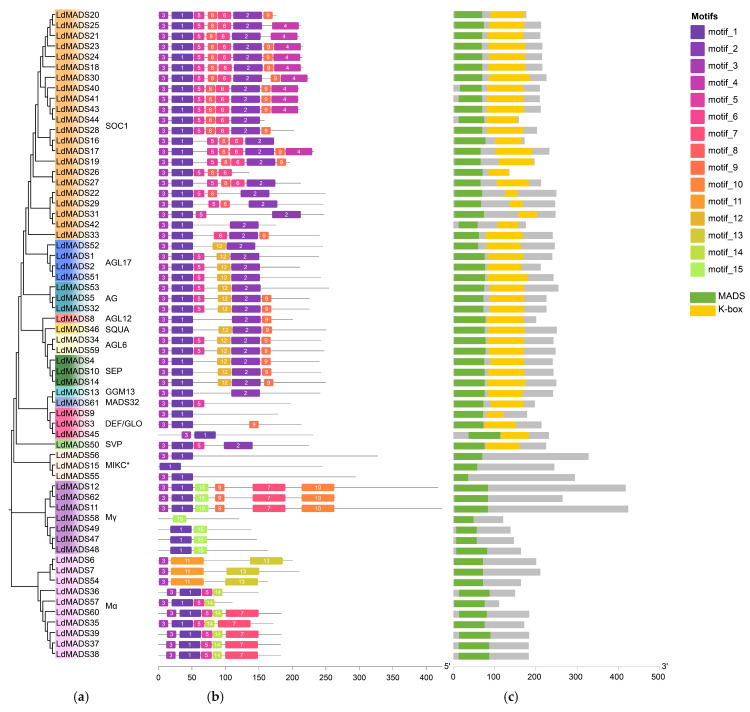 Figure 2
Figure 2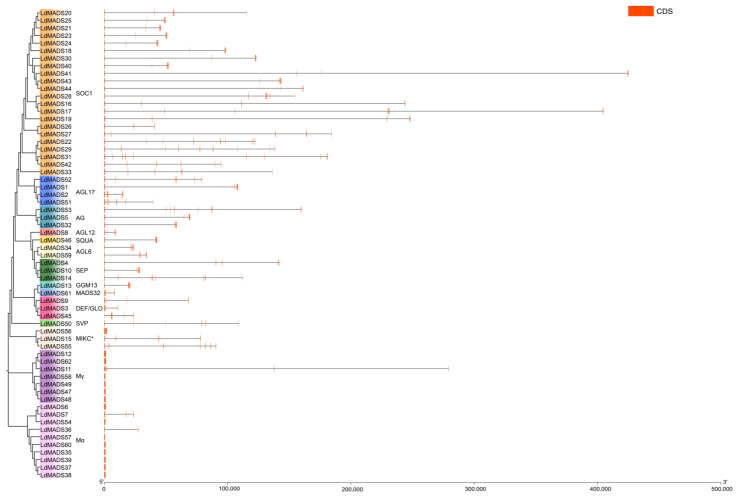 Figure 3
Figure 3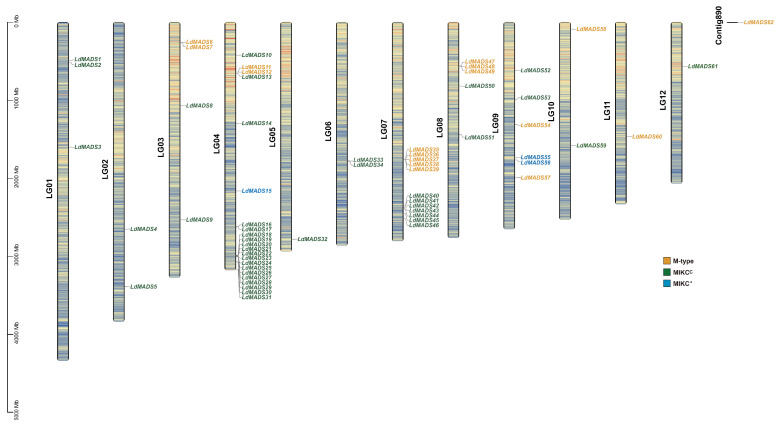 Figure 4
Figure 4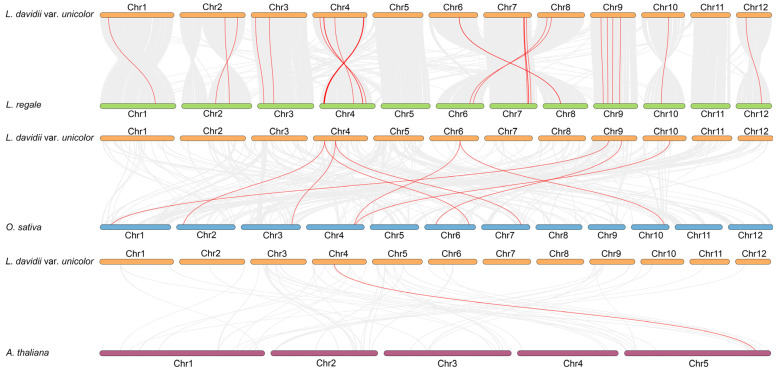 Figure 5
Figure 5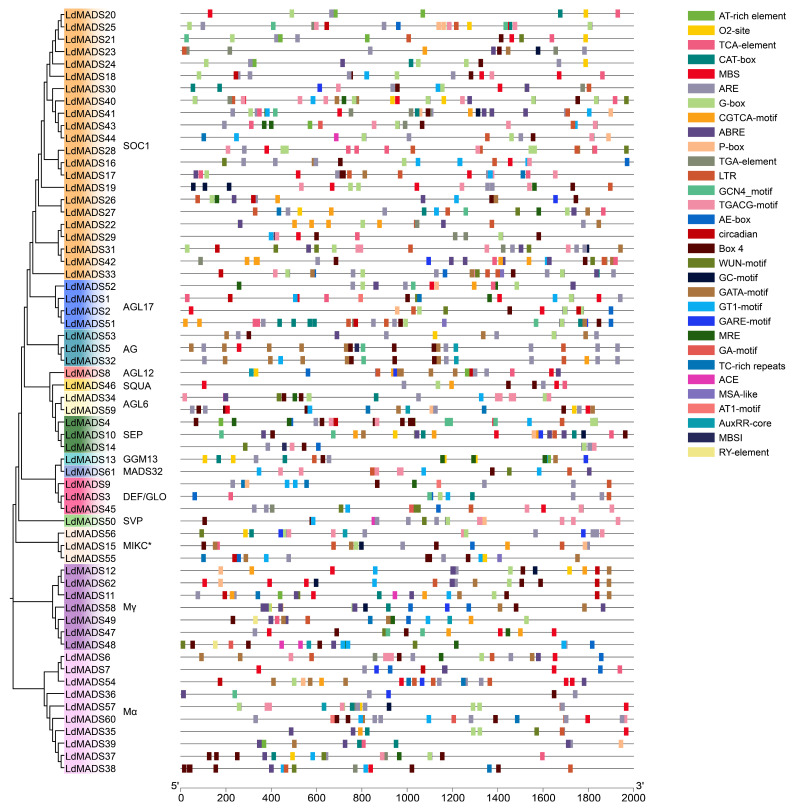 Figure 6
Figure 6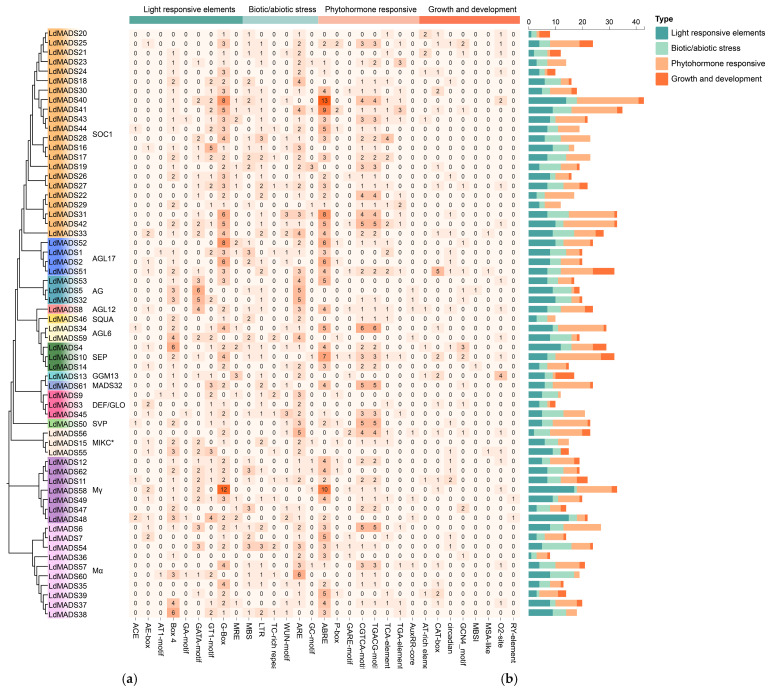 Figure 7
Figure 7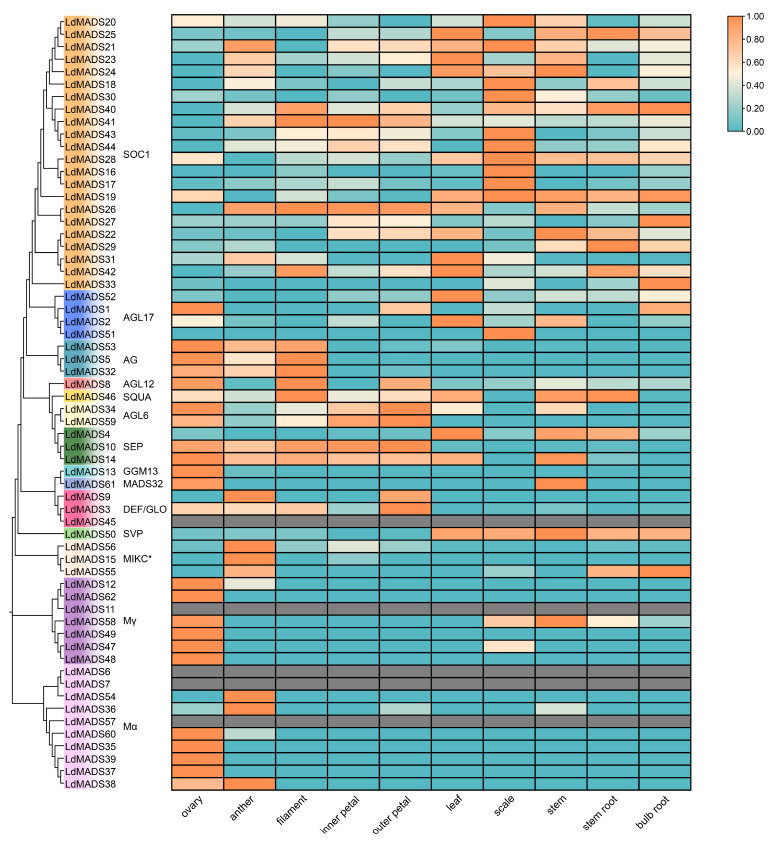 Figure 8
Figure 8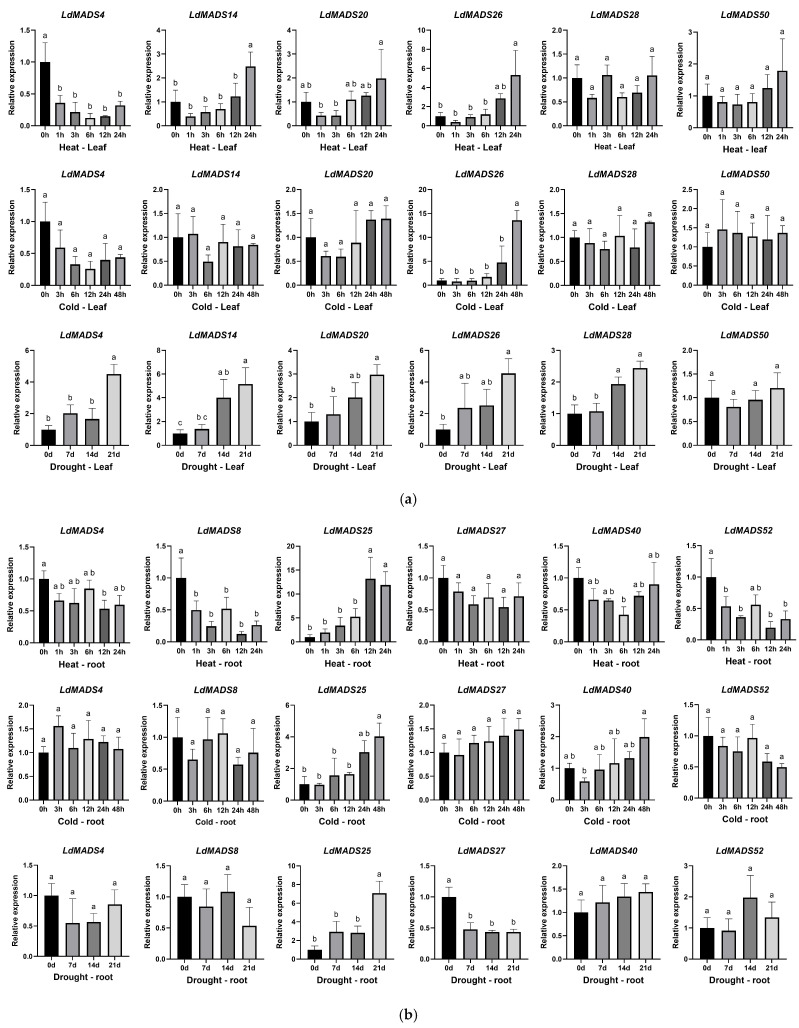 Figure 9
Figure 9- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Molecular Biology Research · Plant Gene Expression Analysis · Plant Reproductive Biology
1. Introduction
Transcription factors (TFs) regulate gene expression and perform diverse functions in higher plants [1,2]. They play a major role in plant development, organogenesis, stress responses, and hormone signaling [3,4,5]. Among eukaryotic TFs, the MADS-box family constitutes one of the most extensive groups [6]. All MADS-box genes encode a conserved MADS domain at the N-terminus of the protein, comprising approximately 60 amino acids [7]. MADS-box genes can be classified into two lineages, Type I and Type II, based on their protein domain structure. Type I MADS TFs, also known as M-type TFs, typically contain only one to two exons [8]. They can be further subdivided into three subfamilies (Mα, Mβ, Mγ) based on phylogeny and conserved motifs in their C-terminal regions [9]. In contrast, Type II MADS TFs are generally composed of 7 exons and 6 introns [10]. They feature four characteristic domains known as the MIKC-type TFs. Extending from the N- to the C-terminus, the domain architecture includes the conserved MADS (M) domain, followed by the intervening (I) domain with lower conservation, the keratin-like (K) domain, and finally the variable Carboxy-terminal (C) domain. Distinct I and K domain features form the basis for subdividing them into MIKC^C^ and MIKC* categories [11]. Moreover, MIKC^C^-type MADS TFs can be grouped into 14 distinctive subfamilies: AG/STK, SEP, AGL6, SQUA, DEF/GLO, SOC1, FLC, AGL12, AGL15, SVP, Bsister (GGM13), AGL17, TM8, and OsMADS32-like [12]. Genes within the same subfamily typically exhibit comparable expression profiles, and their encoded proteins often perform closely associated biological functions [13].
The mechanism of DNA binding is fundamental in determining target gene specificity for TFs. MADS-box proteins form dimers that interact with DNA through their conserved MADS domains. In vitro studies have shown that this dimer typically recognizes a 10 bp consensus motif CC(A/T)6GG, referred to as the CArG-box [14]. Furthermore, the evolutionary acquisition of the I and K domains promotes their oligomerization and facilitates interactions with fellow MADS-box proteins, enabling plant MADS-box TFs to form tetramers and achieve greater functional diversification [15].
In higher plants, MADS-box TFs serve as crucial regulators of numerous biological processes, including floral development, nutrient and energy metabolism, signaling and transduction, dormancy release, seed germination, and fruit development [16,17,18]. The primary functions of M-type transcription factors include gametophyte development, embryogenesis, and other reproductive activities [19]. Type II MIKC* members are predominantly expressed in pollen and are essential for pollen germination and maturation in A. thaliana [20]. In contrast, MIKC^C^-type genes, which are implicated in virtually every stage of plant development, comprise the majority of MADS-box genes with known functions [21]. Tetramerization of MIKC^C^-type proteins represents a key contributor to flowering diversity among angiosperms. This is exemplified by the well-established ABCDE model of floral organ development, which was formulated after extensive studies clarified the critical role of these complexes in specifying floral organ identity [22,23]. In addition, the MADS-box members SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1), AGAMOUS-LIKE24 (AGL24) [24], FLOWERING LOCUS C (FLC) [25], and SHORT VEGETATIVE PHASE (SVP) [26] have also been found to be associated with flowering time and floral transition. Furthermore, FRUITFULL (FUL) has been implicated in fruit ripening and expansion in diverse species [27,28]. To date, research has increasingly demonstrated the participation of MADS-box TFs in various stress responses. Overexpression of SlMBP22 in tomato has been demonstrated to enhance drought tolerance by improving water retention, osmolyte accumulation, and antioxidant capacity [29]. In Oryza sativa, OsMADS22, OsMADS26, and OsMADS55 have been shown to participate in stress responses [30,31]. Notably, the CArG-box motif, recognized by MADS-box proteins, is prevalent in the promoters of numerous defense-related genes. Therefore, elucidating the roles of MADS-box genes under both biotic and abiotic stress provides valuable insights for plant breeding and crop improvement. Despite this potential, the stress-related functions of these genes remain largely unexplored in Lilium davidii var. unicolor.
L. davidii var. unicolor, commonly known as the Lanzhou lily, is a perennial herb and a variant of Lilium davidii. It predominantly grows in the plateau regions south of Lanzhou City, Gansu Province, China, at elevations between 1800 and 2600 m [32]. The major cultivation area is characterized by a typical temperate continental climate. Precipitation is unevenly distributed and infrequent, often resulting in drought during the growing season. Meanwhile, extreme temperature fluctuations between early spring frost and summer expose plants to both high and low temperature stresses. These abiotic stresses, including drought that inhibits bulb expansion and alters sugar metabolism [32,33], heat that causes growth retardation and developmental delays [34,35], and cold that affects lily plants during the seedling and cut-flower stages [36], have become major limiting factors for the yield and quality of Lanzhou lily. Therefore, elucidating the molecular mechanisms underlying MADS-box gene-mediated abiotic stress responses in Lanzhou lily is urgently needed.
Genetic and breeding studies on the Lanzhou lily have been challenging due to the giant genome size of approximately 36.68 Gb [37]. The completion of its chromosome-level genome assembly in 2024 allowed us to perform the genome-wide characterization of MADS-box genes in Lanzhou lily. Furthermore, we assayed the expression changes of selected members under drought, heat, and cold stress conditions, preliminarily identifying potential key LdMADS genes. This study offers a theoretical foundation for enhancing germplasm resources and advancing stress-tolerance studies in Lanzhou lily.
2. Results
2.1. Identification and Physicochemical Properties of LdMADS Genes
In the genome of L. davidii var. unicolor, 62 MADS-box genes were identified. The corresponding 62 amino acid sequences are provided in Supplementary Table S1. These 62 genes were systematically named LdMADS1-LdMADS62 according to their chromosomal locations. The 62 LdMADS proteins range from 110 (LdMADS57) to 423 (LdMADS11) amino acids in length, with molecular weights between 12.449 kDa (LdMADS57) and 48.087 kDa (LdMADS11). Isoelectric point (pI) ranging from 4.92 (LdMADS58) to 10.04 (LdMADS29). Detailed information is provided in Supplementary Table S2.
2.2. Phylogenetic Analysis of LdMADS Genes
To examine the phylogenetic relationships of MADS-box genes in Lanzhou Lily, we constructed separate phylogenetic trees for Type I and Type II by aligning their protein sequences with those of A. thaliana and O. sativa (Figure 1a,b). The phylogenetic results revealed that 17 LdMADS proteins belong to the Type I lineage. These were classified into two subgroups, including 10 Mα and 7 Mγ. Notably, there were no Mβ subgroup members found in the Lanzhou lily genome (Figure 1a). 45 LdMADS proteins were identified as Type II, which included 3 MIKC* and 42 MIKC^C^ members. Furthermore, the MIKC^C^-type LdMADS proteins were distributed into 11 subfamilies, including SEP, AGL6, SQUA, AG, AGL12, SOC1, DEF/GLO, GGM13, AGL17, SVP, and MADS32. No members belonging to the FLC or AGL15 subfamilies were detected (Figure 1b).
Overall, type II proteins in Lanzhou Lily exhibited a quantity similar to that in rice and Arabidopsis, whereas the count of Type I proteins was relatively lower. A significant expansion was observed specifically within the SOC1 subfamily, which contains 22 structurally intact SOC1 proteins in Lanzhou Lily.
2.3. Conserved Motifs and Gene Structure Analysis of LdMADS Genes
The online tool MEME was employed to analyze 62 LdMADS proteins in order to examine the motifs shared among different members and subfamilies, which identified 15 distinct motifs within their amino acid sequences (Figure 2a,b; Table S3). Among these, Motif1 and Motif3 were shared by the majority of LdMADS proteins and together encode the highly conserved M-domain. Motif2, Motif6, Motif9, and Motif12 correspond to K-box and were exclusively found in MIKC proteins (Figure 2b,c). While overall conservation was lower in the C-terminal regions, proteins belonging to the same subfamily tended to possess comparable motif compositions, suggesting a degree of subfamily-specific conservation. These patterns suggest that motif architecture is largely consistent within each subfamily but distinct across different subfamilies, a feature that may reflect their respective functional specializations.
To investigate the structural evolution of the 62 LdMADS genes, we analyzed their exon-intron organization (Figure 3; Table S2). The analysis revealed considerable variation in gene length and intron number among the LdMADS genes, with intron counts ranging from 0 to 10. Specifically, LdMADS53 and LdMADS56 contained the highest number of introns, with 10 each. In contrast, 11 genes (LdMADS6, LdMADS12, LdMADS35, LdMADS37, LdMADS38, LdMADS39, LdMADS48, LdMADS57, LdMADS58, LdMADS60, and LdMADS62) lacked introns entirely and consisted of a single exon. Furthermore, MIKC-type genes generally harbored 3 to 10 introns, whereas M-type genes contained 0 to 3 introns.
Notably, a substantial proportion of LdMADS genes were exceptionally long. Thirty genes (48.39%) exceed 50 kb in length and were therefore classified as ultra-long genes [37]. Considerable variation in gene length was also observed even within the same subfamily. Given that the intron count in LdMADS genes was comparable to that of other plants, the extraordinary gene length is primarily attributable to extremely long introns. Previous studies indicate that genome expansion in lilies results mainly from the proliferation of transposable elements, which dramatically lengthen intergenic and intronic regions, coupled with contributions from whole-genome duplication and tandem duplication events [38].
2.4. Chromosomal Localization and Synteny Analysis
To examine genetic divergence and gene duplication events in the MADS-box family, a chromosomal location map was generated from Lanzhou lily genome data. With LdMADS62 located on an unassembled scaffold, the remaining 61 LdMADS members were distributed across all 12 chromosomes, albeit in a highly uneven pattern (Figure 4). Chromosome 4 stands out with the highest count of 22 genes, while chromosome 7 ranks second with 12 genes. Merely one gene was located on each of chromosomes 5, 11, and 12. Members of the Type II SOC1 subfamily are predominantly clustered on chromosomes 4 and 7. Half of the Type I Mα subfamily members are concentrated on chromosome 7.
Synteny analysis was performed using MCScanX to compare LdMADS genes with their counterparts in O. sativa, Lilium regale, and A. thaliana (Figure 5). A synteny analysis revealed a total of 39 syntenic pairs between the two closely related lily species. Syntenic gene pairs were distributed across all chromosomes with the exception of chromosomes 5 and 11. Chromosome 4 contained the most significant number, accounting for 20 pairs. Conversely, 9 syntenic pairs were detected between Lanzhou lily and O. sativa, while only one pair was found between Lanzhou lily and A. thaliana. This indicates significant genomic divergence during evolution between monocots (lily, rice) and dicots (Arabidopsis). LdMADS14 exhibited collinear relationships across L. regale, Arabidopsis, and rice, suggesting its function may be conserved throughout evolution. Indirect evidence for the potential functional conservation of LdMADS genes across species was provided by syntenic analysis.
2.5. Cis-Acting Elements Analysis of LdMADS Genes
To further analyze the cis-acting elements of LdMADS genes, sequences spanning 2000 bp upstream of LdMADS genes were extracted. 31 elements with clearly defined biological roles were analyzed using the PlantCARE online database (Figure 6 and Figure 7). These elements could be categorized as light-responsive, stress-responsive, hormone-responsive, and growth regulatory elements. Numerous motifs linked to stress responsiveness and phytohormone regulation were abundant in the promoter region.
The analysis indicated that all candidate genes had at least one stress-related cis-acting element, including 49 drought stress response elements (MBS) distributed across 33 LdMADS gene promoters and 52 low-temperature response elements (LTR) distributed across 39 LdMADS gene promoters (Figure 7). Defense and stress response elements (TC-rich repeats), plant injury response elements (WUN-motifs), and oxidative stress response elements (ARE and GC-motif, etc.) were also identified.
The eight hormone response elements identified can be categorized into five groups, including SA (TCA-element), ABA (ABRE), GA (P-box, GARE-motif), MeJA (CGTCA-motif and TGACG-motif), and auxin (TGA-element and AuxRR-core) response elements. Among them, elements responsive to MeJA were particularly abundant. 197 MeJA-responsive elements were found in the promoters of 44 LdMADS genes. Furthermore, it was shown that ABA plays an essential role in the adaptation of nutrient tissues to abiotic stresses, with a total of 178 ABA-responsive elements identified in the promoters of 45 LdMADS genes. This evidence lends robust support to the putative function of LdMADS genes in mediating abiotic stress adaptation at the transcriptional level.
2.6. Tissue-Specific Expression Analysis of LdMADS Genes
Based on public transcriptomic datasets, a heatmap was generated that revealed clear tissue-specific expression profiles and abundance variations for the 62 LdMADS genes among ten lily tissues (Figure 8). The five genes that showed no expression across all tissues comprised four Type I genes (LdMADS6, LdMADS7, LdMADS11, LdMADS57) and one Type II gene (LdMADS45). Ten genes were exclusively expressed in a single tissue. For instance, LdMADS13, LdMADS35, LdMADS37, LdMADS39, LdMADS48, LdMADS49, and LdMADS62 were expressed solely in the ovary; LdMADS15 and LdMADS54 only in the anther; and LdMADS51 only in the scale. Conversely, nine genes, including LdMADS14, LdMADS19, LdMADS20, LdMADS26, LdMADS28, LdMADS40, LdMADS41, LdMADS42, and LdMADS46, were expressed across all ten tissues.
Type I genes showed more tissue-specific expression than Type II genes. In line with earlier findings that Type I MADS-box genes are crucial for plant reproductive development, the majority of Type I genes were only expressed in the ovary or anther. Their relatively low expression levels imply that they may be active only during specific periods of reproduction. All MIKC*-type genes were discovered to be expressed in anthers, with LdMADS55 also expressed in stem roots and bulb roots.
In contrast, MIKC^C^ genes displayed broader expression patterns. Most MIKC^C^ genes were detected in two or more tissues. This was particularly evident for the SOC1 subfamily, in which 7 out of the 22 members were expressed across all tissues examined. A predominant expression pattern for these SOC1 subfamily genes was their high expression in the bulb, a specialized storage organ in the lily. In addition to SOC1, scales also exhibit significant expression of homologs of SVP (LdMADS50) and FUL (LdMADS46). Members of the same subfamily typically exhibit comparable expression profiles. This is exemplified by the three AG subfamily genes, which show pronounced and specific expression in anthers, filaments, and ovaries. Two of the three genes of the SEP subfamily (LdMADS10 and LdMADS14) were strongly expressed in all floral organs, while two genes (LdMADS14 and LdMADS4) exhibited high transcript levels in leaves. The expression patterns of LdMADS genes show clear subfamily specificity and are consistent with earlier research on their roles in floral organ development.
Analysis of the tissue expression data showed that members of the LdMADS gene family exhibit complex and distinct expression patterns, suggesting a potential association with their functional diversification.
2.7. Expression Patterns of LdMADS Genes Under Stress Conditions
To investigate the potential role of the LdMADS gene family in stress response, we combined analyses of root and leaf expression levels and promoter element distributions with prior functional studies of MADS-box genes. Based on this integrated analysis, six candidate genes were selected for further study. In leaves, these were LdMADS4/14 (SEP2), LdMADS20/26/28 (SOC1), and LdMADS50 (SVP). In roots, these were LdMADS4 (SEP2), LdMADS8 (AGL12), LdMADS25/27/40 (SOC1), and LdMADS52 (AGL16). Their expression patterns under cold (4 °C), heat (37 °C), and drought stress were then analyzed by qRT-PCR (Figure 9).
Examination of the six genes expressed in leaves revealed that all but LdMADS50 responded to at least one stress, showing differences in expression patterns. Notably, under low-temperature conditions, only LdMADS26 (a homolog of SOC1) was significantly induced, reaching more than ten times its pre-treatment level after 48 h of treatment. This response was consistent with the presence of an LTR motif in its promoter. In contrast, the expression changes of other genes, including the SVP homolog LdMADS50, did not show a significant change at low temperature. Under high-temperature stress, LdMADS4 expression decreased, reaching its lowest level at 6 h before a slight rebound, while LdMADS14/20/26 exhibited a delayed up-regulation. All screened genes except LdMADS50 responded to drought. LdMADS4/14/20/26/28 demonstrated sustained transcriptional upregulation, indicating an ongoing response process.
Detection of the six genes expressed in roots revealed that two SOC1 members, LdMADS25 and LdMADS40, were up-regulated in response to low temperature. LdMADS40 showed a slight decrease in expression after 3 h of low-temperature treatment, exhibiting a delayed response. Under high-temperature stress, the expression patterns of different LdMADS genes varied greatly. LdMADS4/8/52 expression decreased in general and reached its lowest at 12 h, while LdMADS25 was strongly upregulated, with its peak expression at 12 h. This common time point may indicate that 12 h is a critical period for responding to high-temperature stress. LdMADS40 also responded to high temperature in roots, with expression first decreasing and then increasing. LdMADS25 and LdMADS40 responded to drought stress, but showed opposite expression patterns.
Based on the qRT-PCR results, we observed that SOC1 subfamily members exhibited distinct stress-responsive expression patterns. LdMADS25 (roots) and LdMADS26 (leaves) were strongly upregulated under all three stress conditions, suggesting they may function as positive regulators involved in multiple stress responses. Notably, SOC1 subfamily members were the only genes among our selected candidates that responded to cold stress in both roots (LdMADS25/40) and leaves (LdMADS26). In contrast, the SEP2 homologs LdMADS4 and LdMADS14 were primarily responsive to high temperature and drought, though their specific expression patterns differed. This suggests that the expanded SOC1 members in Lanzhou lily may partly function as positive regulators in mediating various stress responses.
3. Discussion
In this study, 62 MADS-box genes were identified in Lanzhou lily. In accordance with their chromosomal locations, these genes were designated as LdMADS1 to LdMADS62. The LdMADS members were categorized into two major groups: Type I (Mα 10 genes, Mγ 7 genes) and Type II MIKC (MIKC* 3 genes, MIKC^C^ 42 genes). The MIKC^C^ genes were subsequently assigned to 11 distinct subfamilies. The number of MADS-box genes varies significantly across species. For instance, O. sativa has 75 [39], Sorghum bicolor has 65 [40], and A. thaliana has 107 [8]. Although the genome of Lanzhou lily is markedly larger than that of other species, the total number of LdMADS genes is not correspondingly greater. Further analysis revealed a relatively low number of M-type genes and the absence of members from the Mβ subgroup. Unlike the MIKC genes, Type I genes are typically characterized by shorter lengths and have undergone more rapid birth and death during evolution [9,41]. They often expand through small-scale duplication events, making them more prone to silencing or loss in the genome [42]. Nevertheless, a subset of Type I genes plays crucial roles in key reproductive developmental pathways, such as AGL23, AGL61, AGL62, and AGL80 [43,44,45,46]. In L. davidii var. unicolor, multiple homologs of these genes were identified among its Type I members and were found to be specifically expressed in reproductive tissues. This suggests potential conservation of the structure and function of Type I genes across angiosperms. In contrast, Type II MADS-box genes are preferentially retained following whole-genome or large-scale duplication events. This differential retention may explain why the copy number of Type I MADS-box genes exhibits greater variation than that of Type II genes across species [21].
Expansion of specific MADS-box subfamilies has been documented in various plants. For instance, the SOC1 subfamily of Eucalyptus grandis has undergone significant expansion. The subfunctionalization of SOC1 enables adaptation to variable ecological environments spanning the tropics to temperate zones, thereby meeting different needs in flowering regulation [47,48]. In addition, the expansion of FLC and SEP subfamilies has also been observed in some Asteraceae species, such as Taraxacum kok-saghyz [49] and Taraxacum officinale [50]. In Lanzhou Lily, the SOC1 subfamily has undergone a remarkable expansion, with 22 members identified, accounting for nearly half of all its MIKC-type genes. This proportion is substantially higher than that reported in other angiosperms. The expansion pattern of the SOC1 subfamily in Lanzhou Lily appears complex. Beyond two pairs of tandem duplicate genes (LdMADS20/LdMADS21 and LdMADS25/LdMADS26), most SOC1 members form local clusters on chromosomes 4 and 7. These genes are physically proximate yet interspersed by a few other genes, consistent with the characteristics of proximal duplication [51,52]. Compared to other duplication modes, tandem and proximal duplicates often undergo highly selective pressure, exhibit more compact gene structures and broader expression patterns, and serve as a key genetic reservoir for evolving novel functions [51]. Transcriptomic data reveal that the expanded SOC1 genes exhibit broad and diverse expression patterns across the 10 tissues examined in Lanzhou Lily. Most are highly expressed in the bulb, a specialized storage organ, suggesting potential subfunctionalization or neofunctionalization within this subfamily [53]. A particularly notable observation is the absence of FLC subfamily members in the Lanzhou lily genome. In Arabidopsis, FLC is a master regulator of vernalization, which is a low-temperature requirement for flowering that lilies also exhibit [54]. This suggests that functional diversification among the SOC1 subfamily in lilies may compensate for or replace the regulatory role of FLC in the vernalization pathway. Consistent with this hypothesis, SOC1 in Lanzhou lily is highly expressed in overwintering organ bulbs and responds to cold stress. These expanded SOC1 homologs may therefore not only function as integrators of flowering signals but also participate in Lilium-specific developmental processes, such as bulb formation, dormancy release, and vernalization [55].
The evolution of plant MADS-box genes involves the loss and gain of introns [56]. Typically, Type II MIKC members contain more introns than Type I members. Analysis of the gene structures revealed that Type II members contained an average of 6.4 introns, whereas most Type I genes lack introns entirely, except for a few exceptions. The intron number in LdMADS genes is similar to that observed in Arabidopsis, rice, and other plants. However, Lanzhou lily contains a gigantic genome, and this is reflected in gene length. Previous genomic analysis showed that 33.88% of all annotated genes in Lanzhou lily exceed 50 kb in length and are defined as ultra-long genes [37]. Strikingly, this proportion rises to 48.39% within the LdMADS gene family. Despite their ultra-long lengths, the exon structures of these LdMADS genes remain conserved, with average number and length comparable to those found in other plants. Thus, the extensive lengthening of these genes is primarily due to extreme intron elongation. Previous studies indicate that the dramatic increase in lily genome size is largely associated with bursts of transposable elements insertions, which accumulate abundantly in intergenic regions and introns [37]. The preferential accumulation of transposable element sequences in MADS-box introns may have introduced new regulatory elements, potentially adding complexity to the transcriptional regulation of LdMADS genes.
Tissue-specific expression analysis revealed that LdMADS genes exhibit highly divergent and specific expression patterns, which are closely linked to their functional diversity. M-type genes were expressed mainly at low abundance in the ovary and anther, consistent with their conserved function in plant reproductive development. Conversely, MIKCC genes showed a broader expression profile. Of particular note was the SOC1 subfamily, of which 7 of 22 members were detected across all tissues tested. Similarly, homologs of SVP (LdMADS50) and FUL (LdMADS46) were also highly expressed in scales, suggesting their potential common role in lily-specific biological processes, including dormancy release and environmental adaptation. In addition, three AG subfamily genes showed pronounced and specific expression in the anther, filament, and ovary. AG is a representative gene that characterizes the floral organs of class C and is studied to be expressed in stamens and carpels [57]. Two SEP subfamily genes (LdMADS10 and LdMADS14) were highly expressed in all five floral organ tissues. The specialization of floral organ features depends on SEP subfamily members, which are usually defined as class E genes [58]. In conclusion, tissue expression profiling not only confirmed the important function of the MADS-box family in floral development but also suggested its possible role in bulb formation and in adaptation to environmental stress.
Our investigation of promoter regions identified a prevalent abundance of abiotic stress-responsive cis-elements within the LdMADS promoters. Elements associated with hormone signaling, drought, and low-temperature stress were frequently present. Consistent with this putative role in stress adaptation, MADS-box genes are known regulators of abiotic stress responses in other species. As evidenced by studies in A. thaliana, AGL16 acts as a negative regulator of drought resistance by modulating stomatal density and ABA accumulation, and by directly regulating CYP707A3, AAO3, and SDD1 [59]. In Capsicum annuum, a SEP subfamily member, CaMADS, positively regulates abiotic stress responses and is induced by multiple stresses and hormones [60]. Based on prior studies, tissue expression profiles, and promoter element analyses, we validated the expression patterns of selected LdMADS genes under drought, cold, and heat stress.
LdMADS14, a homolog of SEP2, was also found to be significantly up-regulated in leaves under both drought and heat stresses, while LdMADS4 responded to various stresses in both leaves and roots. SOC1 homologs LdMADS25 and LdMADS26 were strongly induced in response to all three stresses in roots and leaves, respectively. The upregulation of LdMADS25 in leaves under cold stress, together with the responsiveness of LdMADS26 and LdMADS40 in roots, further supports the involvement of the expanded SOC1 subfamily in cold response. Notably, although LTR elements were identified in the promoters of multiple candidate genes, only SOC1 homologs exhibited significant upregulation under cold stress. This may suggest that the cold response of MADS-box genes involves complex transcriptional regulation beyond the mere presence of individual cis-elements. Conversely, LdMADS8 (an AGL12 homolog) and LdMADS52 (an AGL16 homolog) were down-regulated in roots under heat stress. Collectively, the results suggest that specific LdMADS genes, especially SOC1 and SEP homologs, may be crucial for enhancing stress tolerance as regulators of multiple stresses in Lanzhou lily, while their precise molecular mechanisms remain to be further investigated.
4. Materials and Methods
4.1. Identification of LdMADS Gene Family Members
The genomic data of Lanzhou lily were acquired from the China National GeneBank Database (https://ftp.cngb.org/pub/CNSA/data5/CNP0005511/CNS1065710/CNA0139751/, accessed on 15 January 2025) [37]. Identification of MADS-box genes within the Lanzhou lily genome was conducted employing HMMER-3.0 software [61]. Corresponding hidden Markov model (HMM) profiles for the SRF (Type I) domain (PF00319) and the MEF2-like (Type II) domain (PF09047) were obtained from the Pfam database [62]. In addition, MADS-box proteins from Arabidopsis and rice served as query sequences for BlastP searches against the predicted proteins. The TAIR website (http://www.arabidopsis.org/) provided the Arabidopsis MADS-box family database [8], while the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/) provided the rice MADS-box family database [39]. The presence of the conserved domain was then confirmed for each candidate protein sequence using the SMART database (https://smart.embl.de). Redundant sequences and those lacking a complete MADS domain were removed from the final dataset. Additionally, we analyzed the physicochemical properties of the identified LdMADS proteins using the Protein Parameter Calc tool in TBtools [63].
4.2. Phylogenetic Analysis of LdMADS Proteins
Using amino acid sequences of the 62 LdMADS genes and known MADS-box sequences from Arabidopsis and rice, a phylogenetic tree was constructed to confirm the identification. Sequence alignment was carried out with the Clustal W algorithm in MEGA11 software. For statistical support, the Neighbour-Joining (NJ) method was applied with 1000 bootstrap replicates. The iTOL online tool (https://itol.embl.de) was used to visualize and annotate the resultant tree.
4.3. Analysis of Protein Conserved Motifs, Domains, and Gene Structures
Conserved motifs in the LdMADS proteins were detected using the online MEME suite (https://meme-suite.org/meme/, accessed on 24 June 2025), with a maximum of 15 motifs configured while retaining all other default parameters. Domain prediction was performed via analysis of the MADS-box protein sequences using the NCBI Conserved Domain Database (CDD). Moreover, the results of the conserved motif analysis, domain predictions, and the genome annotation file (gff3) were merged and visualized using the Gene Structure View function in TBtools.
4.4. Chromosomal Localization and Synteny Analysis of LdMADS Genes
The chromosomal locations of the LdMADS genes were mapped and visualized using the Gene Location Visualize function in TBtools (version 2.423). Synteny analysis between the LdMADS genes and the MADS-box members from rice, L. regale, and Arabidopsis was analyzed using the One Step MCScanX-Super Fast tool.
4.5. Cis-Acting Element Analysis of LdMADS Genes
The promoter region 2000 bp upstream of 62 LdMADS genes was extracted using the Gtf/Gff3 Sequences Extract tool in TBtools. To complete the prediction and analysis of cis-acting elements, the LdMADS gene promoter sequences were uploaded to the PlantCARE database (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 25 June 2025). The resulting element profiles were subsequently counted and classified accordingly.
4.6. Tissue-Specific Expression Profiling of LdMADS Genes
To investigate their potential functions, the expression profiles of LdMADS genes were analyzed across ten tissues. Transcriptomic data for anther, filament, inner tepal, outer tepal, leaf, ovary, stem, stem-derived root, and basal root were obtained from the RNA-seq dataset published by Xu et al. (BioProject ID: CNP0005511) [37]. Data for the scale tissue were sourced from a separate RNA-seq study by Qi et al. (BioProject ID: PRJNA705898) [64].
4.7. Plant Materials and Treatments
Planting was conducted at the agricultural experiment base of Beijing Forestry University (116.34 E, 40.01 N). Uniformly sized bulbs were transplanted into pots (16 cm in diameter) with three bulbs per pot, constituting one biological replicate. Plants were grown for 30 days, after which healthy individuals of consistent size and vigor were selected for the stress treatment.
For the drought stress treatment, potted plants were placed in trays, and each pot was irrigated with 500 mL of tap water until the soil reached full water-holding capacity. The collected osmotic water from the trays was reapplied to ensure a balanced amount. All plants were then transferred to a growth chamber at 25 °C, 60% relative humidity, and a 16/8 h light/dark photoperiod. Watering was subsequently withheld completely. Soil water content was measured, and samples were collected from the upper leaves and stem roots at 0, 7, 14, and 21 days after the start of the treatment.
For the heat stress treatment, plants were placed at 37 °C under identical humidity and photoperiod conditions. Sampling was performed at 0, 1, 3, 6, 12, and 24 h after treatment initiation. For cold stress treatment, plants were placed at 4 °C. Samples were acquired at 0, 3, 6, 12, 24, and 48 h.
Each treatment included three biological replicates. Plant samples were encased in aluminum foil, rapidly frozen in liquid nitrogen, and subsequently preserved at −80 °C for subsequent analysis and RNA extraction.
4.8. qRT-PCR Analysis of the LdMADS Genes
Total RNA was extracted from the samples using the EASY Spin Plus RNA Kit (Aldlab, Beijing, China). Following evaluation of both quantity and quality, sufficient amounts of cDNA were synthesized using a reverse transcription kit (TOYOBO, Shanghai, China). Quantitative real-time PCR (qRT-PCR) reactions were conducted on a CFX Connect instrument (Bio-Rad, Shanghai, China) with SYBR Green Pro Taq HS (Accurate Biology, Changsha, China). The LdActin gene was utilized as a control to standardize expression levels, and primer sequences were presented in Table S4. The relative gene expression of LdMADS genes was calculated using the 2^−ΔΔCt^ method.
5. Conclusions
This study presented the first genome-wide identification of the MADS-box transcription factor family in Lanzhou lily, a species with a giant genome. Two key genomic features were identified: a significant expansion of the SOC1 subfamily and the prevalence of ultra-long genes driven by intron elongation. Combined with a tissue-specific expression heatmap, we propose that these expanded genes may not only regulate flowering time but also perform essential functions in lily-specific processes, including bulb development, dormancy regulation, and stress responses. This shows the functional diversification of this subfamily in the Lanzhou lily. The results of the stress response analyses indicated that some LdMADS members, particularly LdMADS4, LdMADS14, LdMADS25, and LdMADS26, are likely involved in abiotic stress adaptation. Collectively, these findings supply a genetic resource and potential candidate genes for further research on stress tolerance in Lanzhou lily.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Riechmann J.L. Ratcliffe O.J. A genomic perspective on plant transcription factors Curr. Opin. Plant Biol.2000342343410.1016/S 1369-5266(00)00107-211019812 · doi ↗ · pubmed ↗
- 2Singh K. Foley R.C. Onate-Sanchez L. Transcription factors in plant defense and stress responses Curr. Opin. Plant Biol.2002543043610.1016/S 1369-5266(02)00289-312183182 · doi ↗ · pubmed ↗
- 3Messenguy F. Dubois E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development Gene 200331612110.1016/S 0378-1119(03)00747-914563547 · doi ↗ · pubmed ↗
- 4Gahlaut V. Jaiswal V. Kumar A. Gupta P. Transcription factors involved in drought tolerance and their possible role in developing drought tolerant cultivars with emphasis on wheat (Triticum aestivum L.)Theor. Appl. Genet.20161292019204210.1007/s 00122-016-2794-z 27738714 · doi ↗ · pubmed ↗
- 5Dong X. Deng H. Ma W. Zhou Q. Liu Z. Genome-wide identification of the MADS-box transcription factor family in autotetraploid cultivated alfalfa (Medicago sativa L.) and expression analysis under abiotic stress BMC Genom.20212260310.1186/s 12864-021-07911-934362293 PMC 8348820 · doi ↗ · pubmed ↗
- 6Riechmann J.L. Heard J. Martin G. Reuber L. Jiang C. Keddie J. Adam L. Pineda O. Ratcliffe O.J. Samaha R.R. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes Science 20002902105211010.1126/science.290.5499.210511118137 · doi ↗ · pubmed ↗
- 7de Folter S. Angenent G.C. trans meets cis in MADS science Trends Plant Sci.20061122423110.1016/j.tplants.2006.03.00816616581 · doi ↗ · pubmed ↗
- 8ParenicováL. de Folter S. Kieffer M. Horner D. Favalli C. Busscher J. Cook H. Ingram R. Kater M. Davies B. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world Plant Cell 2003151538155110.1105/tpc.01154412837945 PMC 165399 · doi ↗ · pubmed ↗