Molecular Targeting of EGFR, BRAF, and HER2 Signaling in Colorectal Cancer: Contemporary Advances with Panitumumab, Encorafenib, and Tucatinib
Piotr Kawczak, Tomasz Bączek

TL;DR
This review discusses how targeting EGFR, BRAF, and HER2 pathways improves treatment for colorectal cancer patients based on molecular profiles.
Contribution
The paper provides an updated overview of targeted therapies and their clinical outcomes in molecularly defined colorectal cancer subgroups.
Findings
Panitumumab improves survival in RAS/BRAF wild-type, left-sided colorectal cancer when combined with chemotherapy.
Encorafenib combined with EGFR blockade benefits patients with BRAF V600E mutations.
HER2-targeted therapies like tucatinib show durable responses in HER2-amplified colorectal cancer.
Abstract
Metastatic colorectal cancer (mCRC) remains a major cause of cancer-related mortality worldwide. Advances in molecular profiling have transformed the therapeutic landscape, enabling biomarker-driven treatment strategies based on alterations in RAS, BRAF V600E, HER2 amplification, and mismatch repair status. Among these, dysregulation of the epidermal growth factor receptor (EGFR), BRAF, and HER2 signaling pathways represents a central driver of tumor progression and therapeutic resistance. Targeted agents directed against these pathways—including the anti-EGFR monoclonal antibody panitumumab, the selective BRAF inhibitor encorafenib, and the HER2-selective tyrosine kinase inhibitor tucatinib—have substantially expanded treatment options for molecularly defined subgroups of patients with mCRC. Anti-EGFR therapy remains a cornerstone of treatment for patients with RAS/BRAF wild-type,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
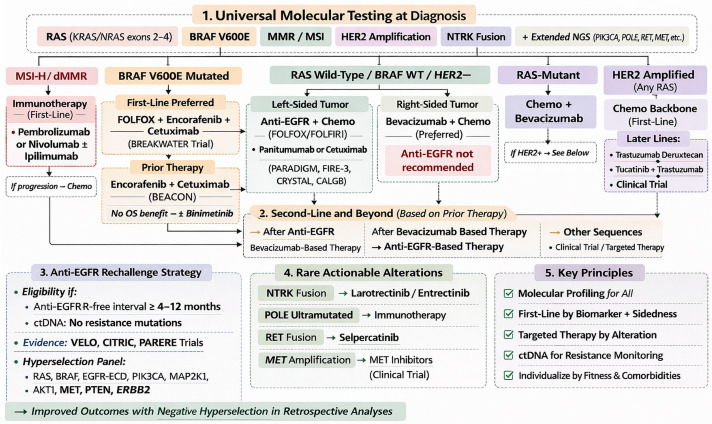 Figure 1
Figure 1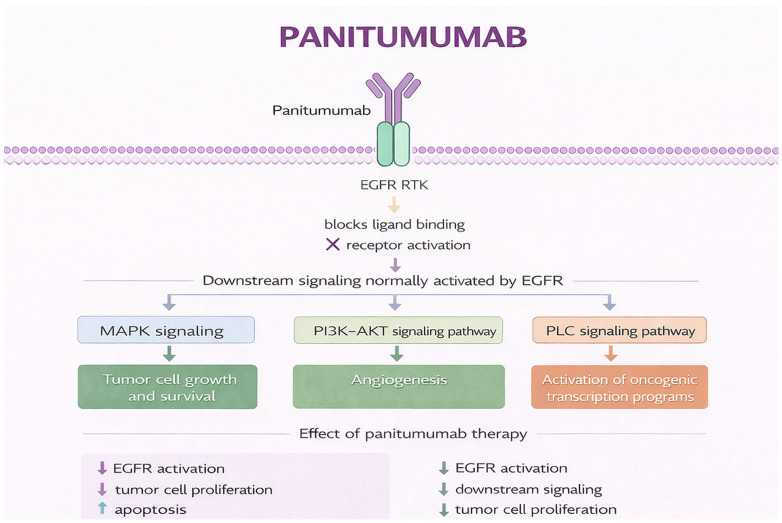 Figure 2
Figure 2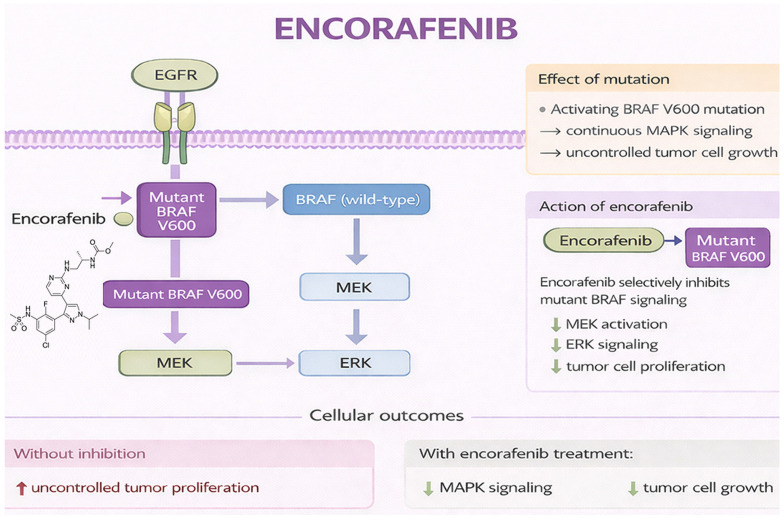 Figure 3
Figure 3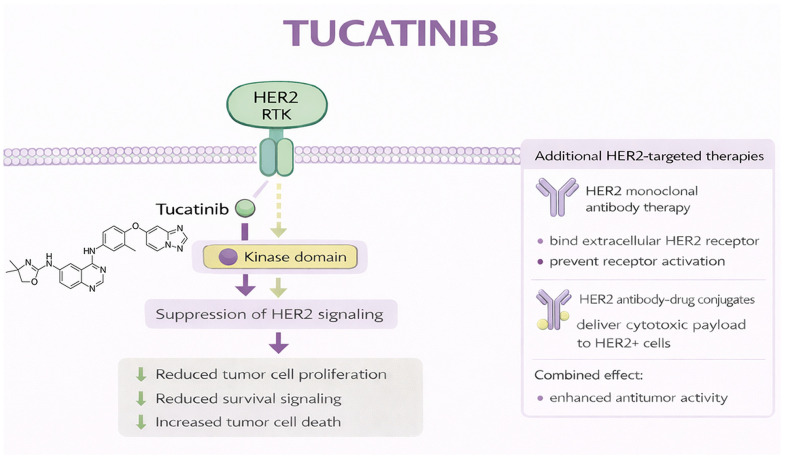 Figure 4
Figure 4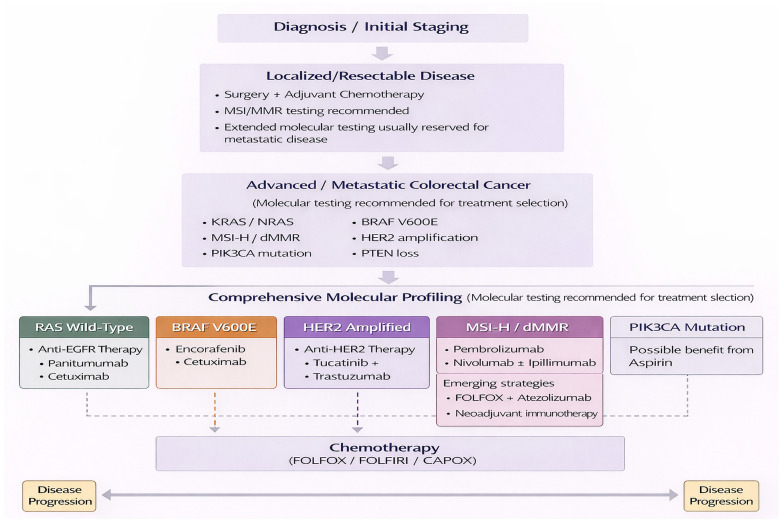 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsColorectal Cancer Treatments and Studies · HER2/EGFR in Cancer Research · Advanced Breast Cancer Therapies
1. Introduction
Colorectal carcinogenesis is driven by recurrent alterations in key oncogenic pathways regulating proliferation, survival, and differentiation [1,2,3,4]. Dysregulation of the EGFR pathway, constitutive activation of RAS–RAF–MEK–ERK (MAPK) signaling, and aberrations within the ERBB receptor family—particularly HER2 (ERBB2)—are central drivers of tumor initiation, progression, and therapeutic resistance [5,6,7,8]. Comprehensive genomic analyses have identified clinically relevant molecular subtypes characterized by actionable alterations in EGFR-related pathways, BRAF V600E mutations, and HER2 amplification [9,10,11,12]. These findings establish colorectal cancer (CRC) as a biologically stratified disease rather than a single histology-defined entity.
This molecular framework has been translated into routine clinical practice through guideline-directed profiling. Current international recommendations require extended RAS (KRAS and NRAS) testing, BRAF mutation analysis, and HER2 assessment in metastatic CRC (mCRC) to guide treatment selection and sequencing [13,14,15,16]. Next-generation sequencing is increasingly used to detect rarer actionable alterations and support precision oncology approaches.
Many of these alterations converge on the MAPK (RAS–RAF–MEK–ERK) and PI3K–AKT signaling pathways, which regulate cellular proliferation and survival. In mCRC, BRAF V600E mutations lead to constitutive MAPK activation, whereas HER2 amplification promotes receptor-mediated signaling that enhances downstream pathway activity. Targeted therapies act at different levels of these networks: anti-EGFR monoclonal antibodies block ligand-dependent receptor activation, BRAF inhibitors suppress constitutively active kinase signaling, and HER2 inhibitors interfere with receptor dimerization and downstream signaling. Because these signaling cascades are well characterized, the following sections focus primarily on their therapeutic implications and mechanisms of resistance rather than detailed pathway biology [17,18].
EGFR targeting remains a cornerstone of therapy for patients with RAS wild-type mCRC. Activating RAS mutations predict lack of benefit—and potential harm—from EGFR inhibition, establishing extended RAS testing as mandatory before therapy [19]. Clinical trials confirmed the efficacy of panitumumab in chemorefractory disease and defined its integration with cytotoxic chemotherapy [20]. Randomized studies further demonstrated that EGFR-targeted therapy provides particular benefit in left-sided, RAS wild-type tumors in the first-line setting, refining patient selection by molecular and clinicopathologic criteria [21,22].
BRAF V600E–mutant CRC represents a biologically distinct subtype with aggressive clinical behavior and historically poor outcomes with conventional chemotherapy. Preclinical and translational studies demonstrated that resistance to BRAF inhibitor monotherapy in CRC is mediated by rapid feedback activation of EGFR signaling, in contrast to melanoma [23]. This insight led to combination strategies incorporating BRAF and EGFR inhibition. The BEACON CRC trial established encorafenib plus cetuximab as the standard of care in previously treated BRAF V600E–mutant mCRC, improving overall survival (OS) and response rates compared with standard chemotherapy. Although the addition of binimetinib increased response rates, it did not improve OS and added toxicity; therefore, it is not considered standard of care [24,25]. Ongoing trials are evaluating encorafenib-based combinations earlier in the disease course, reflecting a shift toward earlier biology-driven intervention in high-risk CRC [26,27].
HER2 amplification and overexpression have emerged as actionable drivers in a subset of CRC, particularly in RAS wild-type tumors, where HER2 signaling also mediates resistance to EGFR-targeted therapy [18,28]. Early studies demonstrated the feasibility and activity of dual HER2 blockade with trastuzumab-based combinations in heavily pretreated HER2-positive mCRC [29,30]. Subsequently, tucatinib, a highly selective HER2 tyrosine kinase inhibitor, combined with trastuzumab, showed clinically meaningful efficacy in chemotherapy-refractory, HER2-positive, RAS wild-type mCRC, leading to regulatory approval and integration into treatment algorithms [31,32,33].
Advances in precision diagnostics have further refined CRC management. Consensus molecular subtyping highlights substantial biological heterogeneity with prognostic and therapeutic implications [7,34,35]. Circulating tumor DNA (ctDNA) analysis enables detection of minimal residual disease, prediction of recurrence, and dynamic monitoring of treatment response and resistance [36,37,38]. Tumors with microsatellite instability–high (MSI-H) or mismatch repair deficiency (dMMR) derive substantial benefit from immune checkpoint inhibitors and should be identified early in treatment planning [39]. Because immunotherapy represents a distinct therapeutic paradigm, its detailed clinical application lies beyond the primary scope of this review, which focuses specifically on EGFR-, BRAF-, and HER2-targeted strategies. Rare actionable alterations, including NTRK fusions and POLE mutations, also warrant testing given the availability of effective targeted therapies [40]. In the context of EGFR-directed therapy, ctDNA analyses have demonstrated the emergence of acquired RAS and EGFR alterations under treatment pressure, enabling real-time assessment of resistance and supporting adaptive strategies such as rechallenge [41,42,43]. Collectively, these developments support an iterative, biomarker-guided treatment paradigm in CRC [44,45,46].
This narrative review summarizes contemporary advances in molecularly targeted therapies directed at the EGFR, BRAF, and HER2 pathways in CRC, with emphasis on panitumumab, encorafenib, and tucatinib as representative agents. Particular attention is given to molecular diagnostics, rational combination strategies, and emerging biomarkers to optimize outcomes in CRC.
A structured literature search was conducted in PubMed and Scopus to identify relevant English-language publications published between January 2006 and January 2026. The starting date was selected to capture early translational investigations of EGFR-targeted therapy in CRC, preceding the regulatory approvals of anti-EGFR monoclonal antibodies and the emergence of biomarker-driven treatment strategies. Search terms included combinations of the following keywords: “colorectal cancer”, “metastatic colorectal cancer”, “panitumumab”, “encorafenib”, “BRAF V600E”, “tucatinib”, “HER2”, “EGFR”, “targeted therapy”, and “rechallenge strategies”. Reference lists of relevant articles and major clinical trial publications were also manually screened to identify additional eligible studies. The initial search identified approximately 450 publications. After title and abstract screening for relevance to targeted treatment strategies in mCRC, approximately 350 articles were selected for full-text evaluation. Ultimately, about 260 studies were included in the final narrative synthesis. Eligible studies comprised phase II and III prospective trials, randomized controlled trials, registration studies, meta-analyses, and practice-changing prospective cohort studies. Priority was given to randomized and registration-directed trials that established the clinical roles of anti-EGFR therapy, BRAF-targeted combinations, and HER2-directed treatment strategies. Large real-world cohorts and retrospective analyses were also included when they provided clinically meaningful insights into treatment effectiveness, resistance mechanisms, or therapeutic sequencing in routine practice. Smaller mechanistic or translational studies were incorporated selectively when they contributed to the biological understanding of EGFR, BRAF, or HER2 signaling pathways. Case reports, non-English publications, and conference abstracts without sufficient methodological detail were excluded. Preclinical-only studies were generally excluded unless they provided important mechanistic insights relevant to treatment efficacy, resistance mechanisms, or biomarker-driven therapeutic strategies. In addition, seminal studies published before 2006 were included when necessary to contextualize the biological rationale for targeted therapies discussed in this review. As this review was designed as a narrative synthesis, study selection emphasized clinical relevance, methodological robustness, and impact on contemporary therapeutic practice, rather than formal systematic review criteria.
Figure 1 presents a therapeutic algorithm for mCRC based on contemporary European Society for Medical Oncology (ESMO) 2023 and National Comprehensive Cancer Network (NCCN) 2024 guidelines and recommendations.
2. Panitumumab
Panitumumab is a fully human IgG2 monoclonal antibody targeting EGFR and represents a key targeted therapy in colorectal malignancies, particularly CRC [47,48,49]. EGFR is a transmembrane receptor tyrosine kinase within the ErbB family that is frequently overexpressed or dysregulated in CRC. Aberrant EGFR signaling promotes sustained proliferation, resistance to apoptosis, angiogenesis, invasion, and metastatic spread [50,51]. Given the epithelial origin of CRC and the dependence of many tumors on EGFR-driven signaling, inhibition of this pathway is both biologically rational and clinically validated [52].
Panitumumab binds the extracellular ligand-binding domain of EGFR, preventing activation by endogenous ligands such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α). This blocks receptor dimerization and autophosphorylation, suppressing downstream signaling through the RAS–RAF–MEK–ERK (MAPK) and PI3K–AKT pathways that regulate proliferation, survival, metabolism, and apoptosis [53,54]. As an IgG2 antibody, panitumumab mediates minimal antibody-dependent cellular cytotoxicity (ADCC); its antitumor activity derives primarily from direct inhibition of signal transduction, distinguishing it mechanistically from IgG1 antibodies such as cetuximab [55,56,57]. Figure 2 illustrates its mechanism of action.
The clinical development of panitumumab paralleled the emergence of biomarker-driven treatment strategies in metastatic CRC (mCRC). Early randomized trials in chemotherapy-refractory disease demonstrated improved progression-free survival (PFS) compared with best supportive care, confirming the therapeutic relevance of EGFR blockade in advanced CRC [59,60,61]. Subsequent translational analyses identified activating KRAS and NRAS mutations as mechanisms of primary resistance, establishing tumor genotype—not EGFR expression—as the key determinant of response [62]. These findings led to regulatory label changes and updates in ESMO and NCCN guidelines mandating extended RAS testing before anti-EGFR therapy [63,64,65].
Randomized trials have since clarified the clinical contexts in which EGFR inhibition provides the greatest benefit. In extended RAS wild-type tumors, anti-EGFR antibodies combined with chemotherapy improve response rates and survival compared with chemotherapy alone [66,67,68]. However, interpretation of these results requires consideration of differences in study design and patient populations. Further analyses demonstrated that primary tumor sidedness is a critical predictive factor. In left-sided RAS wild-type, BRAF wild-type, HER2-negative mCRC, anti-EGFR therapy combined with chemotherapy is generally preferred in the first-line setting, supported by trials including CRYSTAL, FIRE-3, CALGB/SWOG 80405, and PARADIGM [69,70,71,72,73,74]. PARADIGM demonstrated improved OS with panitumumab plus FOLFOX compared with bevacizumab plus FOLFOX in left-sided tumors [21]. In contrast, bevacizumab-based regimens are typically favored in right-sided disease due to inferior outcomes observed with anti-EGFR therapy [75].
Comparative interpretation of key trials highlights important methodological considerations. FIRE-3 suggested an OS advantage for cetuximab over bevacizumab despite similar PFS [76], whereas CALGB/SWOG 80405 reported comparable survival between strategies overall [71]. Differences in chemotherapy backbone, patient selection, molecular testing depth, statistical power, and post-progression therapies likely contributed to these divergent findings. The phase II PEAK study also suggested longer OS with panitumumab compared with bevacizumab in extended RAS wild-type populations, particularly in left-sided tumors [77]. However, the relatively small sample size and phase II design limit definitive conclusions and highlight the need for cautious interpretation. Collectively, these studies support preferential use of anti-EGFR therapy in biologically selected left-sided disease while underscoring the limitations of cross-trial comparisons.
Beyond first-line therapy, panitumumab remains active across later treatment lines in appropriately selected patients. In the second-line setting, panitumumab combined with FOLFIRI improves PFS in RAS wild-type disease [78,79]. In chemotherapy-refractory settings, the ASPECCT trial demonstrated non-inferiority of panitumumab compared with cetuximab monotherapy, confirming comparable efficacy of the two anti-EGFR antibodies [20,80]. Although these results support the continued role of EGFR inhibition in later treatment lines, it should be noted that variations in patient selection and prior therapies across studies may influence observed outcomes.
Anti-EGFR rechallenge has emerged as a potential strategy after a treatment-free interval, particularly when ctDNA analysis demonstrates clearance of resistance mutations [81,82,83,84]. Reported response rates in these studies are encouraging; however, most available evidence derives from small, non-randomized trials or retrospective analyses. Such designs introduce the possibility of selection bias, as patients eligible for rechallenge often represent a subgroup with more favorable tumor biology or slower disease progression. Additionally, heterogeneity in ctDNA assay methodologies, sensitivity thresholds, and definitions of molecular clearance complicates comparisons across studies. Resistance mechanisms—including alterations in RAS, BRAF, EGFR ectodomain, PIK3CA, MAP2K1, AKT1, MET, PTEN, and ERBB2—support the concept of “negative hyperselection” to refine patient selection for rechallenge strategies [82,83,84,85]. Prospective randomized trials are therefore required to confirm the clinical utility of ctDNA-guided rechallenge strategies.
The toxicity profile of panitumumab reflects on-target EGFR inhibition in epithelial tissues. Acneiform rash is the most common adverse event and typically occurs early during treatment. Interestingly, dermatologic toxicity correlates with improved outcomes; pooled analyses demonstrate longer PFS and OS in patients with grade ≥ 2 rash, supporting proactive dermatologic management rather than treatment discontinuation [86,87,88,89]. Additional adverse effects include diarrhea, mucositis, fatigue, and electrolyte disturbances such as hypomagnesemia resulting from impaired renal tubular reabsorption [90,91]. Infusion reactions are less common than with chimeric antibodies, although rare severe toxicities such as interstitial lung disease require prompt discontinuation [92].
Despite its established benefit, several limitations remain. Crossover and heterogeneous post-progression therapies in pivotal trials may confound survival analyses, and frail or elderly patients were often underrepresented. The optimal integration of anti-EGFR therapy with immunotherapy in MSI-H/dMMR tumors and the management of acquired resistance remain areas of ongoing investigation [85].
Within contemporary CRC management, panitumumab is firmly established as a precision therapy restricted to RAS wild-type mCRC, with the strongest evidence supporting its use in left-sided primary tumors. Treatment selection should integrate tumor sidedness, molecular profile, toxicity considerations, comorbidities, and therapeutic goals [71,76,77]. Beyond its clinical benefit, panitumumab exemplifies the broader paradigm of biomarker-driven oncology, demonstrating how molecular stratification can optimize outcomes and advance personalized treatment strategies in CRC [93,94,95,96,97,98,99].
Table 1 summarizes treatment-emergent adverse events and management strategies for panitumumab in CRC, and Table 2 outlines major pivotal clinical trials in mCRC.
3. Encorafenib
Encorafenib is an orally available, small-molecule selective inhibitor of BRAF kinase developed to target tumors harboring activating BRAF mutations, most commonly V600E. It represents a major therapeutic advance in CRC, where BRAF V600E mutations occur in approximately 8–12% of cases and are associated with aggressive clinical behavior, early metastatic spread, chemotherapy resistance, and poor prognosis [112,113,114,115]. BRAF is a serine–threonine kinase and a central component of the RAS–RAF–MEK–ERK (MAPK) pathway, which regulates proliferation, differentiation, survival, and oncogenic transformation [116,117,118].
In CRC, the BRAF V600E mutation leads to constitutive activation of MAPK signaling independent of upstream receptor tyrosine kinase stimulation, driving uncontrolled tumor growth [119,120,121]. However, the biology of BRAF-mutant CRC differs substantially from that observed in melanoma. In colorectal epithelial cells, high basal expression of EGFR enables rapid compensatory activation of EGFR signaling following BRAF inhibition. ERK suppression releases negative feedback on EGFR, resulting in MAPK pathway reactivation through upstream receptor signaling [122]. In contrast, melanoma cells exhibit relatively low EGFR expression, limiting this feedback mechanism. This biologic distinction explains the limited efficacy of BRAF inhibitor monotherapy in CRC and established the rationale for combined BRAF and EGFR blockade [122,123,124].
Encorafenib inhibits mutant BRAF by binding to the ATP-binding pocket of the kinase, suppressing ERK phosphorylation and downstream oncogenic signaling. Compared with earlier BRAF inhibitors, encorafenib demonstrates higher potency, prolonged target engagement, and a longer dissociation half-life, allowing sustained pathway inhibition with improved tolerability [125,126]. Preclinical and early clinical studies showed that combining BRAF inhibition with EGFR blockade prevents adaptive MAPK reactivation, providing the biological rationale for combination strategies in CRC [127,128,129,130]. Figure 3 illustrates its mechanism of action.
The clinical development of encorafenib therefore focused on dual BRAF–EGFR inhibition strategies. Early clinical studies confirmed that BRAF inhibitor monotherapy is ineffective in CRC due to EGFR-mediated feedback activation, establishing the need for combination therapy and, in some approaches, additional MEK inhibition [122,123,132]. This concept was validated in the phase III BEACON CRC trial, which demonstrated that encorafenib combined with cetuximab significantly improved OS, PFS, and response rates compared with irinotecan-based chemotherapy plus cetuximab in previously treated BRAF V600E–mutant mCRC [133,134,135,136].
Importantly, BEACON showed that the doublet regimen of encorafenib plus cetuximab achieved survival outcomes comparable to the triplet regimen that also included the MEK inhibitor binimetinib. Median OS reached 9.3 months with the doublet compared with 5.9 months in the chemotherapy control arm (hazard ratio 0.61), whereas the triplet regimen increased response rates but did not improve survival [133]. The absence of additional survival benefit likely reflects near-maximal MAPK pathway suppression with dual BRAF–EGFR inhibition, while increased toxicity with the triplet regimen may have limited treatment intensity. Consequently, the doublet regimen has been adopted as the preferred standard due to its balance of efficacy and tolerability [133,134,135,136].
Subgroup analyses demonstrated consistent benefit across clinically relevant populations, including patients with right-sided tumors, advanced age, and high metastatic burden—features frequently associated with BRAF-mutant CRC biology [7,135,137]. However, patients with poor performance status were underrepresented in pivotal trials, which may limit generalizability to some real-world populations.
Subsequent analyses and regulatory guidance have confirmed encorafenib plus cetuximab as the standard therapy for previously treated BRAF V600E–mutant mCRC [138]. Real-world studies and expanded-access programs have reported outcomes broadly consistent with trial results, although these observational data should be interpreted cautiously because of potential selection bias and heterogeneity in treatment sequencing [139,140,141].
Recent studies have explored the potential role of encorafenib-based combinations earlier in the treatment course. Early investigations suggested that targeted therapy combinations may be active in untreated disease [142]. More definitive evidence emerged from the phase III BREAKWATER trial, which demonstrated improved survival with the combination of FOLFOX, encorafenib, and cetuximab compared with chemotherapy alone, suggesting that earlier integration of targeted therapy may substantially alter outcomes in BRAF-mutant mCRC [134,143].
The safety profile of encorafenib reflects both its targeted mechanism and its use in combination regimens. Common adverse events include fatigue, nausea, diarrhea, abdominal pain, arthralgia, and dermatologic reactions, most of which are manageable with supportive care or dose adjustment [25,133,134]. Compared with earlier BRAF inhibitors, encorafenib appears to be associated with lower rates of paradoxical MAPK activation and secondary cutaneous malignancies [144,145]. When combined with EGFR inhibitors, overlapping toxicities such as acneiform rash and gastrointestinal symptoms require proactive management but are generally less severe than those associated with cytotoxic chemotherapy [104].
Despite its clinical impact, several limitations remain. Survival improvement in refractory disease remains modest, and optimal sequencing relative to immunotherapy in MSI-H/dMMR BRAF-mutant tumors remains uncertain. In addition, acquired resistance mechanisms—including MAPK reactivation through KRAS mutations, EGFR upregulation, or activation of parallel pathways—highlight the need for ongoing molecular monitoring and development of additional combination strategies [146].
Current international guidelines recommend routine BRAF mutation testing at the time of metastatic CRC diagnosis to identify patients eligible for encorafenib-based therapy [147,148,149]. Ongoing trials evaluating encorafenib combinations in earlier treatment lines and alongside immunotherapy may further refine its role within CRC treatment algorithms [150,151,152,153].
Beyond its clinical application, encorafenib has advanced the understanding of targeted therapy in CRC by demonstrating that effective pathway inhibition requires simultaneous blockade of adaptive resistance mechanisms rather than single-agent targeting [154,155,156]. It therefore represents a paradigm of rational combination therapy and molecular stratification in a historically high-risk CRC subtype [157,158,159].
Table 3 summarizes treatment-emergent adverse events and management strategies for encorafenib in CRC, and Table 4 outlines major pivotal clinical trials in mCRC.
4. Tucatinib
Tucatinib is an orally administered, highly selective small-molecule tyrosine kinase inhibitor (TKI) targeting the intracellular kinase domain of human epidermal growth factor receptor 2 (HER2/ERBB2) [163]. Although HER2 is a well-established oncogenic driver in breast and gastric cancers, its relevance in CRC has become increasingly apparent with advances in molecular profiling and targeted therapy development [18,164,165]. HER2-positive CRC—defined by gene amplification and/or protein overexpression—represents a distinct molecular subtype accounting for approximately 2–5% of metastatic CRC (mCRC) and is characterized by unique biological behavior, resistance patterns, and therapeutic vulnerabilities [29,166,167].
HER2 is a member of the ERBB receptor tyrosine kinase family, which includes EGFR (ERBB1), ERBB3, and ERBB4. Unlike other family members, HER2 lacks a direct ligand and functions primarily as a preferred dimerization partner, forming potent signaling complexes. Activation of HER2-containing dimers stimulates downstream oncogenic pathways, particularly the PI3K–AKT–mTOR and RAS–RAF–MEK–ERK (MAPK) cascades, promoting proliferation, survival, invasion, and metastasis of colorectal epithelial cells [168,169,170]. HER2 amplification or overexpression can also mediate resistance to anti-EGFR therapy in RAS wild-type CRC, providing the biological rationale for HER2-targeted treatment strategies [10,171,172].
Tucatinib was developed as a highly selective HER2 inhibitor designed to minimize off-target EGFR inhibition, thereby reducing dermatologic and gastrointestinal toxicities associated with less selective HER-family TKIs [173,174]. By binding the ATP-binding pocket of the HER2 kinase domain, tucatinib inhibits receptor autophosphorylation and downstream signaling while largely sparing EGFR-mediated physiological processes in normal tissues [173,174,175]. Preclinical studies demonstrated that maximal antitumor activity occurs when tucatinib is combined with trastuzumab, as dual HER2 blockade enhances pathway suppression and promotes tumor cell apoptosis [176]. Figure 4 presents the mechanism of action of tucatinib.
Clinical development in CRC has therefore focused on dual HER2 inhibition strategies. Evidence from several studies established that trastuzumab-based combinations can produce clinically meaningful responses in HER2-positive mCRC that has progressed after standard therapies [178,179]. The pivotal MOUNTAINEER trial provided key clinical validation of this approach, demonstrating durable responses and disease control with tucatinib plus trastuzumab in previously treated HER2-positive, RAS wild-type mCRC [31,33,180]. These findings confirmed HER2 amplification as a therapeutically actionable driver in a subset of CRC and led to regulatory approval of tucatinib in combination with trastuzumab for this indication [33].
Although the activity observed in MOUNTAINEER was clinically meaningful, interpretation of the results should consider several limitations, including the relatively modest sample size, partial single-arm design, and limited long-term follow-up [31]. Furthermore, mechanisms of acquired resistance—such as HER2 kinase domain mutations, activation of parallel pathways (e.g., MET amplification), or downstream MAPK reactivation—may limit response durability. The effectiveness of HER2-targeted therapy in HER2-mutant but non-amplified CRC also remains uncertain.
The clinical success of tucatinib underscores the importance of systematic HER2 testing in mCRC. Current ESMO and NCCN guidelines recommend evaluation of HER2 amplification or overexpression using immunohistochemistry, in situ hybridization, or next-generation sequencing, particularly in patients with RAS wild-type tumors refractory to anti-EGFR therapy [13,14,30,65]. Accurate molecular characterization is therefore essential for identifying patients who may benefit from HER2-directed treatment.
Ongoing research is exploring whether HER2-targeted therapy may be beneficial earlier in the treatment course. The MOUNTAINEER-03 trial is evaluating tucatinib plus trastuzumab combined with mFOLFOX6 as first-line therapy compared with standard chemotherapy regimens [181,182]. Additional data from studies such as the SGNTUC basket trial support the activity and manageable safety of dual HER2 blockade across HER2-altered solid tumors, although these results are not specific to CRC and should be interpreted cautiously [183,184].
Tucatinib-based therapy is generally well tolerated. The most common adverse events include diarrhea, fatigue, nausea, vomiting, and transient elevations of liver enzymes, which are typically low grade and manageable with supportive care or dose modification [31,33,180,185]. Due to its high HER2 selectivity, tucatinib is associated with lower rates of severe diarrhea and dermatologic toxicity compared with earlier HER2-targeted TKIs [173,185]. When combined with trastuzumab, cardiotoxicity remains uncommon but requires routine monitoring according to established HER2-targeted therapy guidelines [186].
Within treatment sequencing, tucatinib plus trastuzumab provides a clinically meaningful option for patients with HER2-positive mCRC who have progressed after standard chemotherapy [181]. Alternative HER2-targeted approaches such as trastuzumab deruxtecan have demonstrated higher response rates but carry a distinct toxicity profile, particularly the risk of interstitial lung disease (ILD) [178]. In the absence of direct comparative trials, treatment selection should consider disease tempo, prior therapy, and safety considerations.
Overall, tucatinib has established a defined role as targeted therapy for HER2-positive, RAS wild-type mCRC after progression on standard treatment. Ongoing trials evaluating tucatinib in earlier treatment lines, in combination with antibody–drug conjugates, and alongside immunotherapy may further refine its position within CRC treatment algorithms [165,187,188,189,190] and clarify optimal sequencing strategies [191,192].
Beyond its clinical application, tucatinib highlights the importance of identifying less common but biologically dominant oncogenic drivers in CRC. Its development illustrates how molecular characterization and rational drug design can translate into effective targeted therapies for selected patient populations, advancing the paradigm of precision oncology in CRC [158,193,194,195,196,197].
Table 5 summarizes treatment-emergent adverse events and their management in CRC, and Table 6 outlines major pivotal clinical trials of tucatinib in mCRC.
5. Future Directions and Contemporary Management of Metastatic Colorectal Cancer
CRC remains a major global health burden despite advances in prevention, diagnosis, and treatment. Management has evolved from stage-based algorithms to complex, multimodal, and molecularly informed strategies [202,203]. Although survival—particularly in metastatic disease—has improved, important limitations persist, including biological resistance, cumulative toxicity, inequitable access, rising costs, and uncertainty regarding optimal sequencing. Further progress will require not only therapeutic innovation but also structural and clinical refinement of current approaches [204,205].
In localized disease, surgical resection remains the cornerstone of curative therapy, often combined with perioperative or adjuvant chemotherapy based on pathologic risk. These strategies reduce recurrence and improve survival but are associated with morbidity, including bowel dysfunction, neuropathy, and long-term quality-of-life impairment [206,207]. In rectal cancer, multimodal treatment incorporating chemoradiotherapy and total mesorectal excision achieves excellent local control; however, radiation-related toxicity and functional sequelae continue to drive interest in de-escalation and non-operative strategies for selected patients [208,209]. These issues underscore the balance between oncologic efficacy and treatment-related harm in early-stage disease.
In mCRC, systemic therapy has expanded substantially. Cytotoxic chemotherapy remains foundational, but integration with targeted agents—including anti-EGFR antibodies, anti-VEGF therapies, BRAF inhibitors, HER2-directed treatments, and immune checkpoint inhibitors—has reshaped outcomes in biologically defined subgroups [65,134,181]. Molecular stratification based on RAS, BRAF, HER2, and mismatch repair status has established mCRC as a model of precision oncology. However, increasing therapeutic complexity has not been matched by clear guidance on optimal sequencing, duration, and combinations in routine practice [210,211,212].
A central limitation in advanced CRC is the near-universal development of therapeutic resistance, driven by secondary genomic alterations, pathway redundancy, tumor heterogeneity, and adaptive signaling feedback [213]. Cross-resistance among biologic agents within shared pathways further complicates sequencing. Primary and acquired resistance to anti-EGFR monoclonal antibodies represents a major challenge in extended RAS wild-type mCRC. The clinical benefit of cetuximab and panitumumab was established in trials such as PRIME and PEAK [19,78], but many patients exhibit intrinsic resistance, and nearly all responders ultimately progress. Resistance commonly arises through reactivation of MAPK signaling, including emergent KRAS, NRAS, and BRAF mutations at progression [41,214,215]. EGFR extracellular domain mutations impair antibody binding [85], while HER2 amplification, MET amplification, PIK3CA mutations, and PTEN loss restore downstream ERK signaling independently of EGFR blockade [10,39,122].
BRAF V600E–mutant mCRC is characterized by aggressive biology and intrinsic resistance to single-agent BRAF inhibition due to rapid EGFR-mediated MAPK reactivation [216]. The BEACON CRC study established encorafenib plus cetuximab as standard therapy after prior treatment [133]. The triplet regimen adding binimetinib did not significantly improve OS compared with the doublet and increased toxicity, suggesting diminishing returns from intensified vertical MAPK inhibition [133]. Future strategies include ERK inhibition, PI3K pathway combinations, and integration with immune checkpoint blockade [25,217]. In MSI-H/BRAF V600E tumors, immune checkpoint inhibitors remain standard, with combination approaches under investigation [218].
HER2 amplification defines a distinct subset of RAS wild-type mCRC. Early dual HER2 blockade demonstrated activity in refractory disease [29,30]. Antibody–drug conjugates have advanced this field; trastuzumab deruxtecan showed clinically meaningful responses in heavily pretreated HER2-positive mCRC in DESTINY-CRC01 [178]. Its mechanism couples HER2 targeting with intracellular delivery of a topoisomerase I inhibitor payload, potentially overcoming prior resistance, though interstitial lung disease (ILD) requires careful monitoring [178]. The MOUNTAINEER trial established tucatinib plus trastuzumab as an effective regimen with favorable tolerability [31]. Direct comparative data between tucatinib-based therapy and trastuzumab deruxtecan are lacking; sequencing decisions should consider disease kinetics, prior HER2 exposure, comorbidities, and pulmonary risk tolerance.
Emerging molecular monitoring strategies may help address therapeutic resistance. CtDNA and other liquid biopsy platforms offer promise for real-time resistance monitoring, yet their integration into routine decision-making remains inconsistent [219]. Longitudinal ctDNA studies demonstrate that resistant RAS-mutant clones decay after anti-EGFR withdrawal, providing a biologic rationale for rechallenge strategies [220,221]. Prospective trials such as CRICKET and CHRONOS showed that ctDNA-confirmed RAS/BRAF wild-type status at progression can identify patients who may benefit from rechallenge, reflecting a dynamic model of clonal evolution rather than static baseline genotyping [220,221]. Response rates of approximately 20–30% have been reported in selected patients, and ongoing randomized trials are refining timing and integration into later-line strategies [220,221].
Immunotherapy is standard first-line treatment for MSI-H/dMMR mCRC [39]. In MSI-H/BRAF V600E tumors, optimal sequencing between checkpoint inhibition and BRAF-targeted therapy remains uncertain. In microsatellite-stable (MSS) mCRC, immunotherapy has shown limited efficacy [218], though oncogene-targeted therapies may modulate the tumor microenvironment and enhance immune responsiveness. Trials are evaluating combinations of BRAF/EGFR inhibition with checkpoint inhibitors in MSI-H disease [25], and EGFR inhibition may enhance antigen presentation and antibody-dependent cellular cytotoxicity (ADCC), supporting combinatorial approaches in MSS tumors [222].
Optimal sequencing of anti-EGFR, anti-VEGF, BRAF-targeted, and HER2-directed therapies remains unresolved. The choice between anti-EGFR and anti-VEGF therapy in first-line extended RAS wild-type left-sided tumors is informed by FIRE-3, CALGB/SWOG 80405, and PEAK [70,71,77], though cross-trial comparisons are limited by heterogeneity. The timing of BRAF-targeted therapy relative to chemotherapy in BRAF V600E tumors remains under study [133,223,224], and prospective comparative data guiding HER2-targeted sequencing are lacking [31,178].
Several unresolved questions limit optimal care. Ideal sequencing and combinations across treatment lines remain undefined, and trial populations often fail to reflect real-world heterogeneity [210,225]. The role of treatment de-escalation after deep response, optimal therapy duration in metastatic and adjuvant settings, and validation of emerging biomarkers—including tumor microenvironment features, epigenetic changes, and microbiome composition—require further investigation [226,227].
Future progress will depend on expanded molecular profiling, including longitudinal liquid biopsy to guide adaptive strategies and detect resistance early [214,228,229]. Rational sequencing and biologically informed combinations should replace empirical escalation to maximize efficacy and limit overlapping toxicity [229,230]. Addressing tumor heterogeneity, lineage plasticity, and stromal–immune interactions is critical to overcoming treatment failure in advanced disease [213,231].
Treatment-related toxicity remains a major challenge. Chemotherapy is associated with cumulative neuropathy, myelosuppression, and gastrointestinal toxicity. Targeted therapies introduce distinct adverse-event profiles—including dermatologic reactions, hypertension, thromboembolic events, and metabolic disturbances—that may impair adherence and quality of life [232,233]. Immunotherapy, while transformative in mismatch repair-deficient CRC, carries risks of severe or irreversible immune-related adverse events [234,235,236]. As survival improves, cumulative treatment burden increasingly shapes patient experience, emphasizing toxicity mitigation and survivorship-focused care.
Cost-effectiveness, biomarker accessibility, and real-world feasibility must accompany efficacy considerations [223,224]. Access disparities persist, particularly regarding comprehensive next-generation sequencing (NGS) required to identify RAS, BRAF, HER2, and other actionable alterations [237,238]. Limited availability and delayed turnaround times may impede optimal selection, especially in community and low-resource settings [237]. Financial toxicity from combination targeted regimens and prolonged therapy further affects adherence, outcomes, and equity [239,240].
Clinical trial populations often underrepresent older adults, patients with poor performance status, and those with significant comorbidities [241,242]. Consequently, real-world toxicity and effectiveness may differ from registration studies. Contemporary mCRC management must therefore integrate molecular efficacy with feasibility, sustainability, and health-system constraints [241,242].
Accessibility and cost are central determinants of outcome. Novel agents and molecular diagnostics impose substantial financial burden, raising concerns about affordability and sustainability [243,244,245,246,247,248]. Disparities in genomic testing, multidisciplinary care, and advanced therapeutics contribute to unequal outcomes globally, particularly in low- and middle-income regions where CRC mortality is highest [249,250,251]. In resource-constrained settings, stepwise molecular testing prioritizing RAS and BRAF analysis before broader NGS panels may improve cost-effectiveness [69]. In elderly or frail patients, treatment decisions must balance toxicity risk, comorbidity burden, and expected survival benefit [13]. For BRAF V600E–mutated mCRC, encorafenib-based therapy provides a chemotherapy-sparing option with demonstrated survival benefit, though supportive and geriatric assessment remain essential [133].
Equally important are strategies to reduce treatment burden and improve quality of life through optimized dosing, integrated supportive care, and survivorship planning. Health-system research must incorporate value-based and cost-effectiveness frameworks to ensure sustainable innovation [243,250]. Moving effective targeted and immunotherapeutic approaches into earlier-stage and oligometastatic disease is under active investigation, aiming to improve cure rates rather than merely prolong survival [39,252,253,254].
Contemporary CRC management reflects substantial scientific progress but remains constrained by resistance, toxicity, access disparities, and unresolved clinical questions. Future advances must emphasize precision medicine, rational therapy integration, toxicity mitigation, and equitable access to translate innovation into durable survival and meaningful quality-of-life gains.
Table 7 summarizes current management approaches cross different stages in CRC, and Figure 5 outlines a biomarker-driven treatment algorithm integrating molecular profiling with clinical staging to guide therapeutic sequencing in mCRC.
6. Conclusions
The clinical integration of molecularly targeted therapies against EGFR, BRAF, and HER2 has firmly established precision oncology as a central pillar of CRC management. Beyond the individual successes of agents such as panitumumab, encorafenib, and tucatinib, their collective development has reshaped how CRC is conceptualized, studied, and treated—shifting therapeutic decision-making toward molecular dependencies rather than anatomical origin alone.
These advances also make clear that effective targeting in CRC requires more than identifying a single oncogenic alteration. The colorectal tumor context—marked by pathway redundancy, adaptive feedback signaling, and pronounced intratumoral heterogeneity—often necessitates rational combinations, thoughtful sequencing, and ongoing molecular reassessment. Experience across EGFR-, BRAF-, and HER2-driven disease highlights the need to move beyond static, one-time biomarker testing toward dynamic care models that account for clonal evolution and treatment-induced resistance.
At the same time, therapeutic progress is inseparable from broader clinical and system-level considerations. Cumulative toxicity, quality-of-life trade-offs, limited durability of benefit, and substantial financial burden increasingly shape real-world outcomes and can constrain the use of otherwise effective therapies. These challenges underscore that innovation must be paired with rigorous evaluation of value, accessibility, and patient-centered outcomes, particularly as treatment options expand and survival improves.
Looking ahead, progress in molecular targeting will depend on deeper biological integration rather than incremental drug addition. Key priorities include refining predictive biomarkers beyond single-gene alterations, embedding longitudinal molecular monitoring into routine practice, and designing trials that directly address sequencing, de-escalation, and earlier intervention. Equally important is improving global equity in access to molecular diagnostics and targeted therapies so that precision oncology is not limited to select populations or healthcare systems.
Targeting EGFR, BRAF, and HER2 provides clear proof that biologically driven therapy can improve outcomes in defined subsets of CRC. The next challenge is not validating these targets, but optimizing their use—biologically, clinically, and systemically—to extend durability, minimize harm, and ensure that progress is sustainable and broadly accessible.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bray F. Laversanne M. Sung H. Ferlay J. Siegel R.L. Soerjomataram I. Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20247422926310.3322/caac.2183438572751 · doi ↗ · pubmed ↗
- 2Sung H. Ferlay J. Siegel R.L. Laversanne M. Soerjomataram I. Jemal A. Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20217120924910.3322/caac.2166033538338 · doi ↗ · pubmed ↗
- 3Abedizadeh R. Majidi F. Khorasani H.R. Abedi H. Sabour D. Colorectal cancer: A comprehensive review of carcinogenesis, diagnosis, and novel strategies for classified treatments Cancer Metastasis Rev.20244372975310.1007/s 10555-023-10158-338112903 · doi ↗ · pubmed ↗
- 4Tang Y.L. Li D.D. Duan J.Y. Sheng L.M. Wang X. Resistance to targeted therapy in metastatic colorectal cancer: Current status and new developments World J. Gastroenterol.20232992694810.3748/wjg.v 29.i 6.92636844139 PMC 9950860 · doi ↗ · pubmed ↗
- 5Siegel R.L. Giaquinto A.N. Jemal A. Cancer statistics, 2024 CA Cancer J. Clin.202474124910.3322/caac.2182038230766 · doi ↗ · pubmed ↗
- 6Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer Nature 201248733033710.1038/nature 1125222810696 PMC 3401966 · doi ↗ · pubmed ↗
- 7Guinney J. Dienstmann R. Wang X. de Reyniès A. Schlicker A. Soneson C. Marisa L. Roepman P. Nyamundanda G. Angelino P. The consensus molecular subtypes of colorectal cancer Nat. Med.2015211350135610.1038/nm.396726457759 PMC 4636487 · doi ↗ · pubmed ↗
- 8Zhai Z. Yu X. Yang B. Zhang Y. Zhang L. Li X. Sun H. Colorectal cancer heterogeneity and targeted therapy: Clinical implications, challenges and solutions for treatment resistance Semin. Cell Dev. Biol.20176410711510.1016/j.semcdb.2016.08.03327578007 · doi ↗ · pubmed ↗