Novel Molecular Markers and Immune-Related Candidate Genes for Blackleg Resistance in Rapeseed: A Genome-Wide Analysis
Ewa Starosta, Tomasz Jamruszka, Justyna Szwarc, Jan Bocianowski, Magdalena Grynia, Janetta Niemann

TL;DR
This study identifies new genetic markers and immune-related genes in rapeseed that help resist blackleg disease, offering tools for breeding more resilient crops.
Contribution
The study introduces novel molecular markers and candidate genes for blackleg resistance in rapeseed using GWAS and transcriptomic analysis.
Findings
Over 104,000 markers were identified, with 61% being SilicoDArTs and 39% SNPs.
33 significant markers (p < 0.01) were associated with blackleg resistance, 76% of which were SilicoDArTs.
Thirteen marker-linked genes were expressed during infection, with seven showing significant differential expression.
Abstract
Rapeseed (Brassica napus L.) faces escalating threats from abiotic and biotic stresses, notably blackleg caused by Leptosphaeria maculans. Due to limited chemical control efficacy and stringent GMO regulations, marker-assisted selection (MAS) leveraging natural genetic variation has become an indispensable strategy for crop improvement. This study identified novel molecular markers for blackleg resistance by integrating genome-wide association study (GWAS) results with high-throughput genotyping by Diversity Arrays Technology sequencing. Phenotypic screening across the population demonstrated a wide spectrum of disease severity (scores 0–6), confirming the segregation of key resistance genes. The DArTseq platform identified nearly 104,000 markers, comprising 61% SilicoDArTs and 39% SNPs. Among the 33 most significant markers associated with resistance (p < 0.01), 76% were SilicoDArTs.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
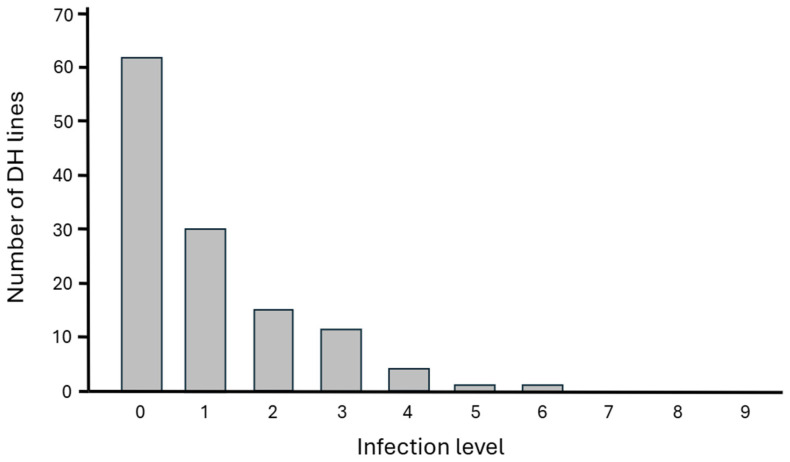 Figure 1
Figure 1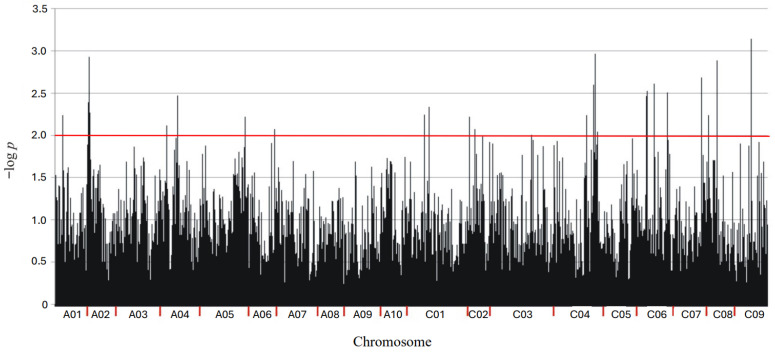 Figure 2
Figure 2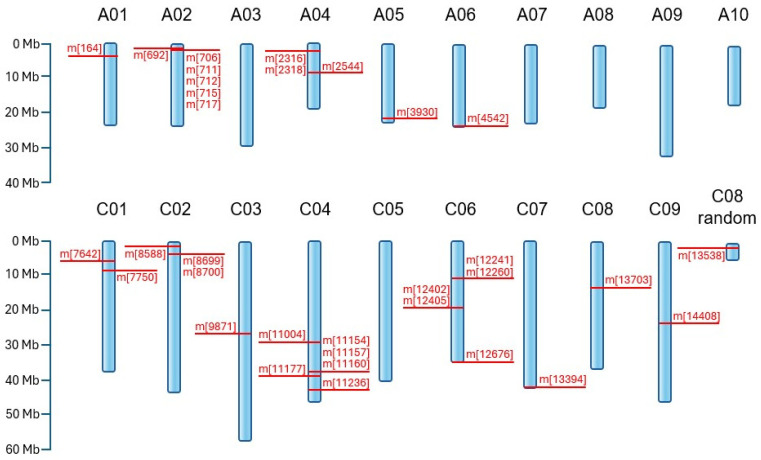 Figure 3
Figure 3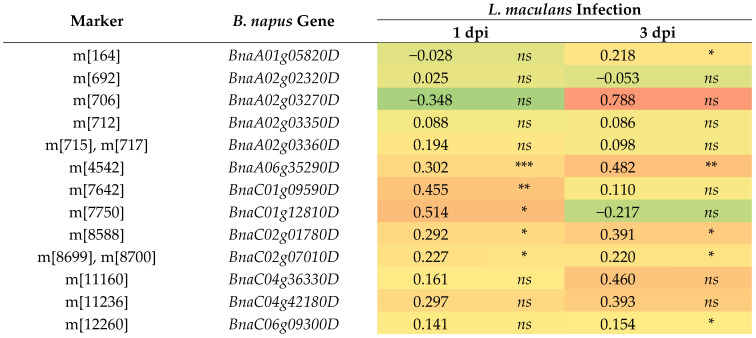 Figure 4
Figure 4- —Ministry of Agriculture and Rural Development, Poland
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant-Microbe Interactions and Immunity · Nitrogen and Sulfur Effects on Brassica · Genomics, phytochemicals, and oxidative stress
1. Introduction
Rapeseed (Brassica napus L.) is considered one of the most significant cultivated plants within the Brassica genus [1]. It belongs to the group of three allotetraploid species formed through hybridization among the core “U’s triangle” species: B. nigra (2n = 2x = 16 = BB), B. rapa (2n = 2x = 20 = AA), and B. oleracea (2n = 2x = 18 = CC) [2]. Moreover, rapeseed is regarded as one of the most important oilseed crops. Contemporary cultivars yield substantial amounts of oil characterized by high nutritional quality. In addition to its use for oil production, rapeseed is also valued as a source of animal feed and as a raw material for biofuel generation [3,4,5]. Initially, rapeseed oil was unsuitable for human consumption because of its high levels of erucic acid and glucosinolates. However, breeding efforts resulted in the development of “double zero” (“00”) cultivars, characterized by very low concentrations of these compounds [6].
Intensive inbreeding in B. napus aimed at improving oil content and quality has significantly reduced its genetic diversity. Consequently, the species has become increasingly susceptible to a wide range of biotic stress factors, including fungi, viruses, and insect pests [7]. Several diseases have become major constraints in rapeseed production. Among them, blackleg is considered one of the most economically important. The disease is caused by a fungal complex of Leptosphaeria maculans and Leptosphaeria biglobosa. The first symptoms of infection appear as oval, gray lesions with black pycnidia spots on cotyledons and leaves. As the disease progresses, similar lesions develop on stems and siliques. In the most advanced stages of infection, a characteristic blackening of the basal stem is observed, which is associated with significant yield losses [8].
Global yield losses in rapeseed caused by this disease are estimated at 10–15%, though in extreme cases, they may reach up to 90%. Various control strategies have been developed and applied in rapeseed cultivation, including agricultural practices such as tillage and crop rotation, as well as biological and chemical protection methods, involving antagonistic microorganisms and fungicide applications [9].
The use of resistant rapeseed varieties remains the most cost-effective and environmentally sustainable strategy for managing blackleg disease. Although numerous studies have investigated the genetic basis of resistance in rapeseed, current knowledge only scratches the surface. The molecular mechanisms underlying plant immunity are highly complex and involve a wide array of genes, ranging from those with minor effects to those with major effects [3,7]. In agriculture, rapeseed varieties carrying well-characterized resistance genes are widely cultivated as a means of protecting crops against L. maculans, the causal agent of blackleg disease. To date, researchers have identified 22 major resistance (R) genes in B. napus and related species [10]. These genes interact in a classic gene-for-gene manner with the corresponding avirulence (Avr) genes of the pathogen, triggering defense responses in the host [11]. However, the intensive use of single resistant cultivars in monoculture, combined with limited rotation of different R genes, has created strong selective pressure on pathogen populations. As a result, new virulent isolates capable of overcoming specific resistance genes have rapidly emerged, leading to a breakdown of R-gene-mediated resistance and a reduction in the long-term effectiveness of genetic resistance in rapeseed breeding programs [10,12].
Therefore, it is crucial to intensify ongoing research aimed at identifying new major resistance (R) genes as well as quantitative trait loci (QTLs) associated with partial resistance to L. maculans [13]. Unlike single R genes, which often lose their effectiveness due to pathogen adaptation, QTLs generally confer a broader and more durable form of resistance by reducing the overall disease severity rather than providing complete immunity. When both types of resistance are strategically combined—major R genes ensuring strong initial protection and QTLs contributing to long-term stability—the resulting cultivars display the highest and most sustainable level of defense against blackleg disease [14]. Such an integrated approach represents a key strategy for maintaining effective crop protection and ensuring stable yields in rapeseed production.
Many breeding programs and stations emphasize the urgent need for reliable and efficient molecular markers to enable rapid and straightforward screening of B. napus germplasm for blackleg resistance. Such tools would greatly facilitate the development of elite cultivars that combine strong disease resistance (conferring both R-genes and QTLs) with high economic and agronomic value.
Marker-assisted selection (MAS) has become one of the main methods of elite germplasm selection. Nevertheless, the highly complex nature of the B. napus genome poses a significant challenge for blackleg resistance research. As an amphidiploid species, B. napus combines two parental genomes (B. rapa and B. oleracea), resulting in extensive genome size, polyploidy, and a high degree of gene redundancy. This complexity complicates the precise identification and mapping of resistance loci, as well as the development of reliable molecular markers tightly linked to blackleg resistance. Consequently, distinguishing functional resistance genes from closely related paralogs and pinpointing their exact chromosomal positions requires advanced genomic tools, large-scale mapping populations, and integrative approaches combining genomics, transcriptomics, and bioinformatics.
Diversity Arrays Technology sequencing (DArTseq) is a cost-effective genotyping-by-sequencing platform that reduces genome complexity using restriction enzymes, followed by adapter ligation, amplification, and high-throughput sequencing. This approach enables the detection of thousands of SNPs and presence–absence variants without requiring a reference genome. Consequently, DArTseq is particularly well suited for genetic diversity analysis, linkage mapping and marker-assisted breeding in crops with complex genomes.
Doubled haploid (DH) rapeseed lines provide an optimal population structure for such analyses due to their complete homozygosity and genetic stability. Their fixed genotypes allow unambiguous evaluation of associations between molecular markers and blackleg resistance. Moreover, the use of DH lines reduces the required population size, which is necessary for reliable detection of qualitative and quantitative resistance loci while maintaining high power for detecting both qualitative and quantitative resistance loci. The integration of DH populations with DArTseq facilitates precise marker–trait association analysis. The genetic uniformity of DH lines ensures clear and reproducible marker signals, thus increasing the reliability of identified markers and supporting their effective application in MAS and resistance gene pyramiding.
In recent decades, numerous molecular markers linked to blackleg resistance have been reported using a wide range of approaches, including biparental linkage mapping, candidate gene analysis, association mapping, and high-throughput genotyping platforms. These studies have led to identification of markers associated with both major resistance (R) genes and quantitative trait loci (QTLs), many of which are located within or near genes involved in plant defense pathways [14,15]. While these findings have substantially advanced the understanding of the genetic basis of resistance, their practical implementation in breeding programs has been comparatively limited. Many reported markers exhibit population specificity, reduced effect sizes in diverse genetic backgrounds, or insufficient validation across environments and germplasm panels [16]. Therefore, only a relatively small proportion of identified markers have proven robust and transferable enough for routine application in MAS.
This study aims to (i) identify novel molecular markers associated with blackleg resistance in rapeseed DH lines by employing next-generation sequencing in combination with physical mapping and (ii) conduct a preliminary validation of their association with disease resistance through in silico functional annotation of candidate genomic regions. The primary objective is to identify SNP and SilicoDArT markers linked to blackleg resistance. By integrating DArTseq genotyping with precise phenotyping of DH lines, we aim to map new resistance loci within the B. napus genome. Furthermore, with functional inference, we aim to provide biologically informed interpretation of marker–trait associations and support their prospective application in resistance breeding programs. These findings will provide robust tools for MAS, enabling the efficient development of rapeseed cultivars with enhanced and durable resistance to L. maculans.
2. Results
2.1. Phenotyping
The level of resistance of the 125 analyzed DH lines was assessed using a 10-point severity scale, where extreme values of 0 and 9 indicate no visible disease damage and a completely damaged plant, respectively. Varying infection levels, ranging between 0 and 6, were observed with an average of 0.96 ± 1.27. Most plants exhibited very low or no infection. None of them were severely affected, i.e., at a level of 7–9 (Figure 1).
2.2. Genotyping
Through DArTseq, a total of 103,898 molecular markers (60,067 SilicoDArT and 43,831 SNP) were obtained. Based on a significance threshold of MAF > 0.25 and less than 10% missing observations as well as chromosomal location, 14,759 markers (10,314 SilicoDArTs and 4445 SNP) were selected and used for association mapping (Figure 2).
A highly significant association (p < 0.01) between 33 markers (25 SilicoDArT and 8 SNP) from this group and plant resistance to Leptosphaeria spp. was observed (Table 1). Chromosomes A02 and C04 contained the highest number of identified markers (6 each), while no marker was found on chromosomes A07 to A10 and C05 (Table 1 and Figure 3).
Out of all the markers identified, 21 markers (16 SilicoDArT and 5 SNP) were intragenic. Nearly all of these markers corresponded to only one gene. The exceptions were the SilicoDArT marker m[715] and the SNP marker m[717], which were found within a subsequent intron and exon of BnaA02g03360D, respectively. Additionally, two SNP markers, m[8699] and m[8700], were localized within an adjacent exon and intron of another gene, BnaC02g07010D (Table 2). Among the 19 genes harboring markers, 15 were identified in the transcriptomic data from Becker et al. (2019) [17], which assessed gene expression changes in response to L. maculans infection. Notably, eight of these genes showed a statistically significant reaction to the pathogen. Additionally, a few others (BnaA02g03270D, BnaC04g36330D, and BnaC04g42180D) exhibited a noticeable though not statistically significant reaction (Figure 4).
3. Discussion
Rapeseed (B. napus L.) is a globally significant crop due to its high-quality oil and protein content [3]. Unfortunately, the expansion of the rapeseed cultivation area leads to an increased risk of production disturbances caused by abiotic and biotic stresses, including pathogenic fungi [7,18]. Given the often-limited scope and low efficacy of agronomical interventions, as well as environmental concerns associated with the utilization of chemical plant protection products, solutions based on genetically mediated plant resistance are being increasingly explored [19,20]. One of the methods for the genetic improvement of economically important plant varieties, including rapeseed, is MAS. It remains a key strategy for B. napus improvement, leveraging natural genetic variation to bypass the regulatory complexities of genome editing while maintaining high breeding efficiency [21]. The efficacy of MAS increases with the advent of newer high-throughput marker detection technologies. Thus, this study describes the procedure and results of the identification of novel molecular markers for blackleg resistance in rapeseed by integrating Genome-Wide Association Study (GWAS) results from phenotypic screening and genotyping via DArTseq.
The use of DH lines in this study eliminated the need to distinguish signals derived from two different alleles (heterozygosity), which can be problematic in NGS analysis. This increases the reliability of read mapping and genotyping, because the fully homozygous nature of DH lines enhances the precision of variant calling, facilitating the subsequent and accurate identification of linked genes and QTLs [22].
The low mean disease severity observed in the analyzed plants could be attributed to a few factors: either the population possesses high innate resistance, or there was potentially low inoculum pressure of the pathogen in the field. Fortunately, the wide range of symptoms that were also observed is crucial for genetic research. The disease severity across the analyzed plant population exhibited high variability, ranging from a score of 0 to 6. This spectrum covers everything from completely healthy plants with no visible disease symptoms (score 0) up to those displaying severe symptoms—specifically, discoloration covers 51% to 75% of the main stem and is accompanied by numerous pycnidia (score 6). The high variability observed confirms the segregation of genes responsible for resistance, which makes the population suitable for genetic mapping, e.g., QTL analysis [23] (Figure 1).
The effectiveness of the DArTseq platform in dissecting complex traits is well-documented across various B. napus studies [24,25,26]. Increasingly, this technique is being utilized to study this species in economically relevant contexts. Examples of such work include studies on pod shatter resistance [27,28], aluminum stress tolerance [29], Mn^2+^ tolerance [30], and blackleg resistance. The latter has been a particularly active area of research, with studies using DArTseq markers to identify and map R genes [31] and GWAS [32].
By employing DArTseq, a level of genomic resolution was achieved that far exceeds the capabilities of traditional marker systems like RFLP or RAPD. This high-throughput platform enabled the simultaneous interrogation of tens of thousands of loci, providing the dense marker coverage necessary to dissect the complex, polygenic nature of blackleg resistance in B. napus. In addition to high efficiency, the integration of sequence-specific data allowed for the direct anchoring of the findings to the reference genome—a critical step for precise marker localization that could be largely infeasible with earlier molecular tools [33].
The DArTseq technology generates molecular markers, which are primarily classified into two types: SilicoDArTs, based on presence or absence in the pool of analyzed genotypes, and SNPs present in the representation. Furthermore, it is also possible to extract Copy Number Variation (CNV) polymorphism information (https://www.diversityarrays.com/) [34]. In this study, over 100 thousand molecular markers were identified, of which 58% were SilicoDArT-type and 42% were SNP-type. The ratios among the 33 most significant markers associated with blackleg resistance with the highest significance (p < 0.01) were 76% and 24%, respectively (Table 1). However, the actual content of markers containing single or multiple SNPs in their structure might be greater. This is due to the lack of precise specification regarding exactly what polymorphism contributes to SilicoDArT, as it can contain SNP or an indel rather than exclusively a fragment presence/absence not linked to a point mutation. A case supporting this possibility, which warrants further investigation, is the pair of identified markers m[715] and m[717]. The SNP for the first marker, m[715], is located in the 9th intron of the gene BnaA02g03360D, while the second marker, m[717], is a SilicoDArT marker whose sequence covers a fragment of the same gene spanning from the 9th exon to the 9th intron. This positional overlap suggests that the two different marker types may linked to the same polymorphism [34] (Table 2).
In this study, the higher frequency of SilicoDArT markers compared to SNPs stems from both the DArTseq methodology and the inherent nature of the DH lines. Because DH lines are fully homozygous, they lack heterozygous loci; consequently, genetic variation is expressed exclusively through fixed allelic differences.
SilicoDArT markers, which operate on a presence/absence basis, are particularly effective here as they detect not only point mutations at restriction sites but also broader structural changes like insertions and deletions. In contrast, while SNP calling in these lines is precise, it is technically more demanding, requiring high read depth to ensure reliability. Therefore, the binary nature of SilicoDArT markers provides a more comprehensive and robust dataset for high-density genetic mapping in homozygous populations.
The molecular markers identified in this study were distributed across multiple chromosomes of both the A and C subgenomes (A01, A02, A04, A05, A06, C01, C02, C03, C04, C06, C07, and C09), highlighting the polygenic and genome-wide nature of blackleg resistance in B. napus. Notably, none of the detected markers were located within the well-characterized A07 resistance cluster, which harbors several major R-genes (Rlm1, Rlm3, Rlm4, Rlm7, and Rlm9), nor within the A10 region containing Rlm2 and LepR-associated loci [35,36,37]. Our results revealed resistance-associated loci distributed across both subgenomes, including multiple C-genome chromosomes (C01, C02, C03, C04, C06, C07, and C09). Previous reports indicate that C-genome loci are frequently associated with moderate, additive effects contributing to partial but potentially more durable resistance [38]. The broad chromosomal distribution observed here suggests a complex genetic architecture involving multiple loci with cumulative effects rather than reliance on single major R-genes.
Among the 19 characterized genes harboring markers, 13 were found in the transcriptomic data from Becker et al. (2019) [17]. Seven of these exhibited a statistically significant change in expression 1 and/or 3 days following infection with L. maculans. Additionally, some genes displayed prominent, though not statistically significant, changes in expression. The reason for the lack of statistical significance, or indeed, the complete absence of a reaction to the tested pathogen, might be that the gene’s activity is primarily restricted to tissue other than the one analyzed (i.e., not the cotyledons), or that the timing of the reaction to the tested stimulus might be different (i.e., earlier or later than the 1- and 3-day time points) [39]. Furthermore, the presence of markers in intergenic regions may indicate that markers are located within regulatory elements (like enhancers or promoters) that control the expression of adjacent or distant genes, thus influencing the plant’s reaction to the pathogen without being a part of the coding sequence itself [40] (Figure 4).
Functional annotation of identified loci suggests that blackleg resistance in B. napus is a multifaceted process involving several key pathways. Stress signaling and transcriptional regulation are pivotal, evidenced by the presence of OBF5 (systemic acquired resistance activation) [41], HSF3 (stress signal transduction) [42], XPO1A (antioxidant defense) [43], and FBS2 (protein degradation) [44,45]. These findings point toward a comprehensive and coordinated defense response (Figure 4 and Table 3).
Additionally, structural and metabolic adaptations appear crucial, with genes linked to cell wall integrity (SFH19) [46], cytoskeleton organization (At5g14790), and sugar transport (At5g17010, VGT2) [47,48] supporting cellular barriers. Finally, the involvement of fundamental maintenance genes, including REIL1 (ribosome remodeling) [49,50], INH3 (dephosphorylation) [51], and At1g51980 (electron transport) [52], highlights the high metabolic cost of this defense. Together, these diverse pathways—from DNA damage response (PP4R2L) [53] to salt-stress factors (GT-4) [54]—indicate a broad-spectrum physiological adaptation that enhances resistance to L. maculans (Figure 4 and Table 3).
The clustering of markers around candidate genes suggests that structural changes within these markers may drive the differences in resistance between the DH lines. These modifications likely alter gene expression or lead to the gain or loss of specific defense receptors. Within a theoretical defense network, these genes may act as key nodes, where structural variations determine how quickly and strongly the plant triggers a resistance response.
While the biological roles of these candidate genes are well-documented in A. thaliana, their specific functions in B. napus during L. maculans infection remain to be experimentally confirmed. These inferred roles provide a strong basis for future functional validation studies, such as virus-induced gene silencing (VIGS) or CRISPR/Cas9-mediated mutagenesis, to definitively link these loci with blackleg resistance.
The ongoing research into blackleg resistance in B. napus exemplifies the crucial intersection of advanced genotyping technologies and effective breeding strategies. By employing innovative methods such as DArTseq and GWAS, the resilience of Brassica crops against key diseases is progressively enhanced, thereby safeguarding yield and contributing to food security. This study successfully identified a substantial repertoire of SilicoDArT and SNP markers, many of which are situated within or adjacent to genes associated with critical stress response pathways. The functional diversity of these genes and their respective proteins—ranging from transcriptional regulation (HSF3 and OBF5) to structural integrity (XPO1A and SFH19)—underscores the multifaceted nature of the plant’s defense mechanisms. These findings not only expand our understanding of the molecular basis of L. maculans resistance but also provide a suite of validated molecular markers for MAS. Consequently, these results offer a practical pathway for breeding resilient rapeseed varieties, ensuring stable yields in the face of escalating biotic pressures.
4. Materials and Methods
4.1. Plant Material
The plant material consisted of 125 DH rapeseed lines (B. napus L.) from Strzelce Plant Breeding Ltd., Borowo Department (Borowo, Poland), IHAR Group, developed via intraspecific crosses of registered varieties with known blackleg resistance to ensure a diverse disease response.
4.2. Field Assessment
Field evaluations were carried out to assess disease severity and determine the blackleg resistance of all analyzed DH lines. The experiments took place at the test fields located in Borowo, Wielkopolska voivodeship, Poland (Strzelce Plant Breeding Ltd., IHAR Group). The study was carried out using a randomized block design and included three replications. The size of a single testing plot was 10 m^2^ with a sowing density of 60 seeds/m^2^. The agricultural practices were optimal for local conditions. Resistance observations were performed on mature plants during the BBCH 70–89 phase. The degree of resistance to blackleg was assessed by rating disease symptoms based on the 0–9 scale developed by Jędryczka (2006) [55]. On this scale, a score of 0 signifies the absence of visible disease symptoms, while a score of 9 indicates a completely damaged plant with numerous pycnidia present on the stems and leaves.
4.3. DNA Extraction
Genomic DNA was isolated from 7-day seedlings using the Genomic Mini AX Plant kit (A&A Biotechnology, Gdańsk, Poland), according to the supplied protocol. Samples exhibiting 260/280 and 260/230 ratios around 1.8 and 2.0, respectively, and concentrations above 100 ng/uL were diluted to a final concentration of 100 ng/uL.
4.4. Genotyping
The genomic DNA isolated from the analyzed DH lines was subjected to DArTseq analysis by Diversity Arrays Technology (University of Canberra, Canberra, Australia). The procedure includes (i) reduction in genome complexity through digestion with appropriate species-specific restriction enzymes, (ii) ligation of resulting fragments with adaptors, (iii) amplification using polymerase chain reaction (PCR), (iv) preparation of gene pool representation by combining equimolar quantities of amplification products from each genotype, and (v) sequencing with Illumina platform (Illumina, San Diego, CA, USA).
Following initial quality assessment via the DArT analytical pipelines (Diversity Arrays Technology, Canberra, Australia), the markers were subjected to stringent selection criteria for the association study. Only those meeting the following requirements were retained: one marker per 69 nt sequence, a minor allele frequency (MAF) > 0.25, and a missing observations fraction < 10%.
4.5. Physical Mapping and Gene Annotation
Marker positions were physically mapped against the Darmor-bzh v4.1 reference genome. Subsequently, the resulting metadata, specifically, the presence/absence and chromosomal localization of the identified SilicoDArTs and SNP markers, were subjected to statistical analysis. Moreover, the distances between the markers and the nearest genes were measured using the JBrowse tool available on the B. napus multi-omics information resource (BnIR, https://yanglab.hzau.edu.cn/BnIR, accessed on 15 January 2026). The functional role of the selected genes regarding their potential contribution to B. napus defense mechanisms against L. maculans was assessed through re-analysis of pre-existing transcriptomic data from Becker et al. (2019) [17].
4.6. Statistical Analysis
Data were further subjected to analysis of variance in a model with random effects of DH lines. Using the method based on the mixed linear model with a population structure calculated by eigenanalysis and modeled by random effects, association mapping was carried out based on SilicoDArT and SNP data and average trait values [56]. The results were analyzed and are shown in GenStat 23.1 [57] using the QSASSOCIATION method, which uses data from a single-environment experiment to conduct a mixed-model marker–trait association analysis (sometimes called linkage disequilibrium mapping). Some kind of genetic relatedness control is required in association mapping investigations to prevent false positives. The RELATIONSHIPMODEL = eigenanalysis option was used to specify the model, which uses the most significant principal components from the molecular marker matrix to infer the underlying genetic substructure in the population [58]. The scores of the significant axes are used as covariables in the mixed model, which is essentially an approximation to the structuring of the genetic variance covariance matrix by a coefficient of coancestry matrix (kinship matrix). A false discovery rate (FDR) adjusted at 0.05 was initially used to determine the p-value threshold for declaring significant marker–trait associations. However, due to the high stringency of FDR-adjusted p-values and the potential risk of type II error, the criterion of selecting the p-values obtained within the bottom 0.1 percentile of the distribution was utilized. Thus, a threshold of p < 0.001 was used to declare significant marker–trait associations. The Benjamini–Hochberg technique was used to adjust p-values for multiple testing in order to determine the significance of the association between blackleg and SilicoDArT and SNP markers.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Friedt W. Tu J. Fu T. Academic and Economic Importance of Brassica napus Rapeseed Compendium of Plant Genomes Springer Cham, Switzerland 201810.1007/978-3-319-43694-4_1 · doi ↗
- 2Chen S. Nelson M.N. Chèver A.M. Jenczewski E. Li Z. Mason A.S. Meng J. Plummer J.A. Pradhan A. Siddique K.H.M. Trigenomic Bridges for Brassica Improvement CRC Crit. Rev. Plant Sci.20113052454710.1080/07352689.2011.615700 · doi ↗
- 3Raboanatahiry N. Li H. Yu L. Li M. Rapeseed (Brassica napus): Processing, Utilization, and Genetic Improvement Agronomy 202111177610.3390/agronomy 11091776 · doi ↗
- 4So K.K.Y. Duncan R.W. Breeding Canola (Brassica napus L.) for Protein in Feed and Food Plants 202110222010.3390/plants 1010222034686029 PMC 8539702 · doi ↗ · pubmed ↗
- 5Del Gatto A. Melilli M.G. Raccuia S.A. Pieri S. Mangoni L. Pacifico D. Signor M. Duca D. Foppa Pedretti E. Mengarelli C. A Comparative Study of Oilseed Crops (Brassica napus L. subsp. Oleifera and Brassica carinata A. Braun) in the Biodiesel Production Chain and Their Adaptability to Different Italian Areas Ind. Crops Prod.2015759810710.1016/j.indcrop.2015.04.029 · doi ↗
- 6Abbadi A. Leckband G. Rapeseed Breeding for Oil Content, Quality, and Sustainability Eur. J. Lipid Sci. Technol.20111131198120610.1002/ejlt.201100063 · doi ↗
- 7Zheng X. Koopmann B. Ulber B. von Tiedemann A. A Global Survey on Diseases and Pests in Oilseed Rape—Current Challenges and Innovative Strategies of Control Front. Agron.2020259090810.3389/fagro.2020.590908 · doi ↗
- 8Turck D. Castenmiller J. De Henauw S. Hirsch-Ernst K.I. Kearney J. Maciuk A. Mangelsdorf I. Mc Ardle H.J. Naska A. Pelaez C. Safety of Rapeseed Powder from Brassica rapa L. and Brassica napus L. as a Novel Food Pursuant to Regulation (EU) 2015/2283 EFSA J.202018 e 0619710.2903/j.efsa.2020.619732760464 PMC 7391831 · doi ↗ · pubmed ↗