Clinical Details of Low-Frequency Hearing Loss Observed in Autosomal Dominant MYO7A-Associated Hearing Loss Patients
Hiromi Koizumi, Shin-ya Nishio, Shin-ichi Usami

TL;DR
This study identifies and characterizes low-frequency hearing loss caused by MYO7A gene variants in Japanese patients, revealing progression to high-frequency hearing loss over time.
Contribution
The study provides detailed clinical insights into the progression of MYO7A-associated low-frequency hearing loss in a large patient cohort.
Findings
60 patients with MYO7A-associated low-frequency hearing loss were identified, showing initial mild-to-moderate low-frequency loss progressing to flat-type severe loss.
The c.1436T>C:p.Leu479Pro variant was identified as a founder mutation in the Japanese population through haplotype analysis.
Abstract
Background/Objectives: MYO7A is known to be the genetic cause of Usher syndrome type 1, as well as autosomal dominant and autosomal recessive non-syndromic hearing loss. In general, autosomal dominant MYO7A-associated hearing loss shows progressive high-frequency, sloping hearing loss. However, several variants are associated with low-frequency hearing loss. MYO7A-associated low-frequency hearing loss is relatively rare, and the clinical details remain unclear. Methods: A total of 18,475 Japanese patients with hearing loss were recruited. Targeted massively parallel sequencing of 158 deafness-related genes was performed, and individuals with variants related to MYO7A-associated low-frequency hearing loss were identified. Results: Among 18,475 hearing loss patients, we identified 60 patients from 44 unrelated families carrying five variants (p.[Asn140Lys; Glu1835Gln], p.Leu479Pro,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
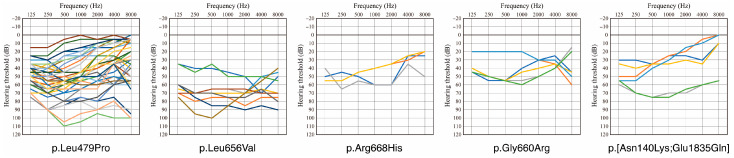 Figure 1
Figure 1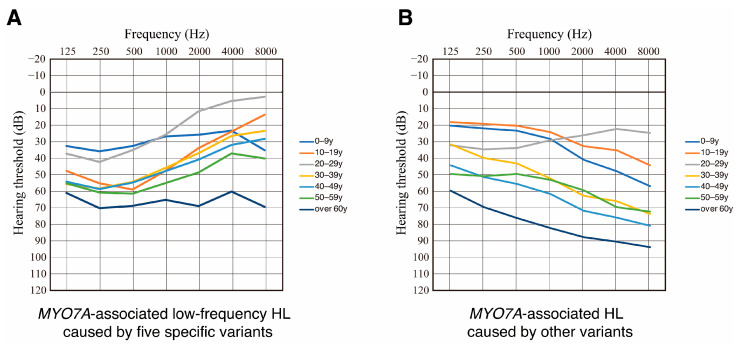 Figure 2
Figure 2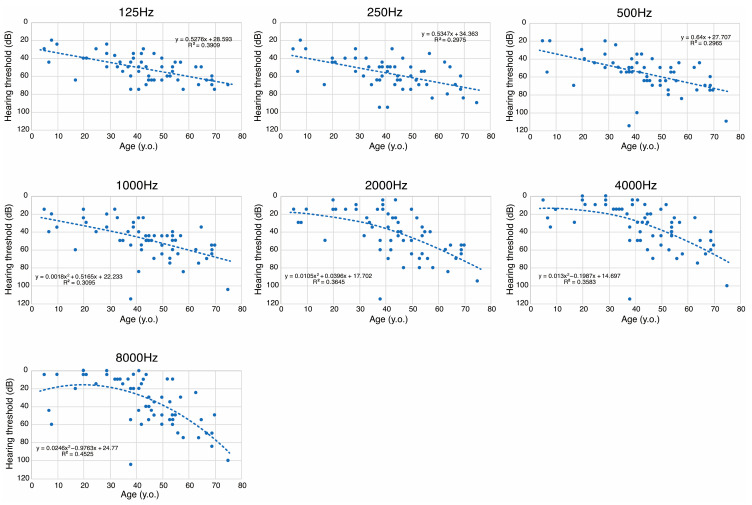 Figure 3
Figure 3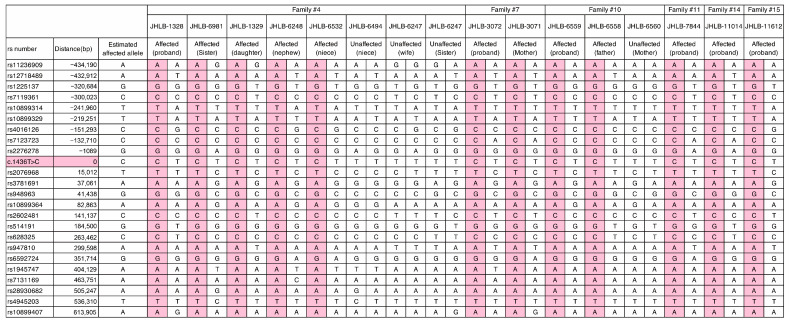 Figure 4
Figure 4- —Ministry of Health, Labor and Welfare of Japan
- —Japan Agency for Medical Research and Development (AMED)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHearing, Cochlea, Tinnitus, Genetics · Genomics and Rare Diseases · Congenital heart defects research
1. Introduction
Hearing loss (HL) is one of the most common sensory disorders, with more than 150 genes identified to date as causative of non-syndromic HL [1]. MYO7A is a frequent genetic cause of Usher syndrome type 1 (USH1B), which causes congenital severe-to-profound bilateral sensorineural HL (SNHL), prepubertal-onset retinitis pigmentosa (PR), and vestibular dysfunction [2]. In addition, MYO7A is known as a causative gene for autosomal dominant non-syndromic HL (ADNSHL, locus DFNA11) and autosomal recessive non-syndromic HL (ARNSHL, locus DFNB2) [3,4].
The MYO7A gene, initially identified as a causative gene for Usher syndrome by Weil et al. in 1995 [1], is located on chromosome 11q13.5, comprises 49 exons, and encodes the unconventional myosin VIIa. Myosin VIIa is expressed in multiple tissues, including the retina, lungs, testes, kidneys, and both the outer and inner hair cells of the inner ear [4]. Within the inner ear, myosin VIIa interacts with SANS and harmonin to form a tripartite protein complex that is critical for mechanoelectrical transduction in stereocilia [5,6,7,8,9,10]. It also plays a pivotal role in maintaining mechanical tension through tip links and facilitating the transport of protein components to the tips of stereocilia [7,10]. The integrity of hair cell stereocilia bundles depends on myosin VIIa function, and its disruption leads to disorganized stereocilia and subsequent HL [11,12].
To date, a total of 882 variants have been reported in association with MYO7A-related HL and Usher syndrome. The majority of these variants have been linked to Usher syndrome, whereas only 35 have been identified as causative of ADNSHL and 49 as causative of ARNSHL [13]. We recently described the clinical characteristics of MYO7A-related HL, including DFNA11, DFNB2, and USH1B [14]. In that study, DFNA11 cases typically showed late-onset progressive SNHL, with onset ranging from the first to the third decade. Most DFNA11 patients initially presented with mild-to-moderate high-frequency sloping SNHL, gradually worsening to a severe-to-profound flat-type configuration with age [14]. DFNB2 cases exhibited congenital or early-onset progressive SNHL. Although the age of onset was consistently within the first decade, severity and audiometric patterns varied across individuals. Around half of those over 30 years of age showed severe-to-profound SNHL, confirming a progressive course [14]. In contrast, USH1B cases uniformly demonstrated congenital or early onset severe-to-profound SNHL. All individuals over 10 years old developed retinitis pigmentosa and/or related visual symptoms [14].
As described above, most DFNA11 cases in our previous study showed high-frequency sloping SNHL. Xia et al. reported the results of a literature review on DFNA11 and found that six of 11 families with DFNA11 showed high-frequency sloping SNHL [15]. However, several patients with specific variants showed low-frequency SNHL. Low-frequency HL is a relatively rare phenotype for DFNA11, and the clinical details, including the patterns of hearing deterioration, remain unclear. We, therefore, collected patients with specific MYO7A variants consistently associated with a low-frequency HL pattern to clarify the clinical details. For this purpose, we analyzed the detailed clinical features (including the audiometric configuration and hearing deterioration at each frequency by age) of DFNA11 cases with low-frequency HL. We also performed haplotype analysis for a recurrent variant (c.1436T>C:p.Leu479Pro) identified in 29 independent families among 18,475 Japanese HL patients.
2. Materials and Methods
2.1. Subjects
A total of 18,475 HL patients from 130 institutions in Japan participated in this study. Clinical information and peripheral blood samples were obtained from each participant. Written informed consent was obtained from all participants (or guardians in the case of minors) prior to participation in the project. This study was performed in accordance with the Declaration of Helsinki. The study protocol was approved by the Shinshu University School of Medicine Ethics Committee (No. 387—4 September 2012, No. 576—2 May 2017, and No. 718—7 March 2022).
2.2. Next-Generation Sequencing and Bioinformatic Analysis
Next-generation sequencing (NGS) analysis for 158 target genes reported to cause non-syndromic HL or syndromic HL was performed. In brief, sequencing libraries were prepared with an Ion AmpliSeq Library Kit 2.0 (ThermoFisher Scientific, Waltham, MA, USA) and Ion AmpliSeq^TM^ Custom Panel (ThermoFisher Scientific) according to the manufacturer’s procedure [16]. Sequencing was performed using the Ion S5 Plus system with the Ion 540 Chip Kit and Ion 540 Kit-Chef (ThermoFisher Scientific). The sequence data were mapped against the human genome sequence (build GRCh37/hg19), and variants were picked up with the Torrent Suite software ver.5.1.6. The annotation of identified variants was performed with ANNOVAR software ver. 2020-06-08 [17]. Variants affecting the amino acid sequence (missense, nonsense, splicing, and insertion/deletion variants) were selected from the identified variants. Variants were further restricted to those with a minor allele frequency of less than 1% in control databases. As control databases, we employed the Genome Aggregation Database (https://gnomad.broadinstitute.org), ToMMo 60KJPN (https://jmorp.megabank.tohoku.ac.jp/, accessed on 10 October 2024), and 333 in-house normal-hearing Japanese controls. All variant filtering was performed with our original database software [18]. Copy number analysis was performed according to our previous report [19]. Sanger sequencing was conducted to confirm the identified variants and family segregation.
The pathogenicity of the selected variants was assessed in accordance with the American College of Medical Genetics (ACMG) standards and guidelines [20] with the ClinGen Hearing Loss Clinical Domain Working Group expert specification [21]. The variants classified as “Likely Pathogenic” or “Pathogenic” were considered to be causative variants. In addition, variants classified as being of “Uncertain Significance” were considered to be causative variants if all of the following conditions were fulfilled: (1) no other candidate variants were identified among the other 157 genes, (2) the allele frequency was extremely low (≤0.00002) in the control populations, (3) most of the in silico prediction scores supported the pathogenic impact, and (4) no contradictory evidence existed regarding the pathogenicity of the identified variant.
2.3. Clinical Evaluation
Clinical data, including sex, age at onset of HL, age and audiometric thresholds at the time of genetic testing, family history, history of tinnitus and vertigo, and hearing loss progression, were obtained through a retrospective review of medical records. Hearing thresholds were assessed using pure-tone audiometry in individuals aged over 5 years. Conditioned orientation reflex audiometry (COR), play audiometry or auditory steady-state response (ASSR) was performed in younger children. The pure-tone average (PTA) was calculated based on the hearing thresholds at four frequencies (500, 1000, 2000, and 4000 Hz). Severity of HL was categorized according to PTA values: normal (<25 dB), mild (>25 dB and ≤40 dB HL), moderate (>40 dB and ≤70 dB HL), severe (>70 dB and ≤90 dB HL), and profound (>90 dB HL). Type of HL were classified into flat, low-frequency ascending, mid-frequency U-shaped, high-frequency gently sloping, and high-frequency steeply sloping [22].
2.4. Haplotype Analysis
In this study, we identified a recurrent variant, c.1436T>C, from 29 independent families. To determine whether this variant resulted from a mutational hotspot or founder mutation, we performed haplotype analysis. The haplotype pattern within the 1 Mbp region surrounding the position of the most frequent variant, c.1436T>C, was characterized using a set of 23 tag single-nucleotide polymorphisms (SNPs). Tag SNPs were selected based on the minor allele frequency for the 1000 Genomes JPT populations using SNPinfo Web Server LD TAG SNP Selection (TagSNP) (https://snpinfo.niehs.nih.gov). SNP genotyping for haplotype analysis was performed by Sanger sequencing.
3. Results
3.1. Identified Variants and Clinical Features of MYO7A-Associated Low Frequency Hearing Loss
In this study, we evaluated the audiogram for 60 cases, including those from our previous study [14], and found that the five variants, p.[Asn140Lys; Glu1835Gln], p.Leu479Pro, p.Leu656Val, p.Gly660Arg, and p.Arg668His, were associated with low-frequency HL.
To elucidate the detailed clinical features of MYO7A-associated low-frequency HL, we selected patients carrying the above-mentioned five variants from the 18,475 HL patient cohort. As a result, we identified 60 patients from 44 unrelated families carrying these five variants associated with low-frequency HL (Table 1). The most prevalent variant was p.Leu479Pro, which was identified in 40 cases from 28 families, followed by p.Leu656Val in nine cases from nine families, p.Arg668His in five cases from three families, p.Gly660Arg in four cases from two families, and p.[Asn140Lys; Glu1835Gln] in two cases from two families. Twenty-two patients were male, and 38 were female. Most of the cases (84.1%, 37/44) were from families with an autosomal dominant family history, which was defined as one or more affected individuals among first-degree relatives. The average onset age of HL for these patients was 28.5 years (range 0 to 64 years old). Most cases (91.7%, 44/48) showed postlingual-onset HL, and only four cases (8.3%, 4/48) showed prelingual-onset HL. Most of the identified variants were only observed in Japanese patients and were not observed in a large-scale control database (gnomAD ver 4.1). On the other hand, p.Arg668His was also observed in Chinese and American patients (Table 2).
Among the 60 patients, audiometric data were available for 57 patients. Most cases showed mild-to-moderate HL: 20 patients showed mild HL (35.1%, 20/57), 29 patients showed moderate HL (51.0%, 29/57), five patients showed severe HL (8.8%, 5/57), and one patient showed profound HL. In addition, two cases showed normal hearing on PTA at the time of genetic testing. Of these 57 patients, 34 patients (59.6%) exhibited low-frequency HL, whereas 21 patients (36.8%) showed flat-type HL, and one patient each (1.8%) exhibited high-frequency HL and mid-frequency HL. Progressive HL and tinnitus were the major subjective symptoms: 70.8% of patients (34/48) were aware of their HL progression, and 61.8% of patients (34/55) had tinnitus. On the other hand, vertigo was a relatively minor symptom, with 28.6% of patients (16/56) reporting episodes of vertigo. As a limitation of this study, information on the progression of HL, tinnitus, and vertigo was based on subjective awareness, and longitudinal objective measurements would be useful to clarify the true prevalence of each symptom.
Overlapping audiograms of the patients identified in this study for each variant are shown in Figure 1. As shown in Figure 1, most individuals with mild-to-moderate HL showed a typical low-frequency HL phenotype. On the other hand, many patients with moderate-to-severe HL showed flat-type HL. Thus, we hypothesized that MYO7A-associated HL caused by these five variants initially presents as mild-to-moderate low-frequency HL that progresses to moderate-to-severe flat-type HL.
3.2. Hearing Deterioration in MYO7A-Associated Low-Frequency Hearing Loss
To elucidate hearing deterioration in patients with MYO7A-associated low-frequency HL, we evaluated the averaged hearing thresholds for each decade of age group for patients with the five specific MYO7A variants (Figure 2A). To highlight the differences in hearing deterioration between MYO7A-associated low-frequency HL and other DFNA11 cases, we also evaluated the averaged hearing thresholds for each decade of age group in DFNA11 patients with other MYO7A variants (Figure 2B). As seen in Figure 2, patients with the five specific variants showed clear low-frequency HL from the second to the sixth decade, with HL later deteriorating at higher frequencies, resulting in flat-type HL. On the other hand, patients carrying other DFNA11 variants showed high-frequency sloping HL that deteriorated across all frequencies.
We also performed a scatter plot analysis of hearing thresholds at each frequency (125 Hz, 250 Hz, 500 Hz, 1000Hz, 2000 Hz, 4000 Hz, and 8000 Hz) by age at audiometric testing (Figure 3). As shown in Figure 3, hearing levels at low frequencies showed progressive deterioration beginning in the first or second decade (>25 dB), with a linear decline at a rate of 0.5–0.6 dB/year. In contrast, hearing levels at higher frequencies remained within the normal range (<25 dB) from the first to the third decade. However, HL beginning in the fourth decade showed more rapid deterioration at higher frequencies than at lower frequencies. Thus, based on the scatter plot analysis results, we clearly demonstrated that patients with MYO7A-associated low-frequency HL initially presented with mild-to-moderate low-frequency HL that progressed with time to moderate-to-severe flat-type HL. It is noteworthy that there was a large variability in the individual progression of HL, and scatter plot analysis showed overall trends for hearing deterioration in MYO7A-associated low-frequency HL.
3.3. Haplotype Analysis for the Recurrent MYO7A Variant Identified in This Study
In this study, we identified 38 cases from 29 unrelated families carrying the same c.1436T>C:p.Leu479Pro variant. It is possible that this recurrent variant may be due to a mutational hotspot or founder mutation. To elucidate this possibility, we carried out haplotype analysis in 16 individuals from six independent families within the 1 Mbp region surrounding the position of c.1436T>C, which was characterized using a set of 23 single-nucleotide polymorphisms (SNPs) (9 sites upstream and 14 sites downstream). Figure 4 shows the haplotype patterns for the six families that carried the c.1436T>C variant. As a result, the six unrelated families were found to share the same haplotype surrounding the c.1436T>C variant, suggesting that this mutation likely occurred and spread as a founder mutation in the Japanese population.
4. Discussion
MYO7A is known to be the genetic cause of Usher syndrome type 1 (USH1B), ADNSHL (DFNA11), and ARNSHL (DFNB2) [3,4]. Most of the previously reported variants were identified from Usher syndrome patients, while DFNA11 cases were relatively rare, and details of the clinical characteristics remain unclear. DFNA11 HL is generally characterized by a high-frequency sloping audiometric configuration with progressive HL [14,23]. However, several variants have been associated with low-frequency HL.
In this study, we analyzed the detailed clinical features, especially hearing deterioration, in DFNA11 patients with low-frequency HL caused by five specific variants (p.Leu479Pro, p.Leu656Val, p.Gly660Arg, p.Arg668His, and p.[Asn140Lys; Glu1835Gln]). As a result, DFNA11 patients with these five specific variants generally presented with postlingual-onset mild-to-moderate low-frequency HL. Hearing thresholds for 125, 250, and 500 Hz deteriorated at a rate of 0.5 to 0.6 dB/year in a linear manner (Figure 3). On the other hand, hearing thresholds at 2000, 4000, and 8000 Hz were within the normal range in the first to third decades, but rapidly deteriorated after the fourth decade and finally progressed to moderate-to-severe flat-type HL (Figure 2 and Figure 3). These estimates of hearing deterioration were based on mean thresholds by decade (Figure 2), scatter plots of different patients (Figure 3), and an analysis of hearing deterioration analysis based on longitudinal observations of the same individuals will be required to clarify the variability between patients carrying each variant.
The encoded protein myosin VIIa is localized in the stereocilia of the inner and outer hair cells and plays a crucial role in sound transduction, but the underlying mechanism for different types of HL (five variants resulting in low-frequency HL, whereas other DFNA11 variants generally cause high-frequency HL) remains unclear. The myosin VIIa protein is composed of a myosin motor domain (65–741 aa), a neck domain with five IQ motifs (745–857 aa), an SAH plus short coiled-coil domain (858–935 aa), and a tail domain that contains the MyTH4 1 domain (1017–1253 aa), FERM 1 domain (1258–1602 aa), SH3 domain (1603–1672 aa), MyTH4 2 domain (1747–1896 aa), and FERM 2 domain (1902–2205 aa) [24]. In a previous report, most MYO7A variants causing DFNA11 or DFNB2 were found to be located in the motor domain, while variants responsible for USH1B were observed across all domains [25].
Among the five variants identified in this study, p.Leu479Pro, p.Leu656Val, p.Gly660Arg, and p.Arg668His are located in the motor domain. In a recent review paper, five different variants, p.Arg616Gln, which was identified from Korean patients; Arg668His, which was identified from Chinese, Japanese, and American patients; Gly671Ser, which was identified from Chinese patients; p.Gly722Arg, which was identified from American patients; and p.Arg853Cys, which was identified from German patients, were reported to be causative of DFNA11 with low-frequency HL [26]. Among them, p.Arg616Gln, Arg668His, Gly671Ser, and p.Gly722Arg are also located in the motor domain. The motor domain can be divided into the N-terminal subdomain, Upper 50 kDa subdomain, Lower 50 kDa subdomain, and converter/lever-arm region. Among the above- mentioned variants identified in this study and previous reports, p.Leu479Pro and p.Arg616Gln are localized in the Lower 50 kDa subdomain, whereas p.Leu656Val, p.Gly660Arg, p.Arg668His, Gly671Ser, and p.Gly722Arg are localized in the converter/lever-arm region. Thus, there might be a genotype-phenotype correlation between the variant localization and type of HL; however, the detailed mechanisms remain unclear. Kallman et al. reported an interesting DFNA11 family in which some patients in a specific branch of the large family showed high-frequency HL, whereas other patients showed low-frequency HL despite carrying the same MYO7A variant. They speculated that a modifier gene may be involved in these phenotypic differences; however, this modifier gene remains unclear [27]. Further studies, including the prediction of protein structure changes, will be needed to elucidate the mechanisms for the different phenotypes observed in DFNA11 patients.
Among cases of hereditary HL, low-frequency HL is known to be classically associated with genes such as the WFS1 gene [28]. In a recent review article on the monogenic causes for low-frequency non-syndromic HL, most cases of WFS1-associated HL (DFNA6/14/38), and some cases of DIAPH1-associated HL (DFNA1), MYO7A-associated HL (DFNA11), TNC-associated HL (DFNA56), and CCDC50-associated HL (DFNA44) showed low-frequency HL [26]. Based on our study, when evaluating patients with low-frequency HL, MYO7A-related HL should also be considered in the interpretation of next-generation sequencing results.
In this study, we identified 38 cases from 29 unrelated families carrying the same c.1436T>C:p.Leu479Pro variant. Recurrent pathogenic variants are generally considered to arise through two principal mechanisms: a founder effect or the presence of mutational hot spots. To distinguish between these mechanisms, haplotype analysis is a widely used and effective approach. To date, several recurrent variants have been shown to result from founder effects, including variants in CDH23 [29], MYO15A [30], and TMC1 [31]. In Japanese patients, founder mutations have also been reported in multiple genes. Haplotype analyses have suggested that p.Arg1939Gln in OTOF [32], c.211delC in KCNQ4 [33], and c.4212+1G>A in LOXHD1 [34] are likely founder variants. In contrast, recurrent variants such as c.5597C>T in TECTA [35] and p.Ala716Thr, p.Lys836Thr, and p.Glu864Lys in WFS1 [28] have been attributed to mutational hot spots. In the present study, we investigated the high prevalence of the MYO7A p.Leu479Pro variant in the Japanese population to determine whether its recurrence is attributable to a founder effect or to a mutational hot spot. Haplotype analysis revealed a shared haplotype among affected individuals from unrelated families, indicating that this variant represents a founder mutation. Further, the p.Leu479Pro variant was not observed in the gnomAD database [36] and has only reported from the Japanese population, which indicates that this variant likely first occurred in a Japanese ancestor. Interestingly, all of the patients with this variant were living on the main island of Japan (Honshu), and no patients were observed on the northern island (Hokkaido) or the southern island (Kyushu), which also supports the idea that this variant arose as a founder mutation and spread from a common ancestor.
5. Conclusions
In this study, we were able to clarify the detailed characteristics of HL for MYO7A-associated low-frequency HL in a significant number of patients. The most notable result of this study was our ability to clarify the details of hearing deterioration at each frequency. Hearing thresholds for 125, 250, and 500 Hz deteriorated at a rate of 0.5 to 0.6 dB/year in a linear manner. On the other hand, hearing thresholds for 2000, 4000, and 8000 Hz rapidly deteriorated after the fourth decade and progressed to moderate-to-severe flat-type HL. In addition, we performed haplotype analysis for the recurrent c.1436T>C:p.Leu479Pro variant identified in this study and determined that this variant was likely derived from a founder mutation that occurred in a common ancestor. The findings of this study will be beneficial in enabling more appropriate treatment for patients with MYO7A-associated low-frequency HL based on the expectation of future hearing deterioration.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Weil D. Blanchard S. Kaplan J. Guilford P. Gibson F. Walsh J. Mburu P. Varela A. Levilliers J. Weston M.D. Defective myosin VIIA gene responsible for Usher syndrome type 1B Nature 1995374606110.1038/374060 a 07870171 · doi ↗ · pubmed ↗
- 2Liu X.Z. Walsh J. Tamagawa Y. Kitamura K. Nishizawa M. Steel K.P. Brown S.D. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene Nat. Genet.199710381197126810.1038/ng 1197-2689354784 · doi ↗ · pubmed ↗
- 3Liu X.Z. Walsh J. Mburu P. Kendrick-Jones J. Cope M.J. Steel K.P. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness Nat. Genet.19971618819010.1038/ng 0697-1889171832 · doi ↗ · pubmed ↗
- 4Hasson T. Heintzelman M.B. Santos-Sacchi J. Corey D.P. Mooseker M.S. Expression in cochlea and retina of myosin VI Ia, the gene product defective in Usher syndrome type 1B Proc. Natl. Acad. Sci. USA 1995929815981910.1073/pnas.92.21.98157568224 PMC 40893 · doi ↗ · pubmed ↗
- 5Boëda B. El-Amraoui A. Bahloul A. Goodyear R. Daviet L. Blanchard S. Perfettini I. Fath K.R. Shorte S. Reiners J. Myosin VI Ia, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle EMBO J.2002216689669910.1093/emboj/cdf 68912485990 PMC 139109 · doi ↗ · pubmed ↗
- 6Bahloul A. Michel V. Hardelin J.P. Nouaille S. Hoos S. Houdusse A. England P. Petit C. Cadherin-23, myosin VI Ia and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids Hum. Mol. Genet.2010193557356510.1093/hmg/ddq 27120639393 PMC 2928128 · doi ↗ · pubmed ↗
- 7Grati M. Kachar B. Myosin VI Ia and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction Proc. Natl. Acad. Sci. USA 2011108114761148110.1073/pnas.110416110821709241 PMC 3136276 · doi ↗ · pubmed ↗
- 8Wu L. Pan L. Zhang C. Zhang M. Large protein assemblies formed by multivalent interactions between cadherin 23 and harmonin suggest a stable anchorage structure at the tip link of stereocilia J. Biol. Chem.2012287334603347110.1074/jbc.M 112.37850522879593 PMC 3460447 · doi ↗ · pubmed ↗