Integrated Computational Analysis Reveals Structurally Destabilizing Missense Variants in the PDX1 Transcription Factor
Elsadig Mohamed Ahmed

TL;DR
This study uses computational methods to identify harmful genetic variants in the PDX1 gene linked to diabetes, focusing on how these variants affect protein structure and stability.
Contribution
The study introduces an integrated computational framework to systematically assess the functional and structural impact of PDX1 missense variants.
Findings
Variants R197G, Y170N, and T151K in PDX1 show consistent predictions of pathogenicity and reduced protein stability.
Molecular dynamics simulations reveal significant structural and dynamic perturbations caused by these variants.
The findings offer a framework for prioritizing PDX1 variants for experimental validation and clinical analysis.
Abstract
Background/Objective: Pancreatic and duodenal homeobox 1 (PDX1) is a key transcription factor required for pancreatic development and maintenance of β-cell function. Genetic variants in PDX1 have been associated with monogenic forms of diabetes, including maturity-onset diabetes of the young type 4 (MODY4). However, the func-tional consequences of many reported non-synonymous single-nucleotide polymorphisms (nsSNPs) in PDX1 remain unclear. In this study, an integrated in silico approach was applied to systematically identify and characterize po-tentially deleterious nsSNPs in the PDX1 gene. Methods: Missense variants were retrieved from public databases and evaluated using multiple sequence- and structure-based prediction tools to assess functional impact, disease association, protein stability, and structural consequences. Variants considered deleterious were further examined through…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
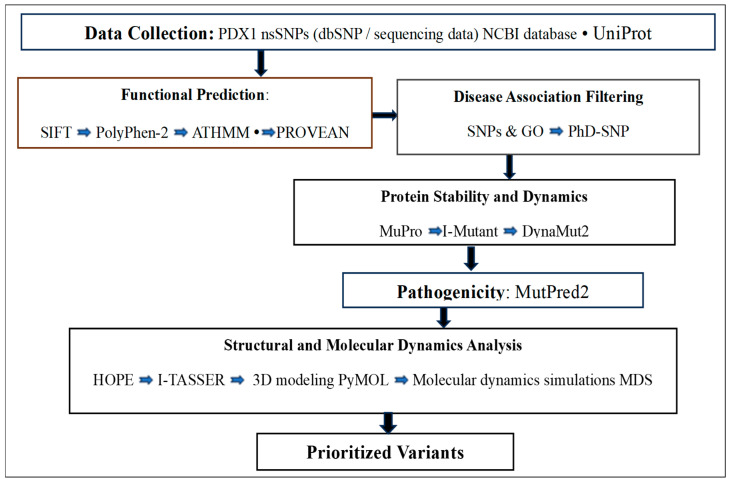 Figure 1
Figure 1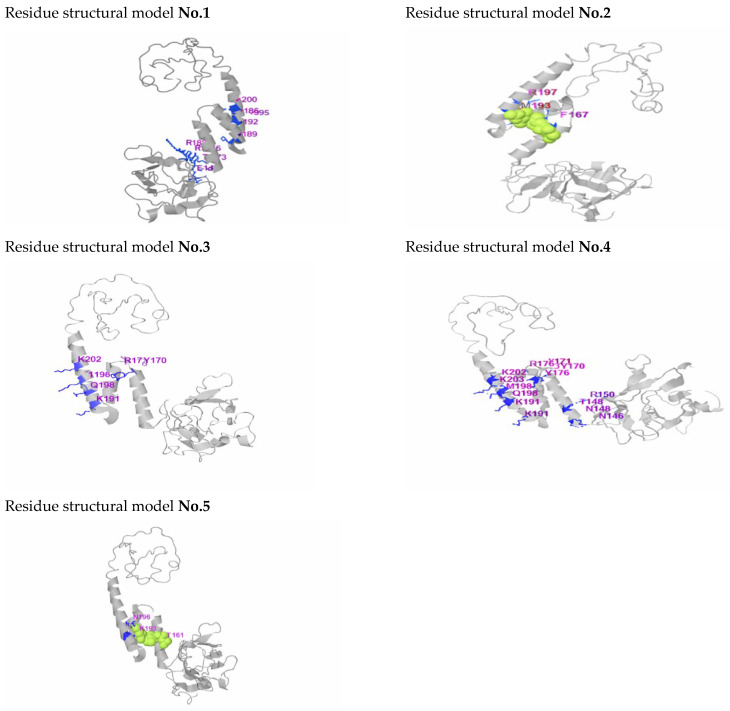 Figure 2
Figure 2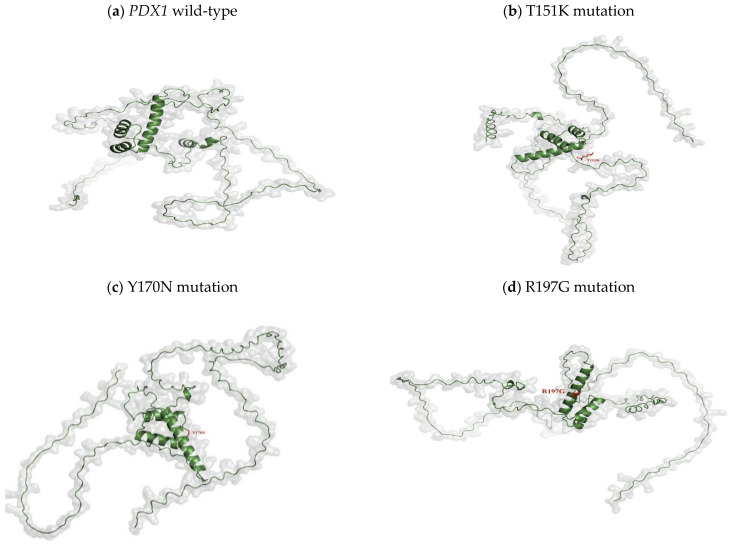 Figure 3
Figure 3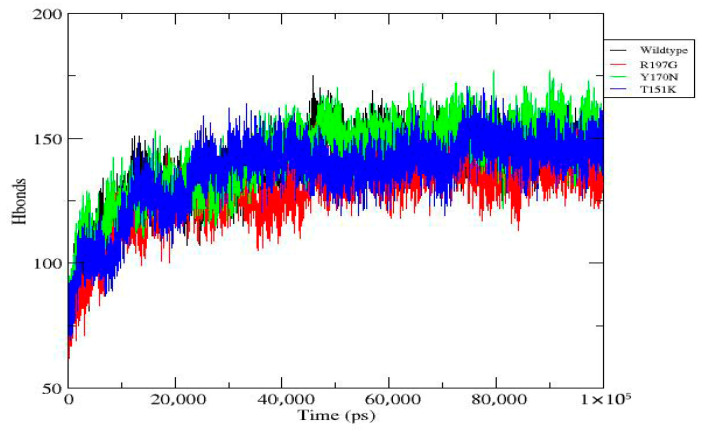 Figure 4
Figure 4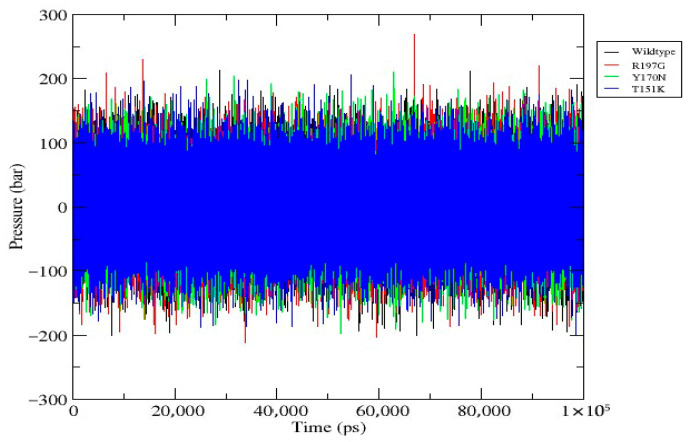 Figure 5
Figure 5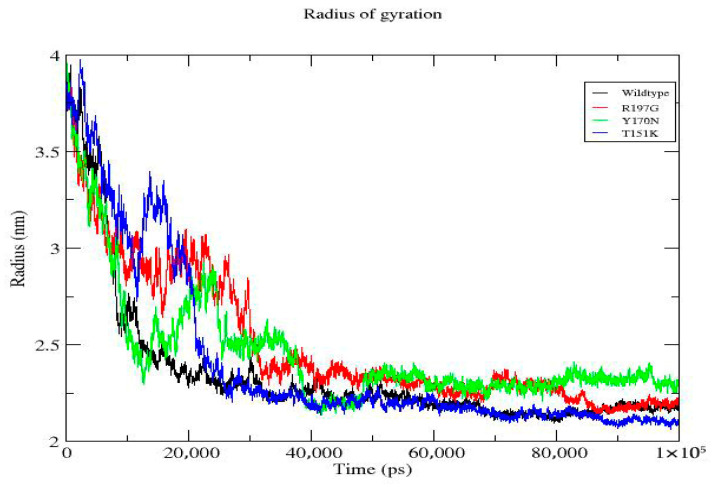 Figure 6
Figure 6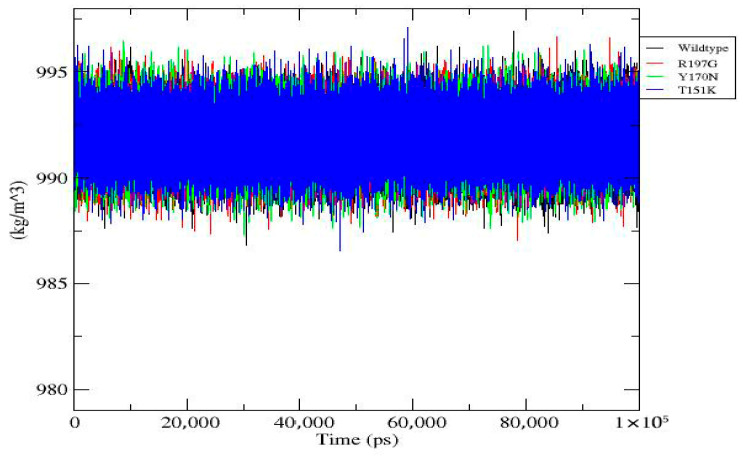 Figure 7
Figure 7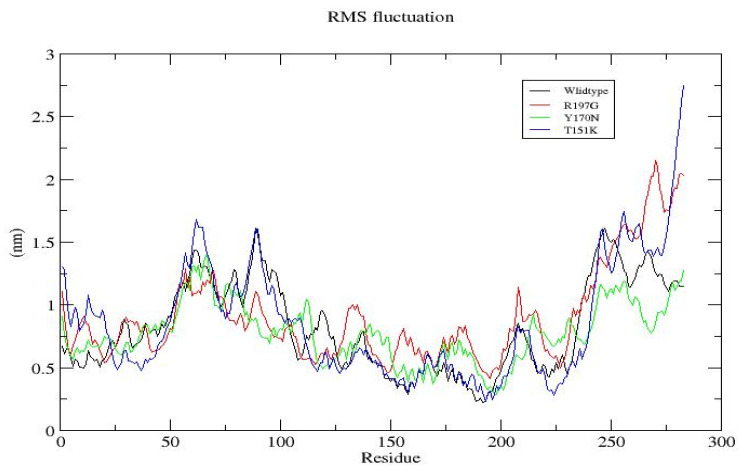 Figure 8
Figure 8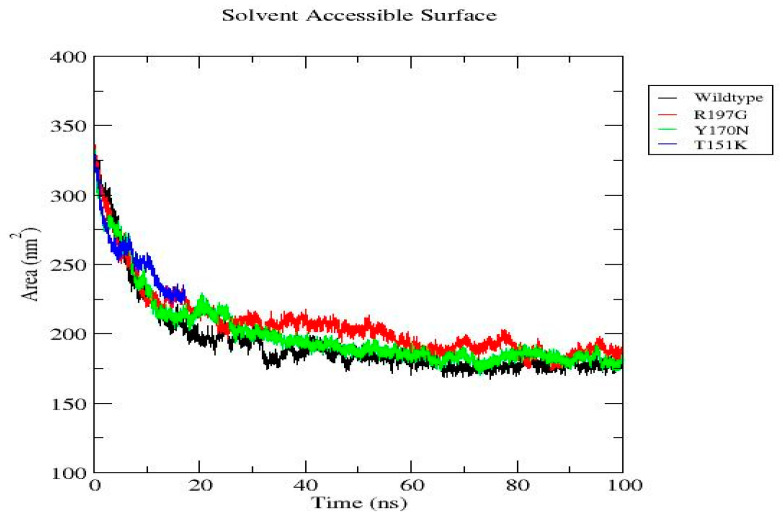 Figure 9
Figure 9 Figure 10
Figure 10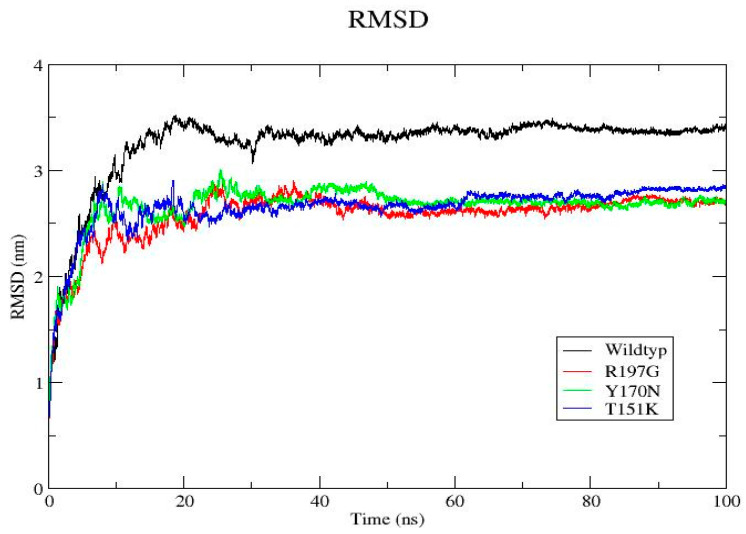 Figure 11
Figure 11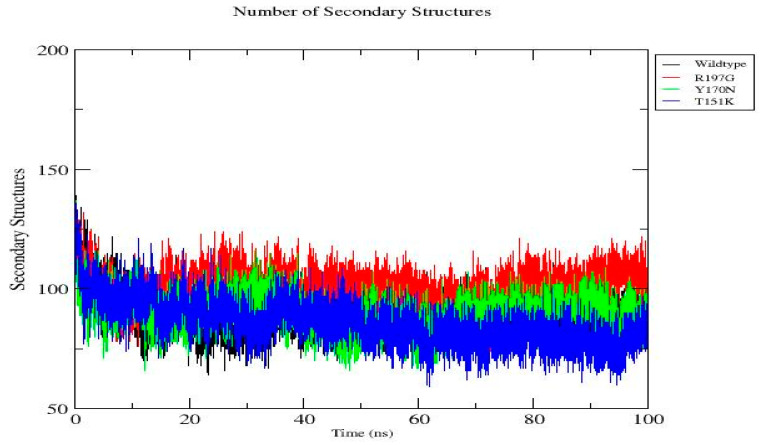 Figure 12
Figure 12Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPancreatic function and diabetes · Genomics and Rare Diseases · Genetic Associations and Epidemiology
1. Introduction
Diabetes mellitus (DM) comprises a heterogeneous group of metabolic disorders characterized by chronic hyperglycemia arising from insufficient insulin secretion, impaired insulin action, or both. Insulin is synthesized and released by pancreatic β-cells within the islets of Langerhans and is essential for maintaining glucose homeostasis. Dysregulation of insulin production or signaling leads to sustained elevations in blood glucose levels and underlies the development of diabetes and its associated complications [1,2,3,4,5,6].
Among the various forms of diabetes, type 1 and type 2 diabetes mellitus are the most prevalent. However, monogenic diabetes represents a distinct and clinically important category caused by pathogenic variants in single genes that directly affect pancreatic β-cell development, function, or survival [3,7]. These monogenic forms differ substantially from polygenic diabetes in their genetic basis, clinical presentation, and therapeutic management [3,7,8].
Maturity-onset diabetes of the young (MODY) is a well-recognized subtype of monogenic diabetes inherited in an autosomal dominant manner and typically manifests during adolescence or early adulthood, often before the age of 25 [3,7]. MODY is caused by mutations in genes that regulate key processes such as β-cell function, insulin biosynthesis, glucose sensing, and transcriptional control of pancreatic genes [7,8,9]. Although MODY is relatively rare, accurate genetic diagnosis is essential because its clinical features may overlap with type 1 or type 2 diabetes, potentially leading to misclassification and suboptimal treatment [3,9].
Multiple MODY subtypes have been identified, each associated with mutations in distinct genes critical for β-cell biology. Among these, MODY type 4 (MODY4) is caused by mutations in the pancreatic and duodenal homeobox 1 (PDX1) gene [5,8,10]. This PDX1 gene, also known as insulin promoter factor 1 (IPF1), encodes a homeodomain-containing transcription factor that plays a central role in pancreatic organogenesis and maturation of β-cells [11,12].
Located on chromosome 13q12.1, the human PDX1 gene encodes a 283–amino acid protein that regulates the transcription of several genes essential for pancreatic function, including insulin, glucokinase, and somatostatin [2,9]. During embryonic development, PDX1 is indispensable for pancreatic formation, while in adult β-cells it is required for insulin gene transcription and glucose-responsive insulin secretion [5,8].
Pathogenic variants in PDX1 give rise to a wide spectrum of clinical phenotypes depending on mutation type and zygosity. Homozygous or compound heterozygous mutations can result in pancreatic agenesis and neonatal diabetes, whereas heterozygous mutations typically cause MODY4, characterized by progressive β-cell function impairment and reduced insulin secretion [13,14,15]. These observations underscore the exceptional sensitivity of PDX1 function to alterations in its protein structure.
Among genetic variants, non-synonymous single-nucleotide polymorphisms (nsSNPs) are of particular importance because they introduce amino acid substitutions that may alter protein folding, stability, DNA-binding affinity, or transcriptional activity [16,17]. In the case of transcription factors such as PDX1, even subtle amino acid changes within functional domains can disrupt regulatory interactions and lead to impaired β-cell gene expression [18,19].
Several PDX1 nsSNPs have been experimentally validated as pathogenic and shown to compromise transcriptional activity, nuclear localization, or protein stability, thereby contributing to β-cell dysfunction and diabetes development [8,10]. However, many reported PDX1 variants remain poorly characterized, and their functional consequences are unknown. Experimental validation of each variant is costly, effective, and time-consuming. This highlights the need for effective computational strategies to prioritize potentially deleterious mutations.
Advances in bioinformatics have led to the development of numerous tools capable of predicting functional, structural, and pathogenic effects of amino acid substitutions [20,21,22,23]. Integrating these multiple predictive approaches improves confidence in variant classification and enables identification of deleterious nsSNPs that necessitate further experimental validation. Such integrated computational pipelines have been successfully applied to pathogenic PDX1 variants and have provided valuable insights into genotype–phenotype relationships [19].
Functionally, pathogenic PDX1 variants can impair DNA binding, disrupt transcriptional regulation, or interfere with nuclear localization, ultimately reducing insulin gene expression and compromising β-cell function [15,24]. From a clinical perspective, accurate identification of pathogenic PDX1 mutations is essential for distinguishing MODY4 from other diabetes subtypes, thereby enabling personalized treatment strategies, including the use of oral hypoglycemic agents instead of insulin therapy when appropriate [25].
Considering the essential function of PDX1 in pancreatic biology and the expanding spectrum of reported missense variants, a systematic and integrative characterization of deleterious nsSNPs is essential. Therefore, the present study employed an integrated in silico framework to screen, prioritize, and characterize nsSNPs in the PDX1 gene. By combining functional prediction, pathogenicity analysis, protein stability assessment, structural modeling, molecular dynamics simulations, and three-dimensional visualization. This work aims to elucidate the structural and functional consequences of deleterious PDX1 variants and provide a rational basis for their prioritization in future experimental and clinical studies. Unlike previous studies that relied primarily on sequence-based pathogenicity prediction, this work integrates multi-tool consensus filtering with structural-dynamic analysis through molecular dynamics simulations, thereby enabling mechanistic evaluation of variant-induced structural and conformational alterations within the PDX1 homeodomain.
2. Materials and Methods
2.1. Retrieval of PDX1 Gene and Protein Information
The PDX1 gene and its corresponding protein sequence were retrieved from the National Center for Biotechnology Information (NCBI) and UniProt databases. The reference protein sequence in FASTA format (283 amino acids) was used as the wild-type template for all subsequent analyses. Structural and functional annotations, including domain organization and residue numbering, were verified to ensure consistency across prediction tools. This computational analysis is consistent with previously published research [19,26].
2.2. Identification of Non-Synonymous Single-Nucleotide Polymorphisms
The nsSNPs associated with the PDX1 gene were collected from the dbSNP database. Only missense variants resulting in amino acid substitutions within the coding region were selected for further evaluation (websites were accessed on 2 July 2025). Variants lacking protein-level annotation or mapping ambiguity were excluded. Minor allele frequency (MAF) information, when available, was recorded to distinguish rare from common variants.
2.3. Functional Impact Prediction of nsSNPs
To assess the potential functional consequences of PDX1 nsSNPs, multiple sequence-based prediction tools were employed. SIFT (accessed on 18 July 2025) was used to evaluate that amino acid substitutions affect protein function based on sequence homology and evolutionary conservation [27]. PolyPhen-2 (accessed on 20 July 2025) was applied to predict the impact of substitutions on protein structure and function using physical and comparative considerations [28]. FATHMM and PROVEAN (accessed on 22 and 24 July 2025, respectively) were additionally used to strengthen prediction confidence [29,30]. Variants that are predicted as deleterious by these multiple tools will be prioritized for downstream analysis.
2.4. Disease Association Analysis
The association of nsSNPs with disease phenotypes was evaluated using SNPs & GO and PhD-SNP servers (accessed on 26 and 28 July 2025, respectively). These tools integrate sequence features, evolutionary information, and functional annotations to classify variants as disease-related or neutral. The Reliability Index (RI) provided by both tools was used to assess the confidence level of each prediction. Variants with higher RI values, indicating greater prediction reliability [31,32], will be prioritized for further analysis.
2.5. Prediction of Protein Stability Changes
The effect of selected nsSNPs on protein stability was examined using I-Mutant and MuPro servers (accessed on 30 July and 2 August 2025, respectively). These tools are used to predict changes upon amino acid substitution by estimating the direction and magnitude of free energy change (ΔΔG) [33,34]. Variants that are predicted to significantly decrease the protein stability will be considered structurally disruptive. To further evaluate the stability alterations and the local conformational effects, the DynaMut2 tool (accessed on 8 August 2025) was used to analyze changes in protein flexibility and vibrational entropy. Variants with scores ≤ −2.5 will be considered protein destabilizing [35] and then will be prioritized for the second step of analysis.
2.6. Structural and Functional Consequence Analysis
MutPred2 (accessed on 12 August 2025) was employed to investigate potential molecular mechanisms affected by the prioritized nsSNPs, including alterations in secondary structure, post-translational modification sites, and functional motifs. Variants with a score > 0.5 are predicted to be pathogenic [36,37]. The HOPE server (accessed on 28 August 2025) was used to generate detailed structural interpretations of amino acid substitutions by analyzing differences in size, charge, hydrophobicity, and residue conservation within the three-dimensional context of the protein [38]. Variants identified as most deleterious using the HOPE tool will be prioritized for downstream analysis.
2.7. Protein Structure Modeling and Visualization
The three-dimensional structure of the wild-type PDX1 protein was generated using homology modeling approaches, with I-TASSER serving as the primary modeling platform (accessed on 2 November 2025). Mutant structures were constructed by introducing selected amino acid substitutions into the wild-type model [39,40,41]. Structural visualization and comparative analysis of wild-type and mutant proteins were performed using PyMOL v.2 (accessed on 12 January 2026) to assess changes in folding, secondary structure elements, and spatial orientation of key residues [42].
2.8. Molecular Dynamics Simulation
MD simulations were performed using GROMACS v2020.6 on Google Collaboratory Pro. The MD simulations were conducted to investigate the dynamic behavior and structural stability of the wild-type PDX1 protein and the selected mutant variants. Variants will be selected based on predefined quantitative thresholds, including ≥3/4 deleterious predictions across functional tools, |ΔΔG| ≥ 1 kcal/mol destabilization, localization within conserved homeodomain regions, predicted ligand-binding residues, high evolutionary conservation, and the DynaMut2 tool criteria (|ΔΔG| ≤ −2.5 kcal/mol destabilization). Variants ranked as most deleterious under these combined criteria were therefore prioritized for MD simulations analysis. The following detailed protocol was applied.
2.8.1. Simulation Setup
In the GROMACS system, the OPLS-AA force field was applied for protein parameterization. The system was solvated using the TIP3P explicit water model. The protein was placed in a cubic simulation box with a minimum distance of 10 Å from the box edge. The system was neutralized with Na^+^ and Cl^−^ ions, and the ionic strength was adjusted to 0.15 M.
2.8.2. Energy Minimization and Equilibration
Energy minimization was carried out using the steepest descent algorithm until the maximum force was below 1000 kJ/mol/nm. Equilibration consisted of a 100 ps NVT ensemble at 300 K using a V-rescale thermostat and a 100 ps NPT ensemble at 1 bar using a Parrinello–Rahman barostat. Heavy atoms were position-restrained during equilibration.
2.8.3. Production Run and Analysis
A 100 ns production simulation was performed using a 2 fs integration time step under periodic boundary conditions. Electrostatic interactions were computed using PME with 1.0 nm cutoffs for both Coulombic and van der Waals forces. Structural analyses included RMSD, RMSF, Rg, SASA, hydrogen bond formation, and secondary structure content [43]. Comparative analyses were based on averaged mean ± SD values.
2.8.4. Secondary Structure Analysis
Secondary structure content of the PDX1 gene wild-type and mutants was quantified using DSSP analysis, and statistical values were calculated by averaging the number of secondary structural elements over the final 20 ns of each trajectory to ensure analysis of equilibrated conformations [44].
2.9. Data Integration and Variant Prioritization
The obtained results were systematically integrated to identify deleterious nsSNPs with the consistent prediction tools. Variants showing functional impact, pathogenicity, protein destabilization, and pronounced structural perturbations undergo detailed discussion. The overall workflow applied to identify and classify the potentially functional nsSNPs in the PDX1 gene is illustrated in Figure 1.
2.10. Integrated Interpretation
To ensure interpretative consistency, effect size metrics from all computational tools were standardized prior to integration. ΔΔG values were uniformly interpreted such that negative values indicate destabilization and positive values indicate stabilization. Tool-specific pathogenicity outputs were mapped into a harmonized classification framework to reduce variability across predictive approaches. Structural alterations were interpreted strictly in relation to predicted stability changes and plausible functional consequences within the PDX1 homeodomain.
3. Results
All findings presented follow the standardized interpretation framework defined in Section 2, with destabilizing variants consistently identified based on negative ΔΔG values and concordant structural perturbations.
3.1. Retrieval and Filtering of PDX1 nsSNPs
A comprehensive search of the dbSNP database identified 4434 single-nucleotide polymorphisms (SNPs) within the coding region of the PDX1 gene. Following annotation and filtering, only 403 nsSNPs resulting in amino acid substitutions were retained for further analysis. Variants with incomplete protein-level information were excluded. Minor allele frequency data indicated that the majority of retained nsSNPs were rare variants, suggesting a higher likelihood of functional relevance.
3.2. Results of Functional Impact Prediction of nsSNPs
The functional consequences of PDX1 nsSNPs were evaluated using SIFT, PolyPhen-2, FATHMM, and PROVEAN. A subset of 91 variants was consistently predicted to be deleterious across these multiple tools, indicating a high probability of functional disruption (Supplementary Table S1). These variants were prioritized for subsequent analyses. Variants predicted as tolerated or benign by most tools were considered less likely to exert significant biological effects.
3.3. Disease Association Analysis Results
Disease relevance of the prioritized nsSNPs was assessed using SNPs & GO and PhD-SNP. Several variants (55 nsSNPs) were classified as disease-associated by both tools, accompanied by moderate to high Reliability Index (RI) scores, reflecting strong confidence in the predictions (Supplementary Table S2). Variants predicted as neutral with low RI values were deprioritized.
3.4. Effect of nsSNPs on Protein Stability
Protein stability analysis using I-Mutant and MuPro revealed that most high-risk (41 nsSNPs) were associated with decreased protein stability. These findings were further supported by DynaMut2 analysis, which demonstrated alterations in protein flexibility and vibrational entropy in 27 nsSNPs (Supplementary Table S3). Collectively, these results suggest that the selected mutations destabilize the PDX1 protein structure.
3.5. Structural and Functional Consequence Analysis Results
MutPred2 analysis indicated that the prioritized 18 nsSNPs may disrupt key molecular features, including secondary structure elements, functional motifs, and potential post-translational modification sites (Table 1). Structural interpretation using the HOPE server revealed that several substitutions (7 nsSNPs) involved changes in residue size, charge, or hydrophobicity, potentially affecting protein folding and local structural integrity, particularly within conserved regions (Table 2).
3.6. Three-Dimensional Structural Modeling
The three-dimensional structure of the wild-type PDX1 protein was successfully modeled using I-TASSER. Mutant models were generated by introducing selected amino acid substitutions into the wild-type structure. The predicted ligand-binding residues for PDX1, as generated by I-TASSER, are listed for the top-ranked structural models (Table 3) and (Figure 2). Based on I-TASSER results and the seven nsSNPs identified through HOPE analysis (Table 2), the T151K, Y170N, and R197G mutations were selected for further investigation, as these missense variants are located within or in close proximity to the ligand-binding site residues. Comparative structural analysis using PyMOL (v.2) revealed noticeable conformational deviations in mutant proteins, including local structural rearrangements and altered residue orientations, especially within functionally important regions (Figure 3).
3.7. Molecular Dynamics Simulation Analysis
In order to be analyzed using MD simulations, variants were selected based on predefined quantitative thresholds. Using the combined criteria, T151K, Y170N, and R197G were ranked as the highest deleterious nsSNPs and therefore were prioritized for molecular dynamics analysis.
MD simulations were performed for the wild-type PDX1 protein and selected mutant variants over a 100 ns timeframe using GROMACS v2020.6. RMSD analysis showed that mutant proteins exhibited greater structural deviations compared to the wild type, indicating reduced stability. RMSF analysis demonstrated increased flexibility at mutation sites and neighboring residues. During the final 100 ns, the wild-type system exhibited an average RMSD of 0.23 ± 0.02 nm, whereas the R197G variant showed 0.31 ± 0.03 nm, indicating increased structural deviation. Additional parameters, including radius of gyration, solvent-accessible surface area, hydrogen bond formation, and secondary structure content, further supported the destabilizing effects of the mutations. Overall, the mutants displayed compromised structural compactness and altered dynamic behavior relative to the wild-type protein (Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11) and (Table 4).
3.8. Secondary Structure Analysis Results
Quantitative DSSP analysis was performed to evaluate the impact of mutations on the PDX1 protein secondary structure stability (Figure 12). Statistical averaging over the equilibrated portion of the trajectories (last 20 ns) revealed clear mutation-dependent differences in secondary structure content. The wild-type protein maintained a stable secondary structure with an average of (89.81 ± 5.43) structured residues, indicating preserved folding throughout the simulation. The R197G mutant exhibited a markedly higher mean secondary structure content (104.27 ± 5.59), suggesting enhanced stabilization or increased ordering relative to the wild type. In contrast, the Y170N mutant displayed a secondary structure profile comparable to the wild type (90.11 ± 6.35), albeit with increased fluctuations, indicating moderate destabilization. Notably, the T151K mutant showed the lowest average number of secondary structural elements (78.40 ± 6.11), accompanied by elevated variability, consistent with significant disruption of secondary structure integrity (Table 5). Overall, these results demonstrate that the investigated mutations exert distinct effects on protein structural stability, with T151K inducing the most pronounced destabilization, while R197G appears to enhance secondary structure formation, potentially altering the protein’s conformational landscape.
4. Discussion
PDX1 is a master transcription factor that plays a fundamental role in pancreatic development and the maintenance of mature β-cell function. Given its central regulatory role, even subtle alterations in PDX1 structure or activity can have profound effects on insulin production and glucose homeostasis. In this study, an integrated in silico framework was applied to systematically evaluate the functional and structural consequences of nsSNPs in the PDX1 gene, with the aim of identifying variants with a high likelihood of pathogenicity.
Initial sequence-based analyses revealed that a subset of nsSNPs was consistently predicted to be deleterious by multiple functional prediction tools. Concordant predictions across SIFT, PolyPhen-2, FATHMM, and PROVEAN suggest that these amino acid substitutions are evolutionarily unfavorable and likely disrupt essential functional features of the PDX1 protein. Such consistency across independent algorithms strengthens confidence in the prioritization of these variants, as reliance on a single predictive method may lead to biased or uncertain conclusions.
Disease association analysis using SNPs & GO and PhD-SNP further supported the pathogenic relevance of the prioritized variants. Variants classified as disease-associated with moderate to high Reliability Index (RI) values indicate a strong likelihood of involvement in diabetes-related phenotypes. These findings are consistent with previous reports demonstrating that pathogenic PDX1 mutations are frequently associated with monogenic forms of diabetes, particularly MODY4, through impaired transcriptional regulation of β-cell–specific genes [5,8,9,10].
Protein stability analysis provides additional insight into the molecular mechanisms underlying variant pathogenicity. Hence, most high-risk nsSNPs were predicted to decrease protein stability, as indicated by I-Mutant and MuPro analyses. Destabilization of PDX1 may result in improper folding, reduced protein half-life, or impaired interaction with DNA and transcriptional cofactors. DynaMut2 analysis further revealed alterations in protein flexibility and vibrational entropy, suggesting that these substitutions may perturb the dynamic equilibrium required for normal PDX1 function.
Structural findings were interpreted in direct alignment with standardized stability metrics to maintain internal consistency. Variants exhibiting negative ΔΔG values and concordant conformational perturbations were consistently described as destabilizing. Functional implications were discussed cautiously, particularly with respect to potential effects on DNA-binding affinity and transcriptional regulation, while avoiding extrapolation beyond computational evidence.
Structural interpretation using MutPred2 and HOPE indicated that several prioritized nsSNPs are likely to affect conserved residues, secondary structural elements, or functional motifs. Changes in amino acid size, charge, or hydrophobicity were predicted to disrupt local structural environments, potentially interfering with DNA-binding capacity or transcriptional activity. Given that PDX1 contains a highly conserved homeodomain essential for target gene recognition, structural perturbations within or near this region may have particularly severe functional consequences.
Three-dimensional structural modeling and comparative analysis of wild-type and mutant proteins R197G, Y170N, and T151K revealed notable conformational deviations in mutant models. These structural changes were further substantiated by molecular dynamics simulations, which demonstrated increased structural instability, altered flexibility, and reduced compactness in mutant proteins compared with the wild-type. Parameters such as RMSD, RMSF, radius of gyration, solvent-accessible surface area, and hydrogen bond dynamics collectively indicated compromised structural integrity and altered dynamic behavior in the mutant systems. Such dynamic instability is likely to impair the ability of PDX1 to maintain stable interactions with DNA and regulatory partners.
Although molecular dynamics simulations suggested relative structural stabilization of the R197G variant, thermodynamic stability does not necessarily equate to functional competence in DNA-binding transcription factors. Subtle alterations in residue orientation, electrostatic interactions, or DNA-contact geometry may impair transcriptional regulation despite apparent structural compactness. This interpretation is aligned with established principles in structural biology, which emphasize that protein stability and functional activity are related but distinct properties [45,46].
In this study, all molecular dynamics simulations were performed on the apo (DNA-unbound) form of PDX1 to assess intrinsic mutation-induced structural stability independent of DNA-mediated stabilization. Analyzing the apo state allows evaluation of whether mutations intrinsically destabilize the homeodomain prior to DNA engagement, which is a prerequisite for functional DNA binding. Simulating the apo form supports the physiological interpretation because DNA binding requires a pre-organized and intrinsically stable homeodomain. Any mutation that disrupts this intrinsic stability is likely to impair its biological DNA-binding function.
Taken together, missense mutations in PDX1 are linked to a wide spectrum of diabetes phenotypes, ranging from pancreatic agenesis and neonatal diabetes to MODY4 and type 2 diabetes. This broad clinical heterogeneity underscores the pronounced sensitivity of PDX1 to amino acid substitutions, particularly within functionally critical domains of the protein. Accumulating evidence indicates that even subtle alterations in the PDX1 homeodomain can result in significant functional impairment [7,8,9]. Notably, several reported variants within this region, including T151M, E164D, and E178G/K, markedly reduce transcriptional activation despite preserved nuclear localization, demonstrating that minor structural perturbations at the protein–DNA interface can profoundly disrupt downstream gene regulation [47,48].
To contextualize our computational findings within existing clinical evidence, we compared the prioritized PDX1 variants with classifications reported in ClinVar and examined consistency with published functional studies. Variants previously described as pathogenic typically affect domains critical for DNA binding or transcriptional activation. Our molecular dynamics analyses identified structural and dynamic perturbations that are mechanistically consistent with impaired transcription factor function, although we do not equate these changes directly with confirmed pathogenicity. Experimental studies done by Wang et al. (2019), published in Molecular Metabolism [47], and another study conducted by Yang et al. (2025), published in JCI Insight [48], demonstrate that missense variants in PDX1 can impair β-cell development, chromatin association, and protein–protein interactions. Collectively, our study provides structural-dynamic evidence that complements established variant interpretation frameworks, such as those proposed by the American College of Medical Genetics and Genomics [49], while emphasizing that functional validation remains essential for definitive clinical classification.
From a clinical standpoint, the identification and characterization of structurally and functionally disruptive PDX1 variants have important implications for the diagnosis and management of monogenic diabetes. Accurate interpretation of pathogenic variants can aid in distinguishing MODY4 from other forms of diabetes, thereby informing personalized treatment strategies and enabling appropriate genetic counseling. Furthermore, computational prioritization of high-risk missense variants represents a low-cost, effective approach for narrowing candidate mutations, guiding experimental validation, and focusing clinical attention on only variants that are most likely to exert significant biological and clinical effects.
Inclusively, this study demonstrates the value of integrating sequence-based pathogenicity prediction, structural modeling, and molecular dynamics simulations to systematically evaluate the functional impact of PDX1 nsSNPs. By combining these complementary in silico approaches, our study provides mechanistic insights into how specific missense variants may destabilize PDX1 structure, alter its dynamic behavior, and impair transcriptional function. Such structural and functional perturbations offer a plausible molecular basis for β-cell dysfunction and the development of diabetes. Collectively, these findings refine current understanding of PDX1 variants’ pathogenicity and establish a rational framework for prioritizing candidate mutations for experimental validation, while underscoring the expanding role of computational analyses in genetic variant interpretation and precision medicine.
5. Conclusions
The present study employed a comprehensive computational strategy to investigate the structural and functional consequences of non-synonymous SNPs in the PDX1 gene. By integrating multiple bioinformatics tools with MD simulations, several high-risk variants were identified that are likely to impair protein stability, structural integrity, and dynamic behavior. These alterations may compromise the transcriptional regulatory functions of PDX1 and contribute to β-cell dysfunction underlying MODY4 and related diabetic phenotypes. These findings underscore the value of integrated in silico analyses for prioritizing pathogenic variants and enhancing understanding of genotype–phenotype relationships. Future experimental and clinical studies are required to validate the predicted effects of the identified variants and to explore their potential implications for genetic diagnosis and personalized management of monogenic diabetes.
6. Study Limitations
While integrated computational approaches enhance predictive robustness, the functional consequences of identified PDX1 variants remain theoretical and require experimental validation through biochemical and cellular assays.
Although this study provides comprehensive insights into the structural and functional consequences of PDX1 missense variants, several limitations should be acknowledged. First, the analyses were based exclusively on computational predictions and MD simulations, which, despite their robustness, cannot fully capture the complexity of intracellular environments, post-translational regulation, or tissue-specific effects. Second, the structural models were generated using homology-based approaches, which may not fully reflect native conformations, particularly in flexible or intrinsically disordered regions of the protein. Third, population-level clinical data correlating specific variants with phenotypic severity were not incorporated, limiting direct genotype–phenotype inference. Consequently, the predicted pathogenic effects should be interpreted as hypotheses that require experimental validation using cellular, biochemical, or patient-derived models.
To address this limitation, we propose the following future analyses: protein–DNA docking of wild-type and mutant PDX1 homeodomains with insulin promoter DNA using established docking platforms (e.g., HADDOCK or similar protein–DNA docking tools). Comparative binding energy estimation to quantify mutation-induced changes in DNA-binding affinity. MD simulations of the protein–DNA complex to evaluate interface stability, hydrogen bond persistence, and recognition helix positioning.
7. Future Perspectives
Future studies should focus on experimental validation of the prioritized PDX1 variants using in vitro and in vivo systems to confirm their effects on protein stability, DNA-binding affinity, transcriptional activity, and β-cell function. Integration of functional assays with patient clinical data and population-scale genomic datasets will further enhance variant classification and clinical relevance. Additionally, extending MD simulations to longer timescales and incorporating protein–DNA or protein–cofactor complexes may provide deeper insight into regulatory mechanisms disrupted by pathogenic variants. From a translational perspective, systematic characterization of PDX1 mutations could improve genetic diagnosis of MODY4 and support precision medicine approaches by informing prognosis and therapeutic decision-making.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shoily S.S. Ahsan T. Fatema K. Sajib A.A. Common genetic variants and pathways in diabetes and associated complications and vulnerability of populations with different ethnic origins Sci. Rep.202111750410.1038/s 41598-021-86801-233820928 PMC 8021559 · doi ↗ · pubmed ↗
- 2Zhang Y. Fang X. Wei J. Miao R. Wu H. Ma K. Tian J. PDX-1: A promising therapeutic target to reverse diabetes Biomolecules 202212178510.3390/biom 1212178536551213 PMC 9775243 · doi ↗ · pubmed ↗
- 3Bazzazzadehgan S. Shariat-Madar Z. Mahdi F. Distinct Roles of Common Genetic Variants and Their Contributions to Diabetes: MODY and Uncontrolled T 2DM Biomolecules 20251541410.3390/biom 1503041440149950 PMC 11940602 · doi ↗ · pubmed ↗
- 4Sami A. Javed A. Ozsahin D.U. Ozsahin I. Muhammad K. Waheed Y. Genetics of diabetes and its complications: A comprehensive review Diabetol. Metab. Syndr.20251718510.1186/s 13098-025-01748-y 40452079 PMC 12128284 · doi ↗ · pubmed ↗
- 5Ebrahim N. Shakirova K. Dashinimaev E. PDX 1 is the cornerstone of pancreatic β-cell functions and identity Front. Mol. Biosci.20229109175710.3389/fmolb.2022.109175736589234 PMC 9798421 · doi ↗ · pubmed ↗
- 6De la Cruz-Concepción B. Flores-Cortez Y.A. Barragán-Bonilla M.I. Mendoza-Bello J.M. Espinoza-Rojo M. Insulin: A connection between pancreatic β cells and the hypothalamus World J. Diabetes 2023147610.4239/wjd.v 14.i 2.7636926659 PMC 10011898 · doi ↗ · pubmed ↗
- 7Schwitzgebel V.M. Many faces of monogenic diabetes J. Diabetes Investig.2014512113310.1111/jdi.12197 PMC 402357224843749 · doi ↗ · pubmed ↗
- 8Abreu G.d.M. Tarantino R.M. da Fonseca A.C.P. de Souza R.B. Soares C.A. Cabello P.H. Rodacki M. Zajdenverg L. Zembrzuski V.M. Campos M.Jr. PDX 1-MODY: A rare missense mutation as a cause of monogenic diabetes Eur. J. Med. Genet.20216410419410.1016/j.ejmg.2021.10419433746035 · doi ↗ · pubmed ↗